Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(2):180-192. doi:10.7150/ijbs.7.180 This issue Cite
Research Paper
Novel and Conserved Micrornas in Dalian Purple Urchin (Strongylocentrotus Nudus) Identified by Next Generation Sequencing
Zhenlin Wei1, 2, Xiaolin Liu1 ![]() , Tingting Feng1, Yaqing Chang3
, Tingting Feng1, Yaqing Chang3
1. College of Animal Science and Technology, Northwest A&F University; Shaanxi Key Laboratory of Agricultural Molecular Biology, Yangling, Shaanxi, 712100, CHINA
2. Biology Science Department, Dezhou University, Dezhou, Shandong, 253023, CHINA
3. College of Life Science and Technology, Dalian Fisheries University, Dalian ,116023, CHINA
Received 2010-11-15; Accepted 2011-2-12; Published 2011-2-16
Abstract
MicroRNAs are regulators in regulation of broad range of phenotypes. The purple urchin, Strongylocentrotus nudus, is one of the most important marine economic animals that widely distributed in the cold seas along the coasts of eastern pacific area. To date, only 45 microRNAs have been identified in a related species, Strongylocentrotus purpurtus, and there is no report on S. nudus microRNAs. Herein, solexa sequencing technology was used to high throughput sequencing analysis of microRNAs in small RNA library isolated from five tissues of S. nudus. Totally, 8,966,865 reads were yielded, 131,015 of which were related to 415 unique microRNAs including 345 deuterostoma conserved and 70 urchin specific microRNAs, as well as 5 microRNA* sequences. The miRNA features including length distribution, end variations and genomic locations were characterized. Annotation of targets revealed a broad range of biological processes and signal transduction pathways that regulated by urchin miRNAs, of which signal transduction mechanisms was the subgroup containing the maximum targets. In addition, the expression of 100 miRNAs in female gonad was confirmed using microRNA microarray analysis. This study provides a first large scale cloning and characterization of S.nudus miRNAs and their potential targets, providing the foundation for further characterization for their role in the regulation of diversity of physiological processes.
Keywords: Strongylocentrotus nudus, microRNA, Target, Solexa.
Introduction
MicroRNAs (miRNAs) are endogenous, small, non-coding RNAs, around 22 nucleotides in length [3]. Upon transcription, pri-miRNA is converted to mature miRNA duplex through a sequential processing by two RNaseIII III enzymes, Drosha and Dicer [12,25]. One strand with lower stability base pairing of the 2-4 nt at the 5' end of the duplex is preferentially associates with RISC and thus becomes active miRNA, and then is guided to target sequences by base-pairing, typically with imperfect complementarity in animals [7,32,36]. MiRNAs bind target messenger RNAs (mRNAs) and block the target's expression by inhibiting translation or by targeting the mRNA for degradation or deadenylation [4]. It has evidenced that miRNAs are involved in a variety of biological processes, such as development [2,9], cell proliferation and death [5], apoptosis and fat metabolism [44], cell differentiation [8], as well as diseases [35], supporting the notion that miRNAs are indispensable players in the control of eukaryotic life/biology. Identification of abundant miRNAs and other small regulatory RNAs in different organisms is critical in improving our understanding of genome organization, genome biology, and evolution [20].
Sea urchins are echinoderms, marine animals that originated more than 540 million years ago, sharing a common ancestor with humans. Sea urchin has been proposed as a model marine animal for physiological and genetic studies, such as sea environment toxicology [33], development, evolution and disease, due to its biological and economic importance and its highly similar genomic sequence to human [14].
