Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
A BRIEF HISTORY
HUMAN DNA AND RNA TUMOR VIRUSES
VIRAL ONCOGENES AND THEIR...
CONTROL OF VIRAL ONCOGENE...
VIRAL NONCODING RNAs AND...
VIRAL ONCOPROTEINS AND CELLULAR...
REMARKS
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):730-755. doi:10.7150/ijbs.6.730 This issue Cite
Review
Viral Oncogenes, Noncoding RNAs, and RNA Splicing in Human Tumor Viruses
Zhi-Ming Zheng ![]()
Tumor Virus RNA Biology Laboratory, HIV and AIDS Malignancy Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
Received 2010-10-31; Accepted 2010-11-27; Published 2010-12-1
Abstract
Viral oncogenes are responsible for oncogenesis resulting from persistent virus infection. Although different human tumor viruses express different viral oncogenes and induce different tumors, their oncoproteins often target similar sets of cellular tumor suppressors or signal pathways to immortalize and/or transform infected cells. Expression of the viral E6 and E7 oncogenes in papillomavirus, E1A and E1B oncogenes in adenovirus, large T and small t antigen in polyomavirus, and Tax oncogene in HTLV-1 are regulated by alternative RNA splicing. However, this regulation is only partially understood. DNA tumor viruses also encode noncoding RNAs, including viral microRNAs, that disturb normal cell functions. Among the determined viral microRNA precursors, EBV encodes 25 from two major clusters (BART and BHRF1), KSHV encodes 12 from a latent region, human polyomavirus MCV produce only one microRNA from the late region antisense to early transcripts, but HPVs appears to produce no viral microRNAs.
Keywords: Human papillomaviruses, Epstein-Barr virus, Kaposi sarcoma-associated herpesvirus, adenovirus, polyomavirus, human T-cell leukemia virus, viral noncoding RNA, viral microRNA, RNA splicing
A BRIEF HISTORY
In 1909, a farmer brought Dr. Francis Peyton Rous, a junior faculty member then at Rockefeller University, a hen that had a breast tumor. Rous performed an autopsy, extracted tumor cells, and injected the cells into other hens, which then developed sarcoma [1]. This was the first experimental proof of an infectious etiologic agent of cancer, and the chicken sarcoma-inducing RNA virus was subsequently named the Rous sarcoma virus. After a half-century debate on whether viruses truly cause cancer, Rous was eventually awarded the Nobel Prize in Medicine and Physiology in 1966 for his discovery of tumor-inducing viruses. It is now estimated that 20%-25% of human cancers worldwide have a known viral etiology [2].
Early pioneering efforts on tumor-inducing viruses were mainly focused on avian and small-animal retroviruses, a group of RNA viruses containing an RNA-dependent DNA polymerase (reverse transcriptase), and it was thought that there were no similar viruses in humans. Demonstration in 1980 of the first human retrovirus, human T-cell leukemia virus type 1 (HTLV-1), which causes adult T-cell leukemia, was therefore a landmark achievement [3-5]. Later, HTLV-2, which is far less pathogenic than HTLV-1, was isolated from a hairy T-cell leukemia [6], but soon was demonstrated not to be the agent of the malignant hematological disease. HTLV-3 [7] and -4 [8] have been discovered recently as new members of the HTLV family in central Africa, but their association with human diseases remains unclear [9].
Another line of investigation was to determine whether human DNA viruses play an etiological role in human cancers similar to the role of animal DNA viruses, several of which can easily transform rodent cells in culture and induce tumors when injected into animals. This effort initially led to associate herpes simplex virus type 2 (HSV2) with the development of cervical cancer [10,11], but subsequently human papillomaviruses (HPVs) were proven to be the causative agent in the development of cervical cancer [12]. Dr. Harald zur Hausen of Germany won the Nobel Prize in 2008 for his discovery in 1983 that HPVs cause cervical cancer.
HUMAN DNA AND RNA TUMOR VIRUSES
Tumor viruses can be classified into two groups based on their genetic material, as summarized in Table 1. Cancer-causing DNA tumor viruses and RNA-containing retroviruses have been extensively investigated, and this review will be focused more on human DNA and RNA tumor viruses, instead of other animal tumor viruses.
Human oncogenic viruses.
1. Human DNA tumor viruses
HPVs
High-risk or oncogenic HPVs are etiological agents of cervical cancer. Among the high-risk HPVs, HPV16 and HPV18 are the principal causes of cervical cancer as well as several other tumor types [13]. A characteristic of infection by these HPVs is that the viral genomes are commonly integrated into the cancer cell genome. Two principal viral oncoproteins involved in cervical carcinogenesis are E6 and E7, which destabilize, respectively, two cellular tumor suppressors, p53 and pRb [14]. HPVs are transmitted primarily through sexual contact, and, as human cancer viruses, have been the best studied of the tumor viruses. The US Food and Drug Administration (FDA) in June 2006 approved Gardasil, the first cancer vaccine, for use in females 9-26 years of age to prevent cervical cancer, precancerous genital lesions, and genital warts caused by HPV6, HPV11, HPV16, and HPV18 [15].
Epstein-Barr virus (EBV)
EBV primarily causes infectious mononucleosis, but also contributes to the pathogenesis of four human tumors: the African form of Burkitt lymphoma, B-cell lymphomas in individuals with immunosuppression, nasopharyngeal carcinoma (NPC) in southeastern Asia, and some kinds of Hodgkin disease. EBV infects B lymphocytes, but does not replicate within the B cells; instead, it transforms them into lymphoblasts, which have an indefinite life span, rendering these cells immortal. EBV encodes a viral oncogene, LMP1 (latent membrane protein-1 or BNLF1). LMP1 is expressed in EBV-associated lymphoma and is essential for B-cell transformation and for disruption of cellular signal transduction [16-18]. Although the EBV nuclear antigen 1 (EBNA1) is one of the earliest viral proteins expressed after infection and is the only latent protein consistently expressed in viral-associated tumors, recent results indicate that EBNA1 is not a viral oncoprotein [19,20]. BARF1 (BamHI-A reading frame-1) is also an early gene but is expressed as a latent gene in most NPCs [21]. Recent studies have suggested that BARF1 may have an important role in NPC oncogenesis [22,23].
Kaposi sarcoma-associated herpesvirus (KSHV)
KSHV or human herpesvirus 8 (HHV8) infection is associated with all forms of Kaposi sarcoma, primary effusion lymphoma or body cavity-based B-cell lymphoma, and multicentric Castleman disease. KSHV encodes a viral G protein-coupled receptor (vGPCR) that presumably functions as a viral oncogene in immortalization of human endothelial cells and induction of angioproliferative tumors [24,25]. KSHV vGPCR is not a split gene and thus has no RNA splicing in its expression.
Human polyomaviruses
The most important member in this family is simian virus 40 (SV40), one of the most common latent viruses of rhesus monkeys. For decades, SV40 which does not infect human has attracted the interest of many investigators in the field of cancer research because of its characteristic tumor antigens (T antigens) that can cause malignant cell transformation. Although two human polyomaviruses, BK virus and JC virus, have been described as oncogenic in rodents and nonhuman primates, whether these two viruses have any roles in human cancer is not clear. Recently, a new human polyomavirus, Merkel cell polyomavirus (MCV), was discovered in ~80% of Merkel cell carcinomas (MCCs) [26]. An established MCC cell line contains monoclonal MCV DNA integration. The integrated MCV DNA encodes a mutant T antigen that prevents autoactivation of integrated virus replication [27]. Interestingly, MCV and two new human polyomaviruses, polyomavirus-6 (HPyV6) and HPyV7, appear to inhabit healthy human skin with 40% detection rate by a rolling cycle amplification technique [28] and approximately 88% of adult subjects without MCC were MCV seropositive [29].
Human adenoviruses
Human adenoviruses are a group of small DNA viruses that commonly cause respiratory infections. Human adenoviruses have not been linked to any human cancer, but some serotypes, such as adenovirus types 2, 5, 12, 18, and 31, are capable of transforming rodent cells in culture and inducing tumors in hamsters or rats. Two viral oncogenes, E1A and E1B, have been identified as responsible for the adenovirus tumorigenicity and thus have served as useful tools for studying many important cellular processes in tumor biology [30].
Hepatitis B virus (HBV)
HBV is endemic in Southeast Asia and sub-Saharan Africa. Epidemiological observations and experimental evidence in animal models have established a clear association between HBV infection and liver cancer. Although the precise role of HBV in causing liver cancer is not yet understood, some compelling evidence suggests that the HBx gene could be a viral oncogene [31], as its protein product can disrupt signal transduction and deregulates cell growth [32]. Recent studies indicate that HBx functions through the inhibition of proteasome activities to enhance HBV replication in vivo [33]. HBx also binds to and enhances the enzymatic activity of phosphatidylinositol 3-kinase class III, an enzyme critical for the initiation of autophagy [34]. HBx is not a split gene and thus there is no RNA splicing in its expression.
2. Human RNA tumor viruses
HTLV-1
HTLV-1-associated adult T-cell leukemia/lymphoma is endemic in the southern islands of Japan, the Caribbean basin, and South Africa. HTLV-1 infects CD4+ T lymphocytes, and infected T cells can be transmitted via sexual intercourse, blood transfusion, or breast feeding. Only about 1 percent of infected individuals will develop leukemia, and then only after a period of 20 to 30 years of asymptomatic infection [35].
All members of the HTLV family differ from other oncogenic retroviruses in that they do not contain viral homologues of cellular proto-oncogenes and do not integrate into specific sites of the human genome to disrupt proto-oncogenes. Although the mechanism of transformation is not clear, the viral oncoprotein Tax, which promotes transcription and cell cycle progression, may be involved in setting up an autocrine (self-stimulating) loop that causes continuous proliferation of infected T cells [36].
Xenotropic murine leukemia virus-related virus (XMRV) in human prostate cancer
Recently, Dong et al claimed that they discovered XMRV as a new human retrovirus associated with prostate cancer. XMRV was isolated from prostate cancer tissue from patients homozygous for reduced enzyme activity of RNase L due to a single amino acid substitution and is susceptible to inhibition by interferon [37]. XMRV infection might be also associated with chronic fatigue syndrome [38-40]. XPR1 (xenotropic and polytropic retrovirus receptor 1) is the receptor for XMRV infection [41]. However, recent reports from other groups appear controversial for a possible role of XMRV in prostate cancer and in chronic fatigue syndrome [42-47].
Hepatitis C virus (HCV)
HCV belongs to the flavivirus family. Its plus-strand RNA genome carries a long open reading frame (ORF) encoding a polyprotein precursor of 3010 amino acids, which can be cleaved into ten different proteins (three structural and seven nonstructural). Although HCV oncogenesis is not well understood, persistent HCV infection is a prerequisite for the development of HCV-associated liver cancer [48]. There are no oncogenes in the viral genome, but liver-specific miR-122 is required for HCV replication [49].
VIRAL ONCOGENES AND THEIR FUNCTIONS
E6 and E7 oncogenes in high-risk HPVs
HPV E6 and E7 from high-risk HPV types have the capacity to immortalize and transform keratinocytes and epithelial cells. Low-risk or non-oncogenic HPV E6 and E7, however, lack such biological activity [50-56]. Biochemically, high-risk E6 but not low-risk E6 interacts with E6AP and the tumor suppressor protein p53 to induce ubiquitination-mediated degradation of p53[57-59]. An E6 F47R mutant is defective for polyubiquitination and degradation of p53 [60,61]. High-risk E7 and not low-risk E7 interacts with the pRb tumor suppressor protein via the LXCXE motif in the E7 CR2 domain to promote cell cycle progression [58,59,62-64]. Thus, interaction with cellular tumor suppressor proteins and perturbation of normal cell cycle control by high-risk E6 and E7 are believed to be the most important influence for malignant conversion [65-67]. In this regard, E6 from HPV16 (16E6), a high-risk type, binds E6AP more strongly and drives the degradation of p53 more efficiently than 18E6 [59]. In contrast, low-risk 11E6 has minimal binding affinity for E6AP [68] and influences the degradation of p53 only weakly in vivo [69]. Under hypoxic conditions, high-risk E6 also inactivates the CYLD tumor suppressor through interactions with CYLD deubiquitinase to allow unrestricted activation of NF-κB [70].