While the knowledge regarding to functions of miRNA are rapidly expanding, there is few report about urchin miRNA population. The reported number of urchin miRNAs is significantly lower than that in other basal deuterostoma animals, such as Ciona intestinalis (332) and Branchiostoma floridae (77), only 45 miRNAs were cloned in S.purpuratus and were deposited in miRbase v16 [42], with addition of 101 miRNAs predicted by Li et al (http://insr.ibms.sinica.edu.tw/ZooMir/). It is likely that further miRNAs remain to be discovered in urchin genome. The efforts to identify new urchin miRNAs provided the foundation for further understanding of miRNA function in the regulation of urchin development and substitute metabolism. Recent establishment of high-throughput technologies and deep sequencing analysis has allowed the identification of miRNAs that are not conserved or are expressed in low levels, such as those found in several sea animals, silkworm, amphioxus and cephalochordates [6,10,13,42].
Here we report the first deep sequencing applied to S.nudus in a mixed small RNA library using RNAs isolated from male and female gonad, muscle adjacent to mouth, gut and tube feet. The newly identified miRNAs significantly advance our knowledge of miRNA population presented in urchin and provide insights into miRNA by investigating their characteristics and information about their target genes.
Materials and Methods
Sample collection
Wild sea urchin Strongylocentrotus nudus with about 5 cm in diameter were collected along the coast of Dalian (38°54'45" N, 121°36'09" E), CHINA. The animals were laboratory acclimated for one week prior to RNA extraction.
RNA isolation and Small RNA library construction
The tissues of male gonad, female gonad, muscle adjacent to mouth, gut and tube feet were excised from animal and immediately put into liquid nitrogen for 12 hours, and then were kept frozen in dry ice during transport. Total RNA was extracted using Trizol reagent (Invitrogen, CA, USA). Approximately 20 μg total RNA was ligated with proprietary 5' and 3' adaptors developed by Illumina Inc and size-fractionated on a 6% TBE urea polyacrylamide gel and the 90 base-pair fraction was excised and recovered. The RNA was converted to single-stranded cDNA using Superscript II reverse transcriptase (Invitrogen, CA, USA) and subsequently amplified in 15 cycles using Illumina's small RNA primer set and Phusion polymerase (New England Lab, USA). The PCR products were size-fractionated and recovered for sequencing with Illumina's Genome Analyzer IIe according to manufacturer's instructions (Illumina, San Diego, CA, USA).
Bioinformatic analysis
After masking of adaptor sequences and removal of contaminated reads the clean reads were filtered for miRNA prediction with ACGT101-miR-v3.5 package (LC Sciences, Houston, USA). First, reads that matched to rRNA, tRNA, snRNA, snoRNA, repeat sequences, and other ncRNAs deposited in Rfam 8.0, as well as to the sequences containing polyA tails, were discarded. The retained 18-26 nt reads were mapped onto the Strongylocentrotus purpuratus genome (strPur2.fa.masked.gz). Sequences with up to two mismatches were retained for miRNA prediction based on 10 features. After a rigorous screening, sequences with three or more copies in frequency were considered as miRNAs. Finally, we attempted to align the predicted microRNAs to all deuterostoma known mature miRNA sequences in miRBase Version 16.0 (http://microrna.sanger.ac.uk) to distinguish novelty. Secondary structure prediction of individual miRNA was performed by MFOLD software (Version 2.38, http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) using the default folding conditions. Multiple sequence alignment was conducted using ClustalW 1.84.
In order to conduct comparative genomic analysis of the newly identified miRNAs, the genome of Ciona intestinalis, Ciona savignyi, Danio rerio and Homo sapiens were selected to map S.nudus miRNAs on these genomes with MapMi software under default parameters [21]. S.nudus miRNA mature sequences were used as custom sequences to search S.purpuratus UTR library to perform targets prediction by the RNAhybrid v2.2 (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid) with parameter settings as follows: f 2,7 -u 3 -v 3 -E e-25. The parameter “f 2,7” forces structures to have a helix from position 2 to 7 with respect to the query, indicting the seed region from 2 to 7 of mature sequence was fully complementary to target UTRs. “-E e-25” indicates the cutoff of hybrid energy is -25kcal.mol-1, only results with equal to or lower than this cutoff will be considered as targets. “-u 3”means max internal loop size is shorter than 3 nucleotides, while “-v 3” means max bulge loop size is shorter than three nucleotides.