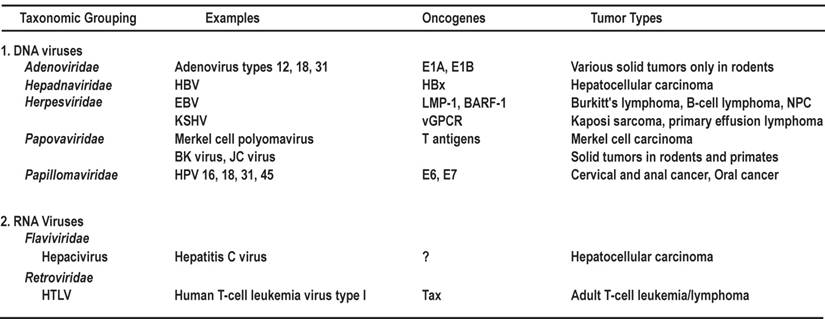
The full-length oncoprotein E6 is a basic nuclear protein (~18 kDa) composed of ~150 amino acid (aa) residues. Similar to E6 proteins encoded by other papillomaviruses, the 16E6 which has been extensively analyzed, contains four zinc-binding motifs (Cys-X-X-Cys) and forms two Cys/Cys fingers that bind zinc directly [71]. 16E6 also contains a PDZ domain-binding motif at its C-terminal extremity [72,73] and three nuclear localization signals (NLSs) [74] (Fig. 1A). Besides the ability to immortalize and transform cells and induce p53 degradation, 16E6 is known to be functionally involved in regulating gene transcription [75,76]. In addition to its effect on p53, 16E6 can interact with other transcription factors and coactivators, including p300/CBP [77,78], IRF-3 [79], and c-Myc [80]. The 16E6-cMyc interaction induces transcription of h-TERT to promote cell immortalization [81,82]. In addition, 16E6 is an RNA binding protein and interacts with cellular splicing factors and RNA via its C-terminal NLS3 to regulate splicing of E6E7 bicistronic RNAs [83]. The multifunctional activity of 16E6 is not restricted to the nucleus, because it can act as a regulator of signal transduction through interacting with cytoplasmic E6BP (Erc55) [84], E6TP [85-87], paxillin [88], TNFR1 [89], protein tyrosine phosphatase H1 [90], and PDZ proteins such as SAP97/hDlg [91,92] and MAGI-1 [73]. Other E6 partner proteins have been summarized recently in a detailed review [14]. Taken together, these interactions suggest that 16E6 and presumably other high-risk E6s can be regarded as multifaceted viral proteins with characteristic and distinct activities in the nucleus and cytoplasm of the cells they infect.
Schematic structures of oncoproteins E6 and E7. A. Protein structure and functions of HPV16 E6. Four zinc-binding motifs are shown as grey boxes. The F47 position in E6 appears to be responsible for p53 destabilization [61]. Nuclear localization signals (NLS) [74] and the PDZ-binding domain in the protein are indicated by underlines. B. Protein structure and functions of HPV16 E7. Relative locations of the regions with sequence motifs similar to a portion of conserved region 1 (CR1) and the entire CR2 of adenovirus E1A are shown with the pRB-binding site LXCXE in the CR2. Grey boxes indicate zinc-binding motifs. CKII, casein kinase II phosphorylation sites. Arrows in this figure and the following figures for individual proteins indicate functions of the protein, not its domains or motifs.
The full-length oncoprotein E7 is a nuclear protein containing ~100 aa residues. The N-terminus of E7 contains sequence similarity to a portion of CR1 and the entire CR2 of adenovirus E1A and related sequences in SV40 T antigen. Oncogenic E7 binds pRB and the related pocket proteins p107 and p130 with high affinity via the LXCXE motif in CR2 (Fig. 1B), whereas low-risk or non-oncogenic E7 binds pRB with much lower efficiency. Oncogenic E7 induces the degradation of pRB by interacting with the cullin 2 ubiquitin ligase complex [93]. The C-terminus of E7 may be involved in zinc-binding [94]. E7 contains a nuclear localization signal in the N-terminal domain (aa 1-37) [95]. In addition to its cellular transformation activities, oncogenic E7 also plays a role in the viral life cycle [96] and affects many other cellular activities in HPV-infected cells. E7 dysregulates the cell cycle by stabilizing p21 [97,98] and upregulating p16 expression [99]. Oncogenic E7 induces mitotic defects and aneuploidy by inducing centrosome abnormalities through its association with the centrosomal regulator γ-tubulin; this inhibits γ-tubulin recruitment to the centrosome [100] and leads to chromosomal instability. More recently, HPV16 E7 expression was shown to complement the requirement of RKO colorectal cancer cells for CDK6, ERBB3, FYN, AAK1, and TSSK2 for cell survival [101]. As both high-risk and low-risk HPV E7s interact with p300 [102], PCAF [103], steroid receptor coactivator 1 [104], and p600 [105], these interactions do not appear to be sufficient to mediate the transformation function of HPV E7. Other E7 partner proteins have been summarized in two recent reviews [14,106].
E1A and E1B in human adenoviruses
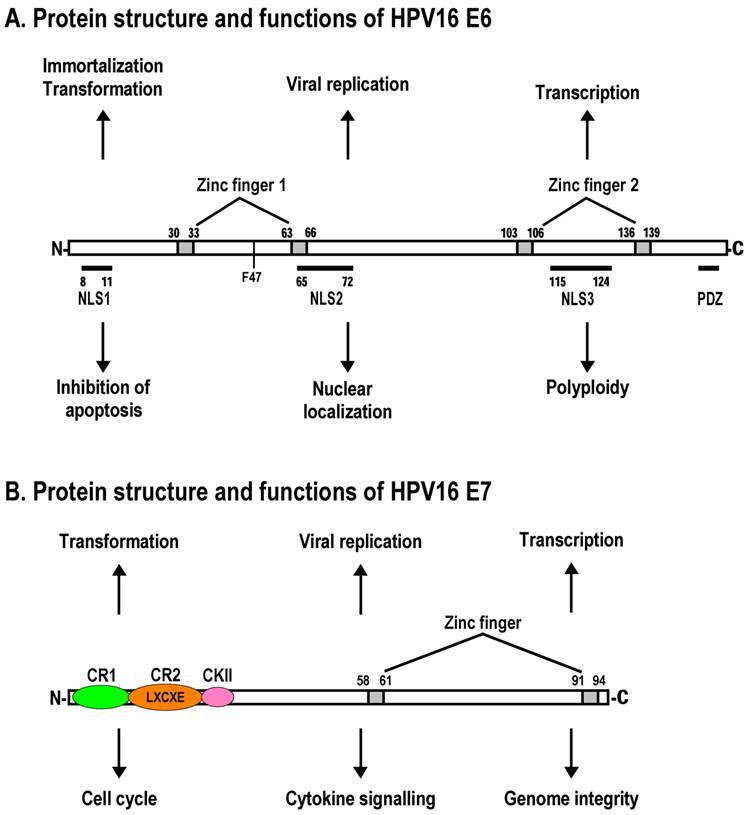
The adenovirus genome encodes two viral oncogenes, E1A and E1B, positioned side-by-side in the left 11.2% of the genome. After an adenovirus infects a human cell, the first viral proteins that are synthesized are products of the E1A region. The full-length E1A protein (289R) is a nuclear protein consisting of 289 aa residues and has four conserved regions: CR1 at the N-terminus, CR2 and CR3 in the middle, and CR4 at the C-terminus (Fig. 2A).
Schematic structures of adenovirus E1A protein and mRNAs. A. Structure of E1A protein (full-length 289R variant) and its biological functions. Four conserved regions (CR1-CR4) in E1A and mapped domains in E1A are diagramed [30]. B. RNA structure and alternative spliced species of E1A pre-mRNA. A bidirectional splicing enhancer (BSE) is shown in exon 2 in green, and cellular splicing factors or regulators that control selection of each splice site are indicated by arrows. The panel is modified from reference [269], with permission. Dotted lines indicate splicing directions.
The expression of a functional full-length E1A is necessary for the transactivation of the other adenovirus early genes, including E1B. E1A also expresses a truncated form or small E1A with 243 aa residues that lacks a CR3 region due to alternative RNA splicing and thus lacks the transactivation activity. Both E1A proteins (289R and 243R) are most abundant during early viral infection and display similar functions in the induction of cellular DNA synthesis, inactivation of pRB (equivalent to the HPV E7 oncoprotein), stimulation of cellular transformation, and promotion of apoptosis through protein-protein interactions. E1A CR2 binds pRB and the related pocket proteins p107 and p130 with high affinity, and together with a lower-affinity binding region within CR1 displaces pRB proteins from E2F transcription factors, resulting in derepression of cell cycle genes. The N-terminus and residues in CR1 which are required for E1A transforming activity bind to p300/CBP, PCAF, GCN5, and p400 to regulate chromatin structure. Recent studies have shown that the E1A-p300/CBP interaction reduces acetylation of cellular histone H3 lysine 18 (H3K18), because CBP and p300 are required for acetylation at H3K18 [107]. E1A binds transiently and in a time-dependent manner to promoters of a large number of genes involved in cell cycle and growth, antiviral activity, and development and differentiation, but this temporal order of E1A binding requires its interactions with p300/CBP and pRB proteins [108].
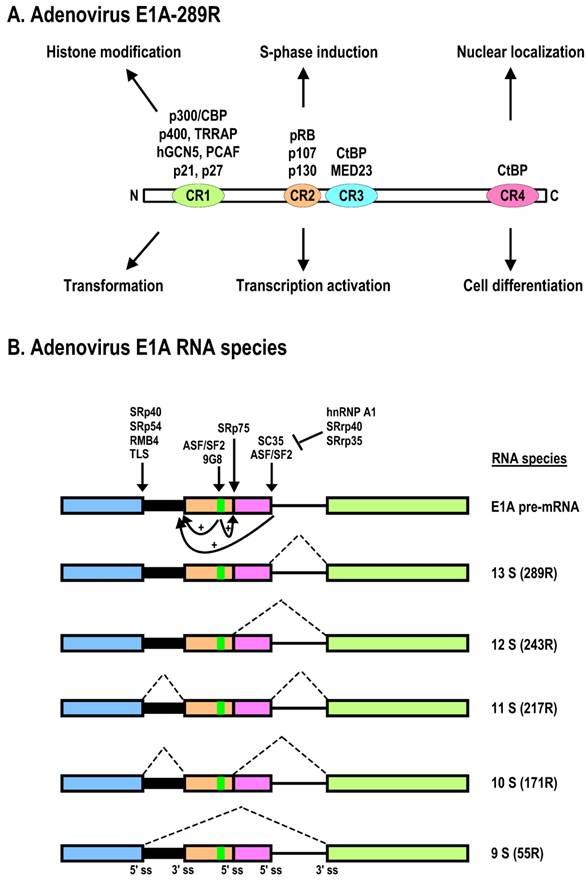
The E1B gene encodes two major proteins, 19K (176R) and 55K (496R), which are both independently capable of promoting cell transformation and uncontrolled cell growth in cooperation with E1A. Both proteins are translated from the same mRNA of ~2.28 kb, but the ORF of E1B-55K is accessed by an internal initiation site and uses an alternative ORF [109]. Thus, E1B-55K has no sequence homology to the E1B-19K protein. The major roles of E1B-19K and E1B-55K in viral infection are suppression of host cell apoptosis and, in rodent cells, promotion of complete transformation in cooperation with E1A. Functional motifs of E1B-55K have been mapped as shown in Fig. 3A. Expression of E1A alone is not sufficient for efficiently transforming primary cells or inducing lung carcinogenesis in transgenic E1A mice, but E1B-19K and E1B-55K can individually cooperate with E1A to produce a fully transformed tumorigenic phenotype and to induce lung carcinogenesis in transgenic mice expressing both E1A and E1B transgenes [110]. This is at least in part a consequence of (1) E1B-19K inhibition of the co-oligomerization of the Bcl-2 family proteins BAK and BAX, leading to blockade of caspase-mediated apoptosis, and (2) the interaction of E1B-55K with p53 [111] for p53 degradation and thus the inhibition of p53 functions [30]. The full-length (55K) E1B is required for mRNA export of viral late mRNA [112].
Schematic structures of adenovirus E1B protein and mRNAs. A. E1B protein structure and its biological functions. NES, nuclear export sequences; NLS, nuclear localization sequences; RNP, ribonucleoprotein motif; CKI/II, casein kinase I/II phosphorylation site. See other reference for more information about E1B [270]. B. RNA structure and alternatively spliced species of E1B pre-mRNA.
Large T and small t antigens in human polyomaviruses
Human polyomaviruses encode two oncoproteins, large T (LT) antigen and small t (st) antigen, due to alternative RNA splicing. Both LT and st are viral nonstructural proteins expressed early in virus infection. Like SV40 T antigens, whose roles in cell transformation have been investigated extensively, human polyomaviral LT inactivates two cellular tumor suppressor proteins, p53 and pRB, by direct protein-protein interactions, and st also inactivates cellular protein phosphatase 2A (PP2A) through protein-protein interactions. Experimentally, coexpression of st enhances the transforming ability of LT [113].
Structurally, LT can be divided into several regions [114]. The N-terminal J domain overlaps with two highly conserved regions (CR1 and CR2) and is the binding site for the Hsc70 family of chaperones, which promote the proper folding of proteins after translation. Both LT and st have this domain with the same aa residues derived from the same exon 1 region, but the remaining parts of each protein are different. A pRB-binding region is close to the J domain in LT and contains an LXCXE motif essential for its interaction with the pRB family members. The C-terminal ATPase/DNA helicase region of LT overlaps with a p53-binding region. Together with a DNA origin-binding domain in the central portion of LT, this region is responsible for viral genome replication (Fig. 4A). LT inactivates p53 via its p53-binding site, but the LT-p53 complex activates insulin-like growth factor I (IGF-I) transcription to promote malignant cell growth [115]. In contrast, st lacks all of the LT domains but contains a unique PP2A-binding site. Of note, binding of st inhibits the phosphatase activity of cellular PP2A, an essential regulator of numerous signaling pathways [116].
Diagrammatic representation of the SV40 large T antigen and its RNA splicing. A. Schematic protein structure of SV40 large T antigen. J, DnaJ domain; OBD, origin DNA-binding domain. B. Alternative splicing of SV40 T antigen pre-mRNA leads to production of Large T, 17K T, and small t mRNAs. Black dots indicate stop codon locations on spliced RNAs.
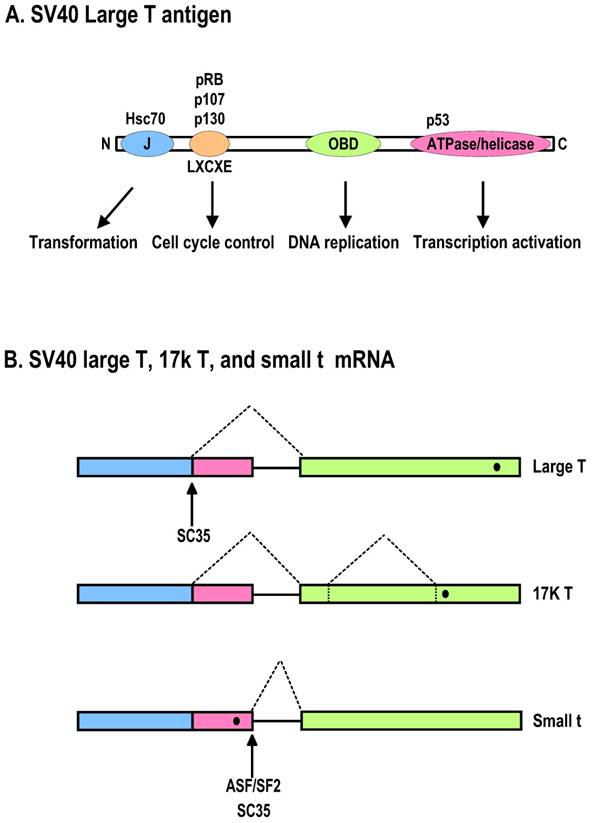
Interestingly, LT mutation appears common in the human MCV genome isolated from MCCs. The MCC tumor-derived LT sequences harbor mutations prematurely truncating exon 2, which encodes the MCV LT ATPase/helicase. Consequently, the resulting MCV LT is deficient in viral replication, whereas pRB targeting is spared in cancer cells [27]. However, recent studies indicate that MCV T antigen expression is necessary for the maintenance of MCV-positive MCC [117].
LMP1 and BARF-1 in EBV
Apparently, several restricted forms of EBV latency occur in the EBV-carrying malignancies, and modulation of LMP1 expression differs greatly depending on the latency form. In B cells with type III latency, the EBV genome expresses six nuclear antigens (EBNA-1 to -6) and three latent membrane proteins (LMP1, -2A, -2B). EBNA-2 and -5 are responsible for LMP1 expression. In type II latency, which is mainly found in Hodgkin lymphoma, T- and NK-lymphoma, and NPC, the EBV genome expresses only EBNA-1 and LMPs. The expression of LMP1 is induced by IL-10 [118] and IL-21 [119]. In Burkitt lymphoma, which has type I latency, only EBNA-1 is expressed.
The oncoprotein LMP1 of EBV is a 62-kD integral membrane protein of 386 aa residues that consists of a short cytoplasmic N-terminus of 24 aa, six transmembrane domains of 186 aa, and a cytoplasmic C-terminus of 200 aa (Fig. 5A) [120]. EBV LMP1 is essential for the immortalization and transformation of human B cells, but its oncogenic activity can be downregulated by a truncated LMP1 (258 aa) [121]. LMP1 without a ligand drives proliferation of EBV-infected B cells by signaling within the B cells similar to the signaling of the cellular CD40 receptor [122]: both activate NF-κB, AP-1, Stat-1, CD83, and CD95 by associating with molecules such as TRAFs and JAK3. LMP1's signaling, however, differs fundamentally from that of CD40, because LMP1 regulates these signaling pathways without itself being regulated by a ligand, as CD40 is. LMP1 levels vary in cells of clonal populations by more than 100-fold, which leads to multiple distinct activities of the oncoprotein. When expressed at intermediate levels, LMP1 signals through NF-κB to promote cell proliferation. When expressed at high levels, LMP1 inhibits general protein synthesis by inducing phosphorylation of eIF2α via activation of PERK kinase, leading to upregulation of activating transcription factor 4 (ATF4) expression, which in turn transactivates LMP1's own promoter [18]. LMP1 activation leads to overexpression of antiapoptotic molecules, such as Bcl-2, Mcl-1, and Bcl-2-related protein A1 (Bfl-1) [123] and blocks p53-mediated apoptosis through the induction of the A20 gene [124]. Complementary to its proliferative function, LMP1 inhibits proapoptotic factors such as Bax [125] and downregulates TCL1 oncogene through miR-29b [126].
Schematic structures of EBV LMP1 protein and its RNAs. A. Full-length LMP1 protein. Both the N-terminal and C-terminal cytoplasmic domains are white boxes and the transmembrane domain is a shaded box. Protein-protein interacting motifs are indicated as colored ovals, and a 30-nt deletion (del.) or duplication (dupl.) region often seen in nasopharyngeal carcinomas or B cells is underlined. TRAF, TNFR-associated factor; TRADD, TNFR-associated death domain protein; JAK3, Janus kinase 3. B. Schematic structure of LMP1 mRNAs. Promoters driving the expression of LMP1 and truncated LMP1 are shown relative to the EBV genome. Black dots indicate the first AUG on each transcript that is used for translation initiation.
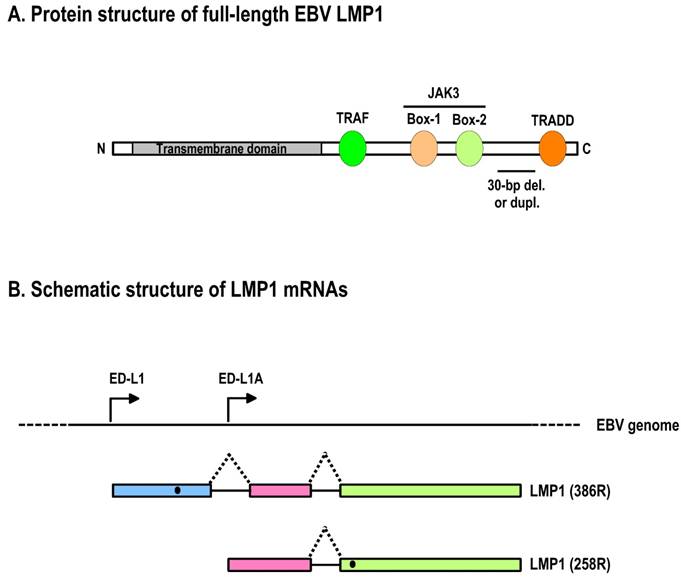
The LMP1 (BNLF1) gene contains three exons and two introns that are located within the BamHI-N region of the EBV genome [120,127]. Two ORFs have been identified based on nucleotide sequences and mRNA mapping of EBV B95-8 [128]. A transcript starting from the ED-L1 promoter, which is located upstream of the first exon, encodes full-length LMP1 (386 amino acids; Fig. 5B) from the first ORF that is abundantly expressed in lymphoblastoid cell lines. There is no alternative RNA splice site in either of its two introns. Another transcript starting from the ED-L1A promoter, which is located within the first intron of the LMP1 gene, encodes the second ORF (Fig. 5B). The translation initiation site of this second ORF is methionine-129 of full-length LMP1 [128], and the ORF thus encodes an amino-terminally truncated form of the LMP1 protein. The truncated LMP1 (258 amino acids) consists of the fifth and sixth transmembrane domains and the cytoplasmic carboxy terminus of full-length LMP1.
Although the B95-8 cell line, an in vitro-immortalized lymphoblastoid cell line, was initially used to characterize truncated LMP1, a number of studies demonstrated that expression of truncated LMP1 was specifically upregulated during the lytic phase of viral replication. Therefore, the truncated form of LMP1 is frequently referred to as lytic LMP1[129]. In contrast to full-length LMP1, truncated LMP1 does not transform rodent cells [130] or alter the phenotypes of human B lymphocytes [131]. The biological activity of truncated LMP1 is its ability to negatively regulate LMP1 signaling pathways [132] and LMP-1-mediated oncogenesis [121].
The BARF1 gene is located in the BamHI-A fragment of the EBV genome and has potential oncogenic activity. The BARF1 gene encodes a 31-kDa early protein, but can be expressed as a “latent” protein, in particular in NPC biopsy samples [21,133]. BARF1 induces malignant transformation of rodent fibroblasts [22], enhances the tumorigenicity of EBV-negative Burkitt lymphoma-derived cell lines [134], and immortalizes primary epithelial cells [135]. BARF1-rEBV-infected NPC cells are tumorigenic in nude mice [23]. A 54-amino-acid region of the N-terminus, which is capable of activating expression of the antiapoptotic protein bcl-2, is essential for the transforming activity [136]. B cells with BARF1 expression exhibit increased levels of c-myc, CD23, and CD2. BARF1 can also be secreted as a soluble receptor for human colony-stimulating factor [134,137]. In this form, it regulates the immune response by inhibiting interferon-α secretion from mononuclear cells and is detectable, along with LMP1, in the serum and saliva of patients with NPC [138]. The secreted BARF1 and LMP1 are mitogenic.
Tax in HTLV
The HTLV-1 Tax protein is a 40-kD nuclear phosphoprotein that consists of 353 aa residues. To distinguish it from the HTLV-2 Tax protein (p37tax or Tax2), the HTLV-1 Tax is also called p40tax or Tax1. Tax1 and Tax2 share many characteristic properties, including in vitro immortalization of lymphocytes. HTLV-1 Tax is a transcriptional activator of the viral promoter and has been implicated in initiating transformation events leading to the development of adult T-cell leukemia, but it is not needed to maintain cell transformation, and the tumor cells from adult T-cell leukemia do not express detectable levels of Tax [139,140]. This makes HTLV-1 Tax different from other cancer viruses, in which a continuous expression of viral oncogene is necessary for maintenance of transformation. However, it is clear that HTLV-1 Tax is oncogenic, because in all reported studies Tax transgenic mice were vulnerable to developing various tumors [141-143]. The emerging concept [144] is that Tax is required to initiate transformation, but viral HBZ protein [145,146] and/or aberrant cellular microRNA expression [147,148] appear to be needed to maintain adult T-cell leukemia when Tax is no longer expressed.
HTLV-1 Tax is a pleiotropic transcription factor that modulates transcription of many cellular genes through direct interactions with transcriptional activators, basal transcription factors, and proteins involved in chromatin remodeling as well as cell cycle progression [149]. Its role in transcription and regulation of cell cycle progression are further supported by its direct presence in nuclear transcription hot spots and nuclear speckles containing SC35 [150] as well as by its direct association with many cell cycle regulators [151]. Tax also associates with proteins involved in post-transcriptional control of mRNAs to further modulate gene expression. Thus, Tax plays an essential role in viral and cellular transcription as well as in cell transformation and oncogenicity. Structurally, HTLV-1 Tax bears multiple protein-binding domains (Fig. 6A), as reviewed recently [152]. Its N-terminal region has a zinc finger motif that is responsible for its interaction with various transcription factors [153-155]. This region overlaps with an NLS. Mutations within this zinc finger affect Tax-mediated CREB transactivation as well as subcellular localization [156].
Schematic structure of the HTLV-1 Tax protein and production of Tax mRNA by alternative RNA splicing. A. Functional regions of HTLV-1 Tax protein. NLS, nuclear localization signal; Zn, zinc finger; LZR, leucine-zipper-like region; NES, nuclear export signal; GLM, Golgi localization motif; SM, secretion motif; PDZ, PDZ-binding domain. B. HTLV-1 Tax expression by alternative RNA splicing. The HTLV-1 genome structure is shown at the top of the panel, with nine species of mRNAs diagramed below. Boxes above RNA exons (solid lines) show an ORF in each spliced mRNA that is used for translation of an individual accessory protein. Introns and splicing directions for each mRNA species are indicated by the dotted lines. Six accessory proteins expressed by alternative RNA splicing all originate from a single pre-mRNA transcribed from the 5' LTR, and their cellular localizations are shown at the right. In addition, HTLV-1 basic leucine zipper factor (HBZ) minus-strand RNA is transcribed from the 3' LTR in an antisense fashion [271,272], and its encoded protein is a nuclear protein [273], as shown at the right. Panel B is modified from reference [151] with permission.