A signal-to noise analysis was performed to estimate the specific of target prediction [1]. Firstly, 10 randomized sequences of each miRNA were generated with the program SHUFFLE from the HMMER package (http://hmmer.janelia.org), maintaining the dinucleotide frequencies of the original miRNAs. The number of predicted targets for these randomly generated miRNAs was compared with the number of predicted targets for the sequenced miRNAs. Specificities and signal-to-noise ratios were calculated for each miRNA, and the averages were defined as specificity and signal-to-noise ratio of the entire prediction. More specifically, if TP is the number of true positives and FP the number of false positives, specificity is defined as specificity=TP/(TP+FP) and signal-to-noise ratio as signal-to-noise = (TP+FP)/FP.
The targets were extracted by an in-house python script and annotated using BLAST to GO database and KOG database. KEGG annotation was conducted by KAAS software. Paraflo(TM) miRNA microarray assay was used to confirm the expression of miRNAs in female gonad (LC Sciences, Houston, USA). Briefly, 460 complementary probes (in triplicate) against miRNAs, corresponding to 415 identified in this research, and 45 known urchin miRNAs, were designed and in situ synthesized using PGR (photogenerated reagent) chemistry. RNA Cy3 labeling, microarray hybridization and array scanning were performed as previously described [19]. Hybridization intensity values were analyzed two-fold: background subtraction and signal normalization using Array-Pro software (Media Cybernetics). We considered candidate miRNAs with signal ≥ 3 × Standard Deviation (SD) of background and spot Coefficient of Variation (CV) < 0.5 to be detectable [27].
Results
Discovery miRNAs in Strongylocentrotus nudus
Sequencing of small RNAs yielded 8,966,865 reads, which were mapped to rRNA, tRNA, sno/snRNA, mRNA, and to other non-coding RNAs were discarded, leaving 787,100 (8.78%) reads that were used for miRNA identification. After mapping to the genome of S.purpuratus, a total of 415 miRNAs were identified, of which 21 conserved between S.nudus and S.purpuratus were termed: snu-miR-92c, -210, -92b, -92a, -124, -10, -29, -2007, 79, 9, 2012, 71, 1, 7, -34, -2002, -375, -182, -184, -31, -2004 and -125, respectively. Except for these 21 miRNAs, other newly isolated miRNAs could be divided into two groups, prefixed with “PN-” and “PC-”, where “PN-” denotes sequences that have other deuterostomia homologous miRNAs rather than urchin genomic cognates and “PC-” denotes sequences that have only urchin genomic cognates. Paraflo(TM) miRNA microarray was used to valid miRNA expression in female gonad. One hundred miRNAs were confirmed to express at different signal values, 68 of which were identified first time in this research and others were known S.purpuratus miRNAs [42]. The top 25 expressed miRNAs were shown in Fig.2C.
Targets and characters of identified S.nudus miRNAs. A: KOG classes of predicted targets; B: conservation profile of identified miRNAs, values on Y axis indicate the number of conserved miRNA between S.nudus and queried specie; C: Top 25 miRNAs with maximum signal detected in female gonad, spu-mir-71, -22 and -31 refer to known S.purpuratus miRNAs which added in to probes.