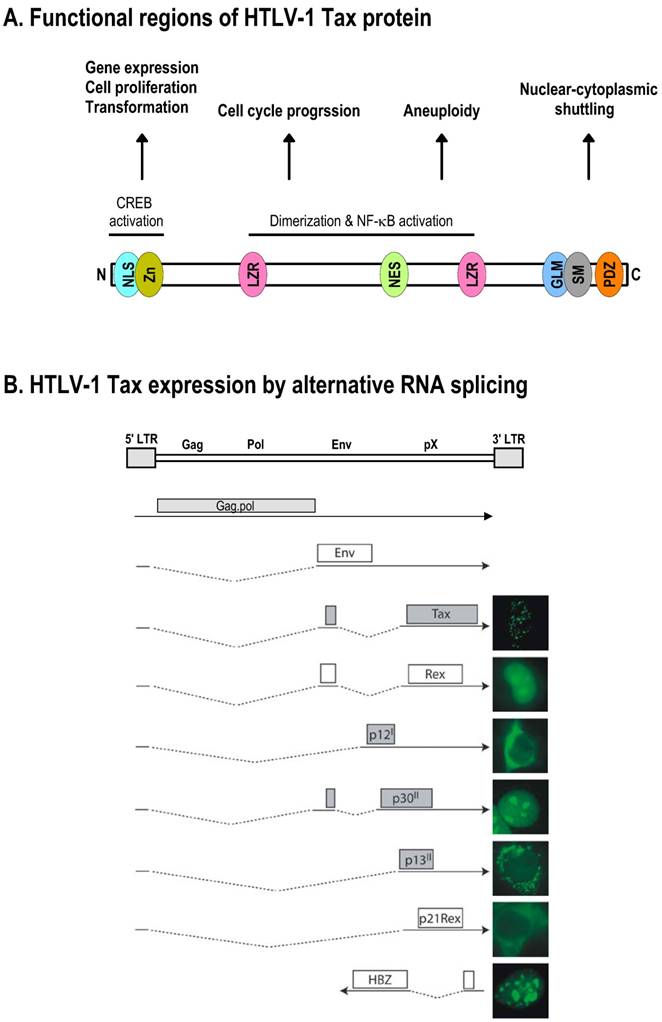
The middle part of Tax bears a region for dimerization that also overlaps with two leucine zipper-like motifs and a nuclear export sequence [157-159]. Substitutions within the first leucine zipper affect Tax interactions with NF-κB and PP2A [160,161]. The C-terminal region of Tax contains an activation domain [162], a Golgi localization and secretion motif [163], an Rb-binding sequence [164], and a PDZ-binding domain [165].
CONTROL OF VIRAL ONCOGENE EXPRESSION BY ALTERNATIVE RNA SPLICING
Regulation of HPV16 and HPV18 E6 and E7 expression by alternative RNA splicing
The E6 and E7 genes are positioned side by side at the beginning of papillomavirus genome. The expression strategy of oncogenic E6 and E7 differs from that of non-oncogenic E6 and E7. In oncogenic HPV16 and HPV18, the two genes are expressed as a bicistronic pre-mRNA from a single promoter immediately upstream of the E6 ORF. In nononcogenic HPV6 and HPV11, the genes are expressed individually from two separate promoters. Another noticeable feature is that oncogenic E6 genes contain an intron, whereas non-oncogenic E6 genes do not. Consequently, a spliced E6 ORF is predominant in oncogenic E6 mRNAs, but does not exist in non-oncogenic E6 mRNAs (Table 2), suggesting an important role of the E6 splicing in viral oncogenesis. Of note, the same promoter that drives oncogenic E6 and E7 expression is also responsible for almost all early gene expression, and the resulting early transcripts are polyadenylated by using the same early poly(A) signal [166,167]. Therefore, these early primary transcripts are all bicistronic or polycistronic, and each has two or more ORFs and contains three exons and two introns [167].
E6 splicing and carcinogenesis of HPVs.
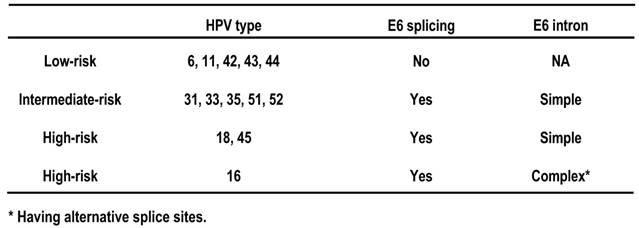
In HPV16, intron 1 and intron 2 of an E6E7 primary transcript each contain three alternative 3' splice sites which can be selected for RNA splicing in virus-infected cells, leading to the production of at least 14 species of mRNA transcripts with various coding potential [167]. Intron 1 of an HPV18 E6E7 primary transcript has a single 3' splice site. Because intron 1 and intron 2 are positioned, respectively, in the E6 ORF and E1 ORF, retention of intron 1 during RNA splicing is necessary for E6 expression, whereas retention of intron 2 is needed for E1 production. E1 functions as a viral DNA helicase essential for viral DNA replication [168]. Splicing of either intron destroys the coding of E6 or E1. Mechanistically, there are only shameful data currently available on how each intron could escape recognition by the cellular splicing machinery in order to produce these two important viral proteins. The cap-binding complex at the RNA 5' end was initially found to promotes the splicing of intron 1 of HPV16 E6E7 transcripts, but the enhanced intron 1 splicing by the cap-binding complex can be restrained by the distance of intron 1 from its RNA 5' cap [169]. Under natural conditions, cellular epithelial growth factor (EGF) pathway regulates the intron 1 splicing of HPV16 E6E7 transcripts via Erk1/2 activation [170]. It is possible that the oncogenic HPVs retain an intron as needed by interfering with the action of the splicing machinery using their own proteins. The findings that both HPV16 E2 and E6 act as RNA-binding proteins that suppress splicing [83] and viral E5 regulation of EGFR expression [171] may help us to understand this striking phenomenon.
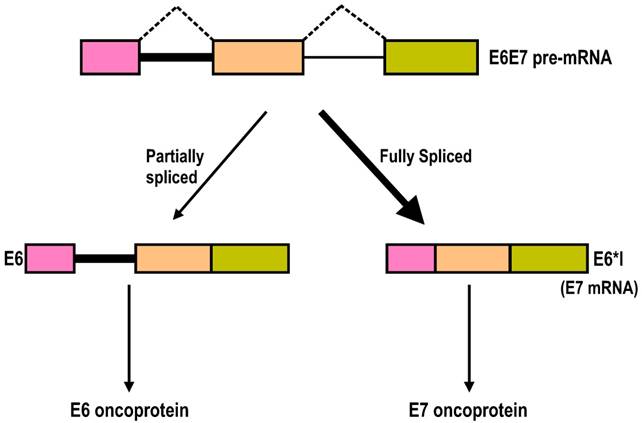
Splicing of intron 1 in the HPV16 and HPV18 E6E7 pre-mRNAs is highly efficient, and the majority of the transcripts in cervical cancer tissues and cervical cancer-derived cell lines are E6*I, a spliced transcript without intron 1. Why then is efficient splicing of intron 1 in the HPV16 or HPV18 E6E7 pre-mRNA needed for viral gene expression, since the splicing harms E6 expression? It has been proposed that splicing of intron 1 in 16E6E7 pre-mRNA might benefit E7 expression [53,172], because the space between the termination of E6 translation and the re-initiation of E7 in an intron 1-containing 16E6E7 mRNA is limited, with only two nucleotides (nts) between them. This limited space between the E6 ORF and the E7 ORF would not give enough room or time for a scanning ribosome to release all of its termination components and to reload all of the necessary translation components to re-initiate translation of the E7 ORF on the same bicistronic mRNA [173]. This hypothesis regarding E7 expression was recently proven in various experiments in my laboratory [169,174]. Splicing of intron 1 creates a frameshift, and the resulting E6*I mRNA obtains a pre-termination codon immediately downstream of the splice junction and, accordingly, creates enough space for the translation termination of E6*I and re-initiation of E7 translation by a scanning ribosome[175]. Mutation of the intron 1 5' splice site to prevent RNA splicing or narrowing the space between the E6*I ORF and E7 ORF from 145 nts to 2 nts significantly decreases E7 expression, demonstrating that E7 is translated from the spliced isoforms of 16E6E7 mRNAs, and splicing of intron 1 from the E6 coding region is essential for the production of the viral oncoprotein E7 (Fig. 7) [169,174].
Intron 1 splicing in the E6 ORF in HPV16 or HPV18 E6E7 pre-mRNA is essential for E7 production. The HPV16 or HPV18 E6E7 pre-mRNA contains three exons (colored boxes) and two introns (lines). The majority of the E6E7 RNA species in cervical cancer tissues and cervical cancer-derived cell lines are fully spliced E6*I mRNAs, which are utilized for E7 translation. However, the presence of a minimal amount of partially spliced E6 with retention of intron 1 has been detected in cervical cancer tissues and cervical cancer-derived cell lines. This RNA species contains an entire E6 ORF and functions as an E6 mRNA for E6 translation [174].
Regulation of adenovirus E1A and E1B expression by alternative RNA splicing
Adenovirus E1A pre-mRNA contains three exons and two introns. The suboptimal feature of E1A intron 1 makes it a minor intron, and its usage for RNA splicing occurs mainly late in infection. Intron 2, which has two alternative 5' splice sites, is a major intron of E1A pre-mRNA and is used constitutively for RNA splicing in every species of E1A mRNA, both early and late in virus infection. Through alternative RNA splicing of intron 2, and inclusion or exclusion of intron 1, the adenovirus E1A pre-mRNA generates five different mRNAs, designated 13S, 12S, 11S, 10S, and 9S mRNA according to their sedimentation coefficients [176].These mRNAs give rise to five distinct proteins of 289 aa residues (13S mRNA), 243 aa (12S mRNA), 217 aa (11S mRNA), 171 aa (10S mRNA), and 55 aa (9S mRNA)[177] (Fig. 2B). The 13S and 12S mRNAs are the two major spliced mRNAs during early virus infection, and their protein products share the same N- and C-terminal aa sequences but differ in the CR3 region, which is unique to the E1A-289R protein. The 9S mRNA accumulates to relatively high levels late in virus infection, at which time the 11S and 10S mRNAs are only minimally expressed [178].
In the past two decades, a number of studies have shown that individual SR and non-SR proteins play roles in the selection of each alternative 5' splice site and in the shift from 13S to 9S RNA production. One interesting finding is that the 13S-to-9S transition can be triggered through titration of the SR proteins 9G8, SC35, and ASF/SF2 by major-late transcripts that accumulate in nuclei late in the infection [179]. ASF/SF2 was the first of the SR proteins found to relate to the selection of the 13S 5' splice site, but its role in this 5' splice site selection can be antagonized by overexpressed hnRNA A1 [180,181], SRrp35, and SRrp40 [182]. ASF/SF2 RNA-binding domain 2 stimulates splicing of the 13S 5' splice site and simultaneously represses 12S 5' splice site splicing [183]. SRp40 [184], SRp54 [185], TLS [186], and RBM4 [187] were found to preferentially activate the 9S 5' splice site, but SRp75 enhances the recognition of the 12S 5'splice site [188]. However, lack of a specific binding site upstream of the 13S and 9S 5' splice sites for these proteins suggests that individual splicing regulators of the 5' splice site selection might function in an RNA sequence-independent manner (Fig. 2B). Identification of a purine-rich ESE (named bidirectional splicing enhancer, BSE) located immediately upstream of the 12S 5' splice site, and demonstration that such an ESE is involved in selection of the 12S 5' splice site and intron 1 3' splice site through binding to the SR proteins 9G8 and ASF/SF2, suggests that at least one of the 5' splice site and 3' splice site in the adenovirus type 2 (ad2) E1A pre-mRNA is preferentially selected in an ESE-dependent manner [189]. The binding of 9G8 to the ESE strongly activates the selection of the 12S 5' splice site, whereas the binding of ASF/SF2 stimulates the usage of the intron 1 3' splice site [189]. Exon definition also plays a role in stimulating the intron 1 3' splice site usage for 9S mRNA production. Of note, the 13S 5' splice site but not the 12S 5'splice site has been described as activating the upstream intron 1 3' splice site, crossing over exon 2 [190].
The E1B gene of adenovirus encodes a pre-mRNA with three exons and two introns (Fig. 3B). Although the E1B intron 1 is suboptimal and large in size, it contains two alternative 3' splice sites. E1B intron 2 is constitutively spliced and small in size, but is used in RNA splicing for production of every species of the E1B mRNA. Through alternative RNA splicing, the primary transcript from the E1B gene produces at least four additional spliced forms of E1B mRNAs of 2.28, 1.26, 1.31, and 1.02 kb to translate five distinct E1B proteins of 496R (E1B-55K), 176R (E1B-19K), 156R, 93R, and 84R (Fig. 3B). E1B-19K and E1B-55K are created from the same 2.28-kb transcript but have two different initiation codons and different ORFs [109], and the two proteins are therefore not related. However, E1B-55K shares the same N-terminal 79 aa residues with E1B-156R, E1B-93R, and E1B-84R translated from exon 1, and the same C-terminal 77 aa residues with E1B-156R from exon 2 [191]. E1B-93 and E1B-84 have unique C-termini due to usage of two alternative 3' splice sites and termination at two different stop codons.
Production of the different E1B mRNAs during infection is regulated over time. While the 2.28-kb form is the main form produced early in infection, the proportion of the shorter spliced mRNAs increases over time, and the E1B-84R mRNA becomes predominant in the late phase. The splicing mechanism of E1B pre-mRNA has been investigated much less thoroughly than the splicing of E1A pre-mRNA, and there is no information on whether RNA cis-elements or splicing factors are involved in the regulation of E1B alternative RNA splicing.
Regulation of polyomaviral T antigen expression by alternative RNA splicing
The primary transcript of polyomaviral T antigen has two exons and one intron. The T antigen intron contains two alternative 5' splice sites and its alternative usage during RNA splicing leads to the production of two spliced mRNAs, LT and st. In this regard, the st intron is relatively weak because of its limiting size and utilizes a different branch point from LT for its splicing [192], giving the LT splicing a competitive advantage relative to st splicing. Whether the two alternative 5' splice sites are regulated by an exonic or intronic splicing enhancer or silencer remains to be identified. Historically, investigation into why the ratio of st to LT mRNAs is much greater in 293 cells than in HeLa cells led to discovery of ASF/SF2 as a splicing factor that promotes st 5' splice site splicing [180,193]. In contrast, overexpressed ASF/SF2 inhibits the selection of the st 5' splice site, and the overexpression of SC35 blocks splicing of both LT and st 5' splice sites and promotes nuclear accumulation of the unspliced pre-mRNA (Fig. 4B) [194]. There is a minor intron within exon 2 of SV40 LT that can be spliced from LT mRNA, leading to production of a 17-kDa T antigen consisting of 135 aa residues, of which 131 correspond to the N-terminus of LT [195,196]. This transcript is detectable in MCC tumors in addition to MCV LT and st transcripts [27].
Regulation of HTLV-1 Tax expression by alternative RNA splicing
HTLV-1 Tax mRNA is derived by alternative RNA splicing from a common pre-mRNA whose transcription is initiated from the 5' LTR of the HTLV-1 genome (Fig. 6B). Thus, Tax mRNA contains a common 5' leader exon as its 5' UTR, as seen in all other spliced HTLV-1 RNA species, and a Tax ORF that spans exon 2 and exon 3. To produce Tax mRNA, the pre-mRNA needs to be double-spliced and precisely controlled, as the pre-mRNA has multiple weak 3' splice sites that can be alternatively utilized during RNA splicing to generate several other species of mRNAs. Unfortunately, we know nothing today about how each splice site is selected and what controls the selection of individual 3' splice sites.
VIRAL NONCODING RNAs AND microRNAs IN VIRAL ONCOGENESIS
Viral noncoding RNAs in viral oncogenesis
Tumor viruses encode several abundant RNA molecules without coding function. These include EBV EBER RNAs [197,198], KSHV PAN RNA (polyadenylated nuclear RNA) [199,200] and herpesvirus saimiri small nuclear RNAs [201,202]. They are all produced from tumor cells with latent or lytic tumor virus infection and are different in size from viral microRNAs. The functions of these viral noncoding RNAs remain to be understood. There is no direct correlation between expression of these RNA molecules during virus transformation and tumorigenesis. However, recent studies show these noncoding RNAs, in particular EBV EBERs, might have some functions related to virus latency, cell survival, and oncogenic potential. It is possible that tumor viruses might utilize these noncoding RNAs to manipulate host-cell gene expression by down-regulation of host microRNAs as reported in herpesvirus saimiri [203].
EBV-encoded EBER RNA molecules that might contribute to oncogenesis have been reported [204]. EBER-1 (166 nts) and EBER-2 (172 nts) are two nonpolyadenylated nuclear RNAs transcribed by the RNA polymerase III system [198,205]. Both are complexed with cellular La protein and have extensive primary sequence similarity to adenovirus VA1 and VA2 [206] and cellular U6 small RNA [198]. EBERs may play a role in the maintenance of malignant phenotypes of Burkitt lymphoma cells by enabling clonability in soft agar; tumorigenicity in immunodeficient mice; upregulated expression of bcl-2 oncoprotein; resistance to apoptosis [207]; and induction of IL-9, IL-10, and IGF [208-210]. EBERs confer resistance to interferon-α and Fas-mediated apoptosis by inhibiting double-stranded RNA-activated protein kinase R through direct binding [211,212].
Viral microRNAs in viral oncogenesis
MicroRNAs are regulatory, noncoding RNAs about 21-23 nucleotides in length and have large-scale effects on the expression of a variety of genes at the post-transcriptional level. Through base-pairing with its targeted mRNAs, a microRNA induces RNA degradation or translational suppression of the targeted transcripts [213,214]. All DNA tumor viruses except human papillomaviruses encode viral microRNAs. EBV encodes up to 25 viral microRNAs precusors from two clusters in the EBV genome [215-217]. Fourteen of the 20 BART (BamHI-A rightward transcripts)-derived microRNAs are produced from BART introns prior to completion of the RNA splicing and are highly expressed in latently infected epithelial cells, but at much lower levels in B cells. In contrast, three BHRF1 (Bam HI fragment H rightward open reading frame 1) miRNAs expressed within introns of EBNA transcripts are found at high levels in B cells with stage III latency but are essentially undetectable in B cells or epithelial cells with stage I or II latency [216,218,219]. It is true that all EBV miRNAs from the BART region are expressed in NPC tissues, whereas EBV miRNAs from the BHRF1 region are not found [220-222]. KSHV encodes 12 viral microRNA precursors, most of which are from a large intron of the KSHV latency-associated region, where KSHV LANA, vCyclin, vFLIP, and Kaposin are transcribed [223,224]. Like EBV microRNAs, KSHV microRNAs are also expressed in B cells with latent KSHV infection. Among the polyomaviruses, MCV encodes one, BK virus generates four, and SV40 and JC virus each produce two microRNAs. All of these microRNAs are produced late during virus infection [225-227] and play a role in degradation of viral early mRNAs. The SV40 microRNAs confer resistance of virus-infected cells to lysis by cytotoxic T cells [225]. Human adenovirus probably produces a microRNA [224]. To date, there has been no report about the production of HPV-derived microRNAs [228,229].
Viral microRNAs may modulate tumorigenesis through various mechanisms during tumor virus infection. For example, recent studies show that EBV BHRF1-3 microRNAs target chemokine CXCL-11/I-TAC for its suppression, which may serve as an immunomodulatory mechanism in AIDS-related diffuse large B-cell lymphomas [230]. EBV miR-BART2 downregulates viral DNA polymerase BALF5 and inhibits the transition from latent to lytic viral replication [231]. EBV miR-BART5 inhibits the expression of the cellular protein PUMA (p53 upregulated modulator of apoptosis) to promote host cell survival and the establishment of latent EBV infection [232]. EBV miR-BART1, -16, and -17 reduce the expression of EBV LMP1 to prevent its overexpression, which would lead to inhibition of cell proliferation and an increased susceptibility to apoptotic stimuli [233]. By systematically introduction of mutations in EBV's precursor miRNA transcripts to prevent their subsequent processing into mature viral miRNAs, a recent study shows that viral miRNAs from the BHRF1 locus inhibit apoptosis and favor cell cycle progression and proliferation during the early phase of infected human primary B cells [234].
KSHV miR-K12-11 shares sequence identity with cellular miR-155 in the seed region, which confers much of the target specificity, and has the potential to regulate multiple transcripts, in particular BACH-1 (BTB and CNC homology 1, a transcription factor), via the same binding sites utilized by the host miR-155 [235,236]. KSHV miR-K12-1, miR-K12-3-3p, miR-K12-6-3p, and miR-K12-11 reduce the expression of THBS1 (thrombospondin 1) by interaction with the THBS1 3' UTR. THBS1 functions in cell-cell and cell-matrix adhesion and possesses both antiproliferative and anti-angiogenic activity [237]. More studies from recent reports indicate miR-K12-5, miR-K12-9 and miR-K12-10 repression of Bcl-2-associated transcription factor 1, an apoptosis-inducing factor [238], miR-K12-3 reduction of LRRC8D (leucine rich repeat containing 8 family, member D), a leucin-rich type III transmembrane protein involved in proliferation and activation of lymphocytes, NHP2L1 (non-histone chromosome protein 2-like 1) [239], and a transcription factor NFIB [240], miR-K12-1 inhibition of p21 [241] and NFκB inhibitor IκBα [242], miR-K12-4-3p suppression of GEMIN8 [239], miR-K12-10a reduction of TWEAKR [243], miR-K12-1, -6, and -11 decrease of MAF (musculoaponeurotic fibrosarcoma oncogene homolog) [244], K12-4-5p targeting retinoblastoma (Rb)-like protein 2 (Rbl2) and miR-K12-5 and -9*suppressing ORF50 mRNA [245,246]. Together, these data suggest mutiple roles for these viral microRNAs in pathogenesis and potentially tumorigenesis in the KSHV-infected host.
VIRAL ONCOPROTEINS AND CELLULAR microRNAs
Although much has been learned about the aberrant expression of cellular microRNAs in various tumor virus-induced cancers, the mechanism of this aberrant expression remains largely unknown. However, several exciting reports suggest that viral oncoproteins may play an important role in this aberrant regulation of cellular microRNA expression, including those oncogenic or tumor suppressive microRNAs.
EBV oncoprotein LMP1 dysregulates the expression of several cellular miRNAs. LMP1 induces miR-146a predominantly through two NF-κB-binding sites in the miR-146a promoter [247]. This observation was verified in a separate study showing that EBV LMP1 activates the miR-146a promoter but not a promoter with a mutation of the NF-κB response elements [248]. EBV LMP1 also induces the production of the BIC (B-cell integration cluster) precursor RNA of cellular miR-155, which is highly expressed in various B-cell lymphomas and may contribute to EBV immortalization of B cells [249,250]. Recent studies have confirmed that LMP1 signals through NF-κB to transactivate miR-155 expression [251] to immortalize B-cells [252].
Tumor-suppressive miR-34a is transactivated by the binding of p53 to a perfect p53-binding site in the miR-34a regulatory region. My laboratory recently observed [253] that expression of miR-34a is reduced in cervical cancer tissues and cervical cancer-derived cell lines containing oncogenic HPVs because the HPV oncoprotein E6 destabilizes the tumor suppressor p53. The reduction of miR-34a expression in organotypic tissues derived from HPV-containing primary human keratinocytes correlates with the expression of viral E6. Knockdown of viral E6 expression in HPV16+ and HPV18+ cervical cancer cell lines by siRNAs leads to increased expression of p53 and miR-34a and a substantial retardation of cell growth. Our study has thus provided new insights into mechanisms by which high-risk HPVs contribute to the development of cervical cancer [253]. Reduced miR-34a expression also occurs in normal cervical tissues and cervical lesions with high-risk HPV infections [254]. In addition, two other reports show that high-risk HPV E6 downregulates miR-218 [255] and E7 reduces miR-203 expression upon keratinocyte differentiation [256], but the mechanisms of how these regulations take place remain unknown.
HTLV-1 Tax is oncogenic in transgenic mice and is required for immortalization of human T-lymphocytes and transformation of rodent fibroblast [149,257]. Recent studies indicate that Tax transactivates miR-146a expression by binding to a single NFκB site in the miR-146a promoter proximal to the transcription start site [148]. Tax may also transactivate miR-130b expression by regulating the expression of transcription factors most likely required for miR-130b promoter to function [147].
REMARKS
It has been almost a century since the discovery of virus-induced cancers. Studies on tumor-causing viruses have led to many landmark breakthroughs that have revolutionized cell biology and the principles of medicine, helped to establish virology as a discipline, and paved the way for most of the subsequent advances and ideas on the molecular basis of viral carcinogenesis. These milestone discoveries include the identification of RNA-dependent DNA polymerase (reverse transcriptase) in Rous sarcoma virus by Howard Temin and David Baltimore [258,259], viral oncogenes and their cellular counterparts (proto-oncogenes) from Rous sarcoma virus by Harold Varmus and Michael Bishop [260,261], and RNA splicing in adenovirus-2 by Philip Sharp and Richard Roberts [262,263].