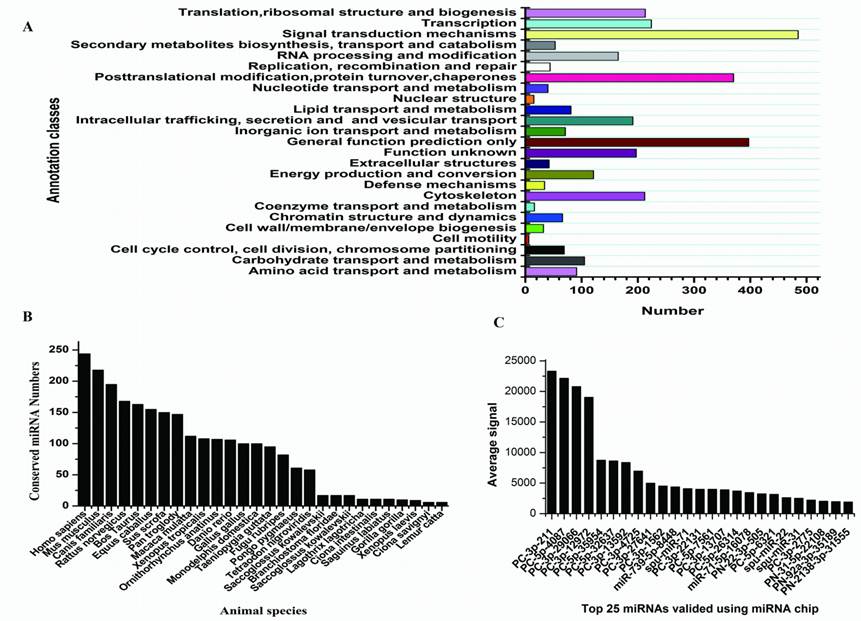
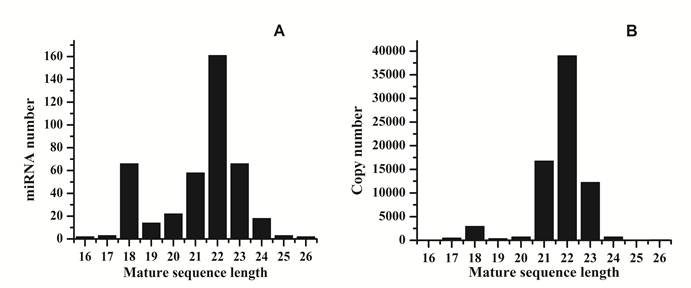
Analysis of 5' and 3' ends nucleotides of these miRNAs revealed that uridine was the most common residue at both ends (account for 49.40% and 62.41%, respectively), a pattern that has been observed for miRNAs sequenced in other organisms [10,27]. The sequencing frequency of the top ten abundantly expressed miRNAs constituted 9.56% of the total miRNA related sequencing reads, suggesting that they may be highly expressed in S.nudus. In contrast, the sequence frequencies of 113 S.nudus miRNAs were extremely low in our library (lower than five copies). It is implied that these miRNAs expressed at very low levels, in limited cell types and/or under limited circumstances, as suggested by Chen et al (2009) [10]. The size of these miRNAs was not evenly distributed. Among these sequences, the number of 22-nt miRNAs was more significant than other shorter or longer sequences (Fig.1 A and B) and account for 38.8% of the total miRNA related reads, and then followed by 23-, 21- and 18-nt miRNAs. This was well consistent with the common size of miRNAs from Dicer digestion products [6]. However, there was a slight difference on the length distribution with other research [10], in which the portion of 18 nt miRNAs was much higher than 20 nt miRNAs (Fig.1A and B).
Length distribution of raw reads and mapped reads. A: distribution of mature sequences of unique miRNAs identified in this research; B: distribution of sequenced reads related to miRNAs.
The secondary structure of miRNA is significant in typical hairpin shape, which is different from siRNA and piRNA is its identical secondary structure, which appears typical hairpin shape. It was observed in the miRNA sequences in the present research (Fig.3C).