Viral strategies for manipulating the expression of cellular genes to enhance viral persistence, viral latency, and survival of infected cells have provided numerous clues to the mechanisms of gene expression and their dysregulation during tumor development. These functions are mainly undertaken by virus-encoded oncogenes, which are present in virtually all well-characterized tumor viruses except the hepatitis C virus. The fact that all characterized viral oncoproteins are “sticky” and interact with cellular proteins in infected cells makes each of them multifunctional and essential for cell immortalization and transformation. By interacting with several dozens or even hundreds of cellular factors, viral oncoproteins from different tumor viruses eventually end up in apparently similar scenarios of viral carcinogenesis by targeting cellular tumor suppressors, deregulating signal transduction pathways, redirecting gene transcription, and/or stimulating anti-apoptotic programs in the host. In the past two decades, the study of cancer biology has been driven by the dynamic networking capacity of individual viral oncoproteins.
RNA splicing plays an important role in regulating viral oncogene expression and is highly conserved among tumor viruses. Through alternative RNA splicing, several species of mRNAs can be derived from a primary transcript of a single viral oncogene to encode different truncated proteins or different oncoproteins. Although various studies over the years have shown that the levels and status of individual cellular splicing factors in virus-infected cells modulate alternative splicing of viral oncogene transcripts, a clear picture of how this modulation might take place during viral oncogene expression has not yet emerged. Because viral RNA transcripts are not naked in the cells and the movement of different sets of RNA-binding proteins on and off a particular RNA molecule is dynamic, it will be very interesting to know how the selection of alternative splice sites in an RNA is precisely defined and triggered. In the case of an HPV16 or HPV18 E6E7 bicistronic transcript, retention of the first intron is needed to express E6, but splicing of this intron promotes E7 translation initiation to produce E7[174]. What is required for the decision to splice this intron or not, resulting in the expression of two different oncoproteins from the same transcript, and when this decision occurs are intriguing questions. A recent finding of EGF pathway that might be involved in this regulation is fascinating [170]. Thus, investigation into these questions will not only help to resolve this puzzle in tumor virology, but will also shed some light on complex biology as a whole.
Noncoding RNAs, including microRNAs and endogenous siRNAs, have profound roles in the regulation of gene expression [264-268]. During latent virus infection, DNA tumor viruses, except papillomaviruses, generate abundant noncoding RNAs. Although we know very little at the present why viruses produce them and what their functions are, the production of these noncoding RNAs by these groups of tumor viruses means that the function of the tumor virus genome has been highly conserved during virus evolution. Noncoding RNAs are not a consequence of virus latency; rather, their function might be necessary for the maintenance of virus latency. Given that each microRNA subtly influences the translation of hundreds of different gene transcripts [264,265], viral microRNAs must be involved in many key biological processes in virus-infected cells during latent infection, leading to establishment of latency in tumor cells. Thus, one can imagine that viral oncogenes do not act alone but perhaps coordinate extensively with other viral products to induce oncogenesis during persistent infection.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. I thank Dr. John Taylor and Dr. Christophe Nicot of University of Kansas Medical Center for their generous permission of figure modification.
Conflict of Interests
The author has declared that no conflict of interest exists.
References
1. Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. 1911;13:397-411
2. Pagano JS, Blaser M, Buendia MA. et al. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol. 2004;14:453-471
3. Poiesz BJ, Ruscetti FW, Gazdar AF. et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77:7415-7419
4. Poiesz BJ, Ruscetti FW, Reitz MS. et al. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature. 1981;294:268-271
5. Miyoshi I, Kubonishi I, Yoshimoto S. et al. Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature. 1981;294:770-771
6. Kalyanaraman VS, Sarngadharan MG, Robert-Guroff M. et al. A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science. 1982;218:571-573
7. Calattini S, Chevalier SA, Duprez R. et al. Discovery of a new human T-cell lymphotropic virus (HTLV-3) in Central Africa. Retrovirology. 2005;2:30
8. Switzer WM, Salemi M, Qari SH. et al. Ancient, independent evolution and distinct molecular features of the novel human T-lymphotropic virus type 4. Retrovirology. 2009;6:9
9. Mahieux R, Gessain A. The human HTLV-3 and HTLV-4 retroviruses: New members of the HTLV family. Pathol Biol (Paris). 2009Mar;57(2):161-6
10. Vonka V, Kanka J, Hirsch I. et al. Prospective study on the relationship between cervical neoplasia and herpes simplex type-2 virus. II. Herpes simplex type-2 antibody presence in sera taken at enrollment. Int J Cancer. 1984;33:61-66
11. Lehtinen M, Koskela P, Jellum E. et al. Herpes simplex virus and risk of cervical cancer: a longitudinal, nested case-control study in the nordic countries. Am J Epidemiol. 2002;156:687-692
12. Durst M, Gissmann L, Ikenberg H. et al. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812-3815
13. zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342-350
14. Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98:1505-1511
15. Lowy DR, Schiller JT. Prophylactic human papillomavirus vaccines. J Clin Invest. 2006;116:1167-1173
16. Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831-840
17. Dirmeier U, Neuhierl B, Kilger E. et al. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by epstein-barr virus. Cancer Res. 2003;63:2982-2989
18. Lee DY, Sugden B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood. 2008;111:2280-2289
19. Humme S, Reisbach G, Feederle R. et al. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci U S A. 2003;100:10989-10994
20. Kang MS, Soni V, Bronson R. et al. Epstein-Barr virus nuclear antigen 1 does not cause lymphoma in C57BL/6J mice. J Virol. 2008;82:4180-4183
21. Decaussin G, Sbih-Lammali F, Turenne-Tessier M. et al. Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Res. 2000;60:5584-5588
22. Wei MX, Ooka T. A transforming function of the BARF1 gene encoded by Epstein-Barr virus. EMBO J. 1989;8:2897-2903
23. Seto E, Ooka T, Middeldorp J. et al. Reconstitution of nasopharyngeal carcinoma-type EBV infection induces tumorigenicity. Cancer Res. 2008;68:1030-1036
24. Montaner S, Sodhi A, Molinolo A. et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23-36
25. Bais C, Van Geelen A, Eroles P. et al. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell. 2003;3:131-143
26. Feng H, Shuda M, Chang Y. et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100
27. Shuda M, Feng H, Kwun HJ. et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105:16272-16277
28. Schowalter RM, Pastrana DV, Pumphrey KA. et al. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509-515
29. Pastrana DV, Tolstov YL, Becker JC. et al. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009;5:e1000578
30. Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24:7673-7685
31. Kim CM, Koike K, Saito I. et al. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317-320
32. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725-12734
33. Zhang Z, Sun E, Ou JH. et al. Inhibition of cellular proteasome activities mediates HBX-independent hepatitis B virus replication in vivo. J Virol. 2010;84:9326-9331
34. Sir D, Tian Y, Chen WL. et al. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107:4383-4388
35. Takatsuki K. Discovery of adult T-cell leukemia. Retrovirology. 2005;2:16
36. Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270-280
37. Dong B, Kim S, Hong S. et al. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci U S A. 2007;104:1655-1660
38. Lombardi VC, Ruscetti FW, Das GJ. et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326:585-589
39. Mikovits JA, Huang Y, Pfost MA. et al. Distribution of xenotropic murine leukemia virus-related virus (XMRV) infection in chronic fatigue syndrome and prostate cancer. AIDS Rev. 2010;12:149-152
40. Lo SC, Pripuzova N, Li B. et al. Detection of MLV-related virus gene sequences in blood of patients with chronic fatigue syndrome and healthy blood donors. Proc Natl Acad Sci U S A. 2010;107:15874-15879
41. Yan Y, Liu Q, Wollenberg K. et al. Evolution of Functional and Sequence Variants of the Mammalian XPR1 Receptor for Mouse Xenotropic Gammaretroviruses and the Human-Derived Retrovirus XMRV. J Virol. 2010;84:11970-11980
42. Hohn O, Krause H, Barbarotto P. et al. Lack of evidence for xenotropic murine leukemia virus-related virus(XMRV) in German prostate cancer patients. Retrovirology. 2009;6:92
43. Switzer WM, Jia H, Hohn O. et al. Absence of evidence of xenotropic murine leukemia virus-related virus infection in persons with chronic fatigue syndrome and healthy controls in the United States. Retrovirology. 2010;7:57
44. Aloia AL, Sfanos KS, Isaacs WB. et al. XMRV: A New Virus in Prostate Cancer? Cancer Res. 2010 [Epub ahead of print]
45. Danielson BP, Ayala GE, Kimata JT. Detection of xenotropic murine leukemia virus-related virus in normal and tumor tissue of patients from the southern United States with prostate cancer is dependent on specific polymerase chain reaction conditions. J Infect Dis. 2010;202:1470-1477
46. Henrich TJ, Li JZ, Felsenstein D. et al. Xenotropic murine leukemia virus-related virus prevalence in patients with chronic fatigue syndrome or chronic immunomodulatory conditions. J Infect Dis. 2010;202:1478-1481
47. Barnes E, Flanagan P, Brown A. et al. Failure to detect xenotropic murine leukemia virus-related virus in blood of individuals at high risk of blood-borne viral infections. J Infect Dis. 2010;202:1482-1485
48. Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127:S62-S71
49. Jopling CL, Yi M, Lancaster AM. et al. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577-1581
50. Band V, Dalal S, Delmolino L. et al. Enhanced degradation of p53 protein in HPV-6 and BPV-1 E6-immortalized human mammary epithelial cells. EMBO J. 1993;12:1847-1852
51. Hawley-Nelson P, Vousden KH, Hubbert NL. et al. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8:3905-3910
52. Munger K, Phelps WC, Bubb V. et al. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417-4421
53. Sedman SA, Barbosa MS, Vass WC. et al. The full-length E6 protein of human papillomavirus type 16 has transforming and trans-activating activities and cooperates with E7 to immortalize keratinocytes in culture. J Virol. 1991;65:4860-4866
54. Storey A, Banks L. Human papillomavirus type 16 E6 gene cooperates with EJ-ras to immortalize primary mouse cells. Oncogene. 1993;8:919-924
55. Pim D, Storey A, Thomas M. et al. Mutational analysis of HPV-18 E6 identifies domains required for p53 degradation in vitro, abolition of p53 transactivation in vivo and immortalisation of primary BMK cells. Oncogene. 1994;9:1869-1876
56. Liu Y, Chen JJ, Gao Q. et al. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol. 1999;73:7297-7307
57. Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775-784
58. Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76-79
59. Scheffner M, Werness BA, Huibregtse JM. et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129-1136
60. Nomine Y, Masson M, Charbonnier S. et al. Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol Cell. 2006;21:665-678
61. Ristriani T, Fournane S, Orfanoudakis G. et al. A single-codon mutation converts HPV16 E6 oncoprotein into a potential tumor suppressor, which induces p53-dependent senescence of HPV-positive HeLa cervical cancer cells. Oncogene. 2009;28(5):762-72
62. Dyson N, Howley PM, Munger K. et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934-937
63. Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620-4624
64. Gonzalez SL, Stremlau M, He X. et al. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583-7591
65. Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20:7874-7887
66. Munger K, Basile JR, Duensing S. et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888-7898
67. Song S, Liem A, Miller JA. et al. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267:141-150
68. Huibregtse JM, Scheffner M, Howley PM. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol. 1993;13:4918-4927
69. Storey A, Thomas M, Kalita A. et al. Role of a p53 polymorphism in the development of human papillomavirus- associated cancer. Nature. 1998;393:229-234
70. An J, Mo D, Liu H. et al. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell. 2008;14:394-407
71. Kanda T, Watanabe S, Zanma S. et al. Human papillomavirus type 16 E6 proteins with glycine substitution for cysteine in the metal-binding motif. Virology. 1991;185:536-543
72. Thomas M, Dasgupta J, Zhang Y. et al. Analysis of specificity determinants in the interactions of different HPV E6 proteins with their PDZ domain-containing substrates. Virology. 2008;376:371-378
73. Zhang Y, Dasgupta J, Ma RZ. et al. Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J Virol. 2007;81:3618-3626
74. Tao M, Kruhlak M, Xia S. et al. Signals That Dictate Nuclear Localization of Human Papillomavirus Type 16 Oncoprotein E6 in Living Cells. J Virol. 2003;77:13232-13247
75. Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991;67:547-556
76. Desaintes C, Hallez S, Van Alphen P. et al. Transcriptional activation of several heterologous promoters by the E6 protein of human papillomavirus type 16. J Virol. 1992;66:325-333
77. Patel D, Huang SM, Baglia LA. et al. The E6 protein of human papillomavirus type 16 binds to and inhibits co- activation by CBP and p300. EMBO J. 1999;18:5061-5072
78. Zimmermann H, Degenkolbe R, Bernard HU. et al. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol. 1999;73:6209-6219
79. Ronco LV, Karpova AY, Vidal M. et al. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061-2072
80. Gross-Mesilaty S, Reinstein E, Bercovich B. et al. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc Natl Acad Sci U S A. 1998;95:8058-8063
81. Liu X, Dakic A, Chen R. et al. Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J Virol. 2008;82:11568-11576
82. Veldman T, Liu X, Yuan H. et al. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc Natl Acad Sci U S A. 2003;100:8211-8216
83. Bodaghi S, Jia R, Zheng ZM. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology. 2009;386(1):32-43
84. Chen JJ, Reid CE, Band V. et al. Interaction of papillomavirus E6 oncoproteins with a putative calcium- binding protein. Science. 1995;269:529-531
85. Gao Q, Srinivasan S, Boyer SN. et al. The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol Cell Biol. 1999;19:733-744
86. Gao Q, Singh L, Kumar A. et al. Human papillomavirus type 16 E6-induced degradation of E6TP1 correlates with its ability to immortalize human mammary epithelial cells. J Virol. 2001;75:4459-4466
87. Singh L, Gao Q, Kumar A. et al. The high-risk human papillomavirus type 16 E6 counters the GAP function of E6TP1 toward small Rap G proteins. J Virol. 2003;77:1614-1620
88. Tong X, Howley PM. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc Natl Acad Sci U S A. 1997;94:4412-4417
89. Filippova M, Song H, Connolly JL. et al. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J Biol Chem. 2002;277:21730-21739
90. Topffer S, Muller-Schiffmann A, Matentzoglu K. et al. Protein tyrosine phosphatase H1 is a target of the E6 oncoprotein of high-risk genital human papillomaviruses. J Gen Virol. 2007;88:2956-2965
91. Kiyono T, Hiraiwa A, Fujita M. et al. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:11612-11616
92. Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94:6670-6675
93. Huh K, Zhou X, Hayakawa H. et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737-9747
94. Liu X, Clements A, Zhao K. et al. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem. 2006;281:578-586
95. Knapp AA, McManus PM, Bockstall K. et al. Identification of the nuclear localization and export signals of high risk HPV16 E7 oncoprotein. Virology. 2009;383:60-68
96. McLaughlin-Drubin ME, Bromberg-White JL, Meyers C. The role of the human papillomavirus type 18 E7 oncoprotein during the complete viral life cycle. Virology. 2005;338:61-68
97. Jian Y, Schmidt-Grimminger DC, Chien WM. et al. Post-transcriptional induction of p21cip1 protein by human papillomavirus E7 inhibits unscheduled DNA synthesis reactivated in differentiated keratinocytes. Oncogene. 1998;17:2027-2038
98. Noya F, Chien WM, Broker TR. et al. p21cip1 Degradation in differentiated keratinocytes is abrogated by costabilization with cyclin E induced by human papillomavirus E7. J Virol. 2001;75:6121-6134
99. Kotake Y, Cao R, Viatour P. et al. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49-54
100. Nguyen CL, Eichwald C, Nibert ML. et al. Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J Virol. 2007;81:13533-13543
101. Baldwin A, Li W, Grace M. et al. Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells. Proc Natl Acad Sci U S A. 2008;105:16478-16483
102. Bernat A, Avvakumov N, Mymryk JS. et al. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871-7881
103. Avvakumov N, Torchia J, Mymryk JS. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene. 2003;22:3833-3841
104. Baldwin A, Huh KW, Munger K. Human papillomavirus E7 oncoprotein dysregulates steroid receptor coactivator 1 localization and function. J Virol. 2006;80:6669-6677
105. DeMasi J, Huh KW, Nakatani Y. et al. Howley PM. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc Natl Acad Sci U S A. 2005;102:11486-11491
106. McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2008
107. Horwitz GA, Zhang K, McBrian MA. et al. Adenovirus small e1a alters global patterns of histone modification. Science. 2008;321:1084-1085
108. Ferrari R, Pellegrini M, Horwitz GA. et al. Epigenetic reprogramming by adenovirus e1a. Science. 2008;321:1086-1088
109. Bos JL, Polder LJ, Bernards R. et al. The 2.2 kb E1b mRNA of human Ad12 and Ad5 codes for two tumor antigens starting at different AUG triplets. Cell. 1981;27:121-131
110. Yang Y, McKerlie C, Lu Z. et al. In vivo potential effects of adenovirus type 5 E1A and E1B on lung carcinogenesis and lymphoproliferative inflammation. J Virol. 2008;82:8105-8111
111. Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;357:82-85
112. Gonzalez R, Huang W, Finnen R. et al. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J Virol. 2006;80:964-974
113. Hahn WC, Dessain SK, Brooks MW. et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111-2123
114. Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729-7745
115. Bocchetta M, Eliasz S, De Marco MA. et al. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008;68:1022-1029
116. Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008;27:137-146
117. Houben R, Shuda M, Weinkam R. et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84:7064-7072
118. Kis LL, Takahara M, Nagy N. et al. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood. 2006;107:2928-2935
119. Konforte D, Simard N, Paige CJ. Interleukin-21 regulates expression of key Epstein-Barr virus oncoproteins, EBNA2 and LMP1, in infected human B cells. Virology. 2008;374:100-113
120. Fennewald S, van Santen V, Kieff E. Nucleotide sequence of an mRNA transcribed in latent growth-transforming virus infection indicates that it may encode a membrane protein. J Virol. 1984;51:411-419
121. Pandya J, Walling DM. Oncogenic activity of Epstein-Barr virus latent membrane protein 1 (LMP-1) is down-regulated by lytic LMP-1. J Virol. 2006;80:8038-8046
122. Kilger E, Kieser A, Baumann M. et al. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998;17:1700-1709
123. D'Souza BN, Edelstein LC, Pegman PM. et al. Nuclear factor kappa B-dependent activation of the antiapoptotic bfl-1 gene by the Epstein-Barr virus latent membrane protein 1 and activated CD40 receptor. J Virol. 2004;78:1800-1816
124. Fries KL, Miller WE, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J Virol. 1996;70:8653-8659
125. Grimm T, Schneider S, Naschberger E. et al. EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood. 2005;105:3263-3269
126. Anastasiadou E, Boccellato F, Vincenti S. et al. Epstein-Barr virus encoded LMP1 downregulates TCL1 oncogene through miR-29b. Oncogene. 2010;29:1316-1328
127. Baer R, Bankier AT, Biggin MD. et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310:207-211
128. Hudson GS, Farrell PJ, Barrell BG. Two related but differentially expressed potential membrane proteins encoded by the EcoRI Dhet region of Epstein-Barr virus B95-8. J Virol. 1985;53:528-535
129. Pandya J, Walling DM. Epstein-Barr virus latent membrane protein 1 (LMP-1) half-life in epithelial cells is down-regulated by lytic LMP-1. J Virol. 2004;78:8404-8410
130. Wang D, Liebowitz D, Kieff E. The truncated form of the Epstein-Barr virus latent-infection membrane protein expressed in virus replication does not transform rodent fibroblasts. J Virol. 1988;62:2337-2346
131. Wang D, Liebowitz D, Wang F. et al. Epstein-Barr virus latent infection membrane protein alters the human B-lymphocyte phenotype: deletion of the amino terminus abolishes activity. J Virol. 1988;62:4173-4184
132. Erickson KD, Martin JM. The late lytic LMP-1 protein of Epstein-Barr virus can negatively regulate LMP-1 signaling. J Virol. 2000;74:1057-1060
133. Seto E, Yang L, Middeldorp J. et al. Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J Med Virol. 2005;76:82-88
134. Sheng W, Decaussin G, Ligout A. et al. Malignant transformation of Epstein-Barr virus-negative Akata cells by introduction of the BARF1 gene carried by Epstein-Barr virus. J Virol. 2003;77:3859-3865
135. Danve C, Decaussin G, Busson P. et al. Growth transformation of primary epithelial cells with a NPC-derived Epstein-Barr virus strain. Virology. 2001;288:223-235
136. Guo X, Sheng W, Zhang Y. [Malignant transformation of monkey kidney epithelial cell induced by EBV BARF1 gene and TPA]. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2001;15:321-323
137. Strockbine LD, Cohen JI, Farrah T. et al. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J Virol. 1998;72:4015-4021
138. Houali K, Wang X, Shimizu Y. et al. A new diagnostic marker for secreted Epstein-Barr virus encoded LMP1 and BARF1 oncoproteins in the serum and saliva of patients with nasopharyngeal carcinoma. Clin Cancer Res. 2007;13:4993-5000
139. Kinoshita T, Shimoyama M, Tobinai K. et al. Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86:5620-5624
140. Furukawa Y, Osame M, Kubota R. et al. Human T-cell leukemia virus type-1 (HTLV-1) Tax is expressed at the same level in infected cells of HTLV-1-associated myelopathy or tropical spastic paraparesis patients as in asymptomatic carriers but at a lower level in adult T-cell leukemia cells. Blood. 1995;85:1865-1870
141. Coscoy L, Gonzalez-Dunia D, Tangy F. et al. Molecular mechanism of tumorigenesis in mice transgenic for the human T cell leukemia virus Tax gene. Virology. 1998;248:332-341
142. Portis T, Grossman WJ, Harding JC. et al. Analysis of p53 inactivation in a human T-cell leukemia virus type 1 Tax transgenic mouse model. J Virol. 2001;75:2185-2193
143. Hall AP, Irvine J, Blyth K. et al. Tumours derived from HTLV-I tax transgenic mice are characterized by enhanced levels of apoptosis and oncogene expression. J Pathol. 1998;186:209-214
144. Jeang KT. HTLV-1 and adult T-cell leukemia: insights into viral transformation of cells 30 years after virus discovery. J Formos Med Assoc. 2010;109:688-693
145. Satou Y, Yasunaga J, Yoshida M. et al. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A. 2006;103:720-725
146. Arnold J, Zimmerman B, Li M. et al. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood. 2008;112:3788-3797
147. Yeung ML, Yasunaga J, Bennasser Y. et al. Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res. 2008;68:8976-8985
148. Pichler K, Schneider G, Grassmann R. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology. 2008;5:100
149. Grassmann R, Aboud M, Jeang KT. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene. 2005;24:5976-5985
150. Semmes OJ, Jeang KT. Localization of human T-cell leukemia virus type 1 tax to subnuclear compartments that overlap with interchromatin speckles. J Virol. 1996;70:6347-6357
151. Taylor JM, Nicot C. HTLV-1 and apoptosis: role in cellular transformation and recent advances in therapeutic approaches. Apoptosis. 2008;13:733-747
152. Boxus M, Twizere JC, Legros S. et al. The HTLV-1 Tax interactome. Retrovirology. 2008;5:76
153. Caron C, Rousset R, Beraud C. et al. Functional and biochemical interaction of the HTLV-I Tax1 transactivator with TBP. EMBO J. 1993;12:4269-4278
154. Goren I, Semmes OJ, Jeang KT. et al. The amino terminus of Tax is required for interaction with the cyclic AMP response element binding protein. J Virol. 1995;69:5806-5811
155. Harrod R, Kuo YL, Tang Y. et al. p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J Biol Chem. 2000;275:11852-11857
156. Smith MR, Greene WC. Characterization of a novel nuclear localization signal in the HTLV-I tax transactivator protein. Virology. 1992;187:316-320
157. Basbous J, Bazarbachi A, Granier C. et al. The central region of human T-cell leukemia virus type 1 Tax protein contains distinct domains involved in subunit dimerization. J Virol. 2003;77:13028-13035
158. Xiao G, Sun SC. Activation of IKKalpha and IKKbeta through their fusion with HTLV-I tax protein. Oncogene. 2000;19:5198-5203
159. Gatza ML, Marriott SJ. Genotoxic stress and cellular stress alter the subcellular distribution of human T-cell leukemia virus type 1 tax through a CRM1-dependent mechanism. J Virol. 2006;80:6657-6668
160. Yin MJ, Christerson LB, Yamamoto Y. et al. HTLV-I Tax protein binds to MEKK1 to stimulate IkappaB kinase activity and NF-kappaB activation. Cell. 1998;93:875-884
161. Fu DX, Kuo YL, Liu BY. et al. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem. 2003;278:1487-1493
162. Semmes OJ, Jeang KT. Definition of a minimal activation domain in human T-cell leukemia virus type I Tax. J Virol. 1995;69:1827-1833
163. Alefantis T, Jain P, Ahuja J. et al. HTLV-1 Tax nucleocytoplasmic shuttling, interaction with the secretory pathway, extracellular signaling, and implications for neurologic disease. J Biomed Sci. 2005;12:961-974
164. Kehn K, Fuente CL, Strouss K. et al. The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene. 2005;24:525-540
165. Rousset R, Fabre S, Desbois C. et al. The C-terminus of the HTLV-1 Tax oncoprotein mediates interaction with the PDZ domain of cellular proteins. Oncogene. 1998;16:643-654
166. Smotkin D, Wettstein FO. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc Natl Acad Sci U S A. 1986;83:4680-4684
167. Zheng ZM, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci. 2006;11:2286-2302
168. Lin BY, Makhov AM, Griffith JD. et al. Chaperone proteins abrogate inhibition of the human papillomavirus (HPV) E1 replicative helicase by the HPV E2 protein. Mol Cell Biol. 2002;22:6592-6604
169. Zheng ZM, Tao M, Yamanegi K. et al. Splicing of a Cap-proximal Human Papillomavirus 16 E6E7 Intron Promotes E7 Expression, but can be Restrained by Distance of the Intron from its RNA 5' Cap. J Mol Biol. 2004;337:1091-1108
170. Rosenberger S, De Castro AJ, Langbein L. et al. Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc Natl Acad Sci U S A. 2010;107:7006-7011
171. Suprynowicz FA, Krawczyk E, Hebert JD. et al. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J Virol. 2010;84:10619-10629
172. Smotkin D, Prokoph H, Wettstein FO. Oncogenic and nononcogenic human genital papillomaviruses generate the E7 mRNA by different mechanisms. J Virol. 1989;63:1441-1447
173. Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1-34
174. Tang S, Tao M, McCoy JP Jr. et al. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol. 2006;80:4249-4263
175. Kozak M. Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol. 1987;7:3438-3445
176. Stephens C, Harlow E. Differential splicing yields novel adenovirus 5 E1A mRNAs that encode 30 kd and 35 kd proteins. EMBO J. 1987;6:2027-2035
177. Brockmann D, Esche H. The multifunctional role of E1A in the transcriptional regulation of CREB/CBP-dependent target genes. Curr Top Microbiol Immunol. 2003;272:97-129
178. Akusjarvi G, Stevenin J. Remodelling of the host cell RNA splicing machinery during an adenovirus infection. Curr Top Microbiol Immunol. 2003;272:253-286
179. Himmelspach M, Cavaloc Y, Chebli K. et al. Titration of serine/arginine (SR) splicing factors during adenoviral infection modulates E1A pre-mRNA alternative splicing. RNA. 1995;1:794-806
180. Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68:365-375
181. Caceres JF, Stamm S, Helfman DM. et al. Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994;265:1706-1709
182. Cowper AE, Caceres JF, Mayeda A. et al. Serine-arginine (SR) protein-like factors that antagonize authentic SR proteins and regulate alternative splicing. J Biol Chem. 2001;276:48908-48914
183. Dauksaite V, Akusjarvi G. The second RNA-binding domain of the human splicing factor ASF/SF2 is the critical domain controlling adenovirus E1A alternative 5'-splice site selection. Biochem J. 2004;381:343-350
184. Screaton GR, Cáceres JF, Mayeda A. et al. Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO J. 1995;14:4336-4349
185. Zhang W-J, Wu JY. Functional properties of p54, a novel SR protein active in constitutive and alternative splicing. Mol Cell Biol. 1996;16:5400-5408
186. Lerga A, Hallier M, Delva L. et al. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001;276:6807-6816
187. Lai MC, Kuo HW, Chang WC. et al. A novel splicing regulator shares a nuclear import pathway with SR proteins. EMBO J. 2003;22:1359-1369
188. Yomoda J, Muraki M, Kataoka N. et al. Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A. Genes Cells. 2008;13:233-244
189. Bourgeois CF, Popielarz M, Hildwein G. et al. Identification of a bidirectional splicing enhancer: differential involvement of SR proteins in 5' or 3' splice site activation. Mol Cell Biol. 1999;19:7347-7356
190. Popielarz M, Gattoni R, Stevenin J. Contrasted cis-acting effects of downstream 5' splice sites on the splicing of a retained intron: the adenoviral E1A pre-mRNA model. Nucleic Acids Res. 1993;21:5144-5151
191. Lewis JB, Anderson CW. Identification of adenovirus type 2 early region 1B proteins that share the same amino terminus as do the 495R and 155R proteins. J Virol. 1987;61:3879-3888
192. Noble JC, Pan ZQ, Prives C. et al. Splicing of SV40 early pre-mRNA to large T and small t mRNAs utilizes different patterns of lariat branch sites. Cell. 1987;50:227-236
193. Ge H, Manley JL. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell. 1990;62:25-34
194. Wang J, Manley JL. Overexpression of the SR proteins ASF/SF2 and SC35 influences alternative splicing in vivo in diverse ways. RNA. 1995;1:335-346
195. Zerrahn J, Knippschild U, Winkler T. et al. Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J. 1993;12:4739-4746
196. Sompayrac L, Danna KJ. The SV40 T-antigen gene can have two introns. Virology. 1985;142:432-436
197. Lerner MR, Andrews NC, Miller G. et al. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci U S A. 1981;78:805-809
198. Howe JG, Shu MD. Epstein-Barr virus small RNA (EBER) genes: unique transcription units that combine RNA polymerase II and III promoter elements. Cell. 1989;57:825-834
199. Sun R, Lin SF, Gradoville L. et al. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1996;93:11883-11888
200. Zhong W, Ganem D. Characterization of ribonucleoprotein complexes containing an abundant polyadenylated nuclear RNA encoded by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J Virol. 1997;71:1207-1212
201. Lee SI, Murthy SC, Trimble JJ. et al. Four novel U RNAs are encoded by a herpesvirus. Cell. 1988;54:599-607
202. Cook HL, Lytle JR, Mischo HE. et al. Small nuclear RNAs encoded by Herpesvirus saimiri upregulate the expression of genes linked to T cell activation in virally transformed T cells. Curr Biol. 2005;15:974-979
203. Cazalla D, Yario T, Steitz JA. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science. 2010;328:1563-1566
204. Nanbo A, Takada K. The role of Epstein-Barr virus-encoded small RNAs (EBERs) in oncogenesis. Rev Med Virol. 2002;12:321-326
205. Howe JG, Steitz JA. Localization of Epstein-Barr virus-encoded small RNAs by in situ hybridization. Proc Natl Acad Sci U S A. 1986;83:9006-9010
206. Rosa MD, Gottlieb E, Lerner MR. et al. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol Cell Biol. 1981;1:785-796
207. Komano J, Maruo S, Kurozumi K. et al. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol. 1999;73:9827-9831
208. Kitagawa N, Goto M, Kurozumi K. et al. Epstein-Barr virus-encoded poly(A)(-) RNA supports Burkitt's lymphoma growth through interleukin-10 induction. EMBO J. 2000;19:6742-6750
209. Yang L, Aozasa K, Oshimi K. et al. Epstein-Barr virus (EBV)-encoded RNA promotes growth of EBV-infected T cells through interleukin-9 induction. Cancer Res. 2004;64:5332-5337
210. Iwakiri D, Sheen TS, Chen JY. et al. Epstein-Barr virus-encoded small RNA induces insulin-like growth factor 1 and supports growth of nasopharyngeal carcinoma-derived cell lines. Oncogene. 2005;24:1767-1773
211. Nanbo A, Inoue K, Adachi-Takasawa K. et al. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt's lymphoma. EMBO J. 2002;21:954-965
212. Nanbo A, Yoshiyama H, Takada K. Epstein-Barr virus-encoded poly(A)- RNA confers resistance to apoptosis mediated through Fas by blocking the PKR pathway in human epithelial intestine 407 cells. J Virol. 2005;79:12280-12285
213. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15-20
214. Farh KK, Grimson A, Jan C. et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817-1821
215. Pfeffer S, Zavolan M, Grasser FA. et al. Identification of virus-encoded microRNAs. Science. 2004;304:734-736
216. Edwards RH, Marquitz AR, Raab-Traub N. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J Virol. 2008;82:9094-9106
217. Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006;12:733-750
218. Cai X, Schafer A, Lu S. et al. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006;2:e23
219. Xing L, Kieff E. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J Virol. 2007;81:9967-9975
220. Zhu JY, Pfuhl T, Motsch N. et al. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol. 2009;83:3333-3341
221. Cosmopoulos K, Pegtel M, Hawkins J. et al. Comprehensive profiling of Epstein-Barr virus microRNAs in nasopharyngeal carcinoma. J Virol. 2009;83:2357-2367
222. Chen SJ, Chen GH, Chen YH. et al. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS ONE. 2010;5:e12745
223. Cullen BR. Viruses and microRNAs. Nat Genet. 2006;38(Suppl):S25-S30
224. Gottwein E, Cullen BR. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe. 2008;3:375-387
225. Sullivan CS, Grundhoff AT, Tevethia S. et al. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature. 2005;435:682-686
226. Seo GJ, Fink LH, O'Hara B. et al. Evolutionarily conserved function of a viral microRNA. J Virol. 2008;82:9823-9828
227. Seo GJ, Chen CJ, Sullivan CS. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology. 2009;383:183-187
228. Wang X, Tang S, Le SY. et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3:e2557
229. Cai X, Li G, Laimins LA, Cullen BR. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J Virol. 2006;80:10890-10893
230. Xia T, O'Hara A, Araujo I. et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008;68:1436-1442
231. Barth S, Pfuhl T, Mamiani A. et al. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008;36:666-675
232. Choy EY, Siu KL, Kok KH. et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. 2008;205:2551-2560
233. Lo AK, To KF, Lo KW. et al. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A. 2007;104:16164-16169
234. Seto E, Moosmann A, Gromminger S. et al. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010;6:e1001063
235. Gottwein E, Mukherjee N, Sachse C. et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096-1099
236. Skalsky RL, Samols MA, Plaisance KB. et al. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81:12836-12845
237. Samols MA, Skalsky RL, Maldonado AM. et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3:e65
238. Ziegelbauer JM, Sullivan CS, Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet. 2009;41:130-134
239. Dolken L, Malterer G, Erhard F. et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe. 2010;7:324-334
240. Lu CC, Li Z, Chu CY. et al. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 2010;11:784-790
241. Gottwein E, Cullen BR. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol. 2010;84:5229-5237
242. Lei X, Bai Z, Ye F. et al. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12:193-199
243. Abend JR, Uldrick T, Ziegelbauer JM. Regulation of TWEAKR expression by KSHV microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol. 2010;84:12139-12151
244. Hansen A, Henderson S, Lagos D. et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 2010;24:195-205
245. Lu F, Stedman W, Yousef M. et al. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol. 2010;84:2697-2706
246. Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe. 2009;6:570-575
247. Cameron JE, Yin Q, Fewell C. et al. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J Virol. 2008;82:1946-1958
248. Motsch N, Pfuhl T, Mrazek J. et al. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 (LMP1) Induces the Expression of the Cellular MicroRNA miR-146a. RNA Biol. 2007;4:131-137
249. Lu F, Weidmer A, Liu CG. et al. Epstein-Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J Virol. 2008;82:10436-10443
250. Costinean S, Zanesi N, Pekarsky Y. et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024-7029
251. Gatto G, Rossi A, Rossi D. et al. Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic Acids Res. 2008;36:6608-6619
252. Linnstaedt SD, Gottwein E, Skalsky RL. et al. Virally Induced Cellular MicroRNA miR-155 Plays a Key Role in B-Cell Immortalization by Epstein-Barr Virus. J Virol. 2010;84:11670-11678
253. Wang X, Wang H-K, McCoy JP. et al. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA. 2009;15:637-647
254. Li B, Hu Y, Ye F. et al. Reduced miR-34a expression in normal cervical tissues and cervical lesions with high-risk human papillomavirus infection. Int J Gynecol Cancer. 2010;20:597-604
255. Martinez I, Gardiner AS, Board KF. et al. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene. 2008;27:2575-2582
256. Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84:5212-5221
257. Anderson MD, Ye J, Xie L. et al. Transformation studies with a human T-cell leukemia virus type 1 molecular clone. J Virol Methods. 2004;116:195-202
258. Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970;226:1209-1211
259. Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211-1213
260. Stehelin D, Varmus HE, Bishop JM. et al. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170-173
261. Spector DH, Varmus HE, Bishop JM. Nucleotide sequences related to the transforming gene of avian sarcoma virus are present in DNA of uninfected vertebrates. Proc Natl Acad Sci U S A. 1978;75:4102-4106
262. Berget SM, Moore C, Sharp PA. Spliced segments at the 5' terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A. 1977;74:3171-3175
263. Chow LT, Gelinas RE, Broker TR. et al. An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell. 1977;12:1-8
264. Selbach M, Schwanhausser B, Thierfelder N. et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58-63
265. Baek D, Villen J, Shin C. et al. The impact of microRNAs on protein output. Nature. 2008;455:64-71
266. Babiarz JE, Ruby JG, Wang Y. et al. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773-2785
267. Watanabe T, Totoki Y, Toyoda A. et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539-543
268. Tam OH, Aravin AA, Stein P. et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534-538
269. Zheng ZM. Regulation of alternative RNA splicing by exon definition and exon sequences in viral and Mammalian gene expression. J Biomed Sci. 2004;11:278-294
270. Flint SJ, Gonzalez RA. Regulation of mRNA production by the adenoviral E1B 55-kDa and E4 Orf6 proteins. Curr Top Microbiol Immunol. 2003;272:287-330
271. Gaudray G, Gachon F, Basbous J. et al. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76:12813-12822
272. Murata K, Hayashibara T, Sugahara K. et al. A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol. 2006;80:2495-2505
273. Hivin P, Frederic M, Arpin-Andre C. et al. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J Cell Sci. 2005;118:1355-1362
Author contact
![]() Corresponding author: zhengtnih.gov
Corresponding author: zhengtnih.gov