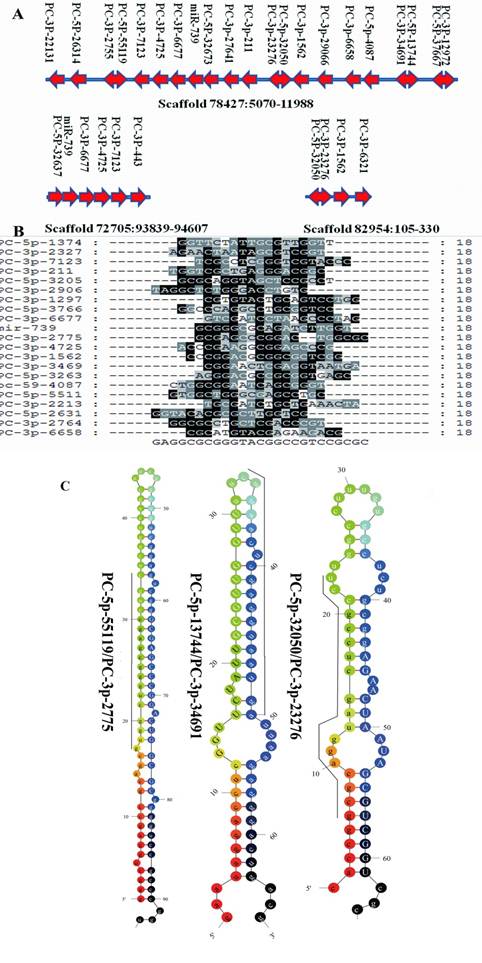
Clustered profile of S. nudus microRNAs in 3 selected scaffolds. A: miRNA gene clusters in urchin. The location of microRNAs in 3 scaffolds were showed. Rightward arrow indicate the locatzation o f mature sequences at the positive strand, while leftward arrow indicate at minus strand. Double arrow represented miRNA and their miRNA* were located in the same pre-miRNA sequences at 5' and 3' termini respectively. B: Sequences alignment of 21 miRNAs clustered in scaffold78427, no significant sequences similiarity was found in these miRNAs. C: Three microRNAs and their microRNA*s, upper-case letters refer to mature sequences, while underlined leters refer to miRNA* sequences.
MiRNA clusters
MiRNAs are often present in the genome as clusters where multiple miRNAs are aligned in the same orientation and transcribed as a polycistronic structure, allowing them to active cooperatively [3]. The majority miRNAs identified in this research are located to single-copy loci in S. purpuratus genome. There were 95 S. nudus miRNAs that could be mapped to S. purpuratus genome, which locating into 104 unique scaffolds, respectively. The most miRNA enriched scaffold78427 harbored 21 unique miRNAs, which located in a region of approx. 5000 nt on the minus strand. Sequence alignment of 21 miRNAs clustered in scaffold78427 revealed no significant homology to each other, suggesting that the majority of clustered miRNAs were unique individuals. ClustalW analysis on the sequences revealed no homology to known miRNAs, except miR-739, suggesting that 20 miRNAs of this cluster seem to be an urchin innovation. To prove this point of view, the corresponding genome sequences in scaffold78427, nucleotides from 5200 to 12000, were extracted and BLAT against the genomes of three deuterostomia lineages (lancelet, sea squirt and human) to look for homology regions. The results showed only short fragments were found homologous to the query sequences and computational prediction in these genome regions failed to recognize any miRNA.
Interestingly, 10 different miRNAs were thought to be cut from 5 unique pre-miRNA sequences, commonly accepted as miRNA and miRNA* (Fig.3A), respectively [39]. It was previously inferred that mature miRNAs are more abundant that miRNA* in living cells, however, Okamura showed a function of miRNA* as regulatory RNAs [34], and the stronger expressions of miRNA* compared to the mature miRNA have been reported in various organisms [41]. A similar abundance of miRNA and miRNA* was detected herein, although both miRNA and miRNA* appeared with low frquency. For PC-3p-2775 and PC-5p-55119, which located in the same pre-miRNA, 143 copies for both were detected. These results suggested that both S. nudus miRNA and miRNA* are important biological regulators.
Conserved and Urchin specific miRNAs
With the identification of hundreds of new sea urchin miRNAs, we could investigate whether they are conserved miRNAs across the deuterostomia lineages. The majority of reported S.nudus miRNAs are conserved across species, because alignment results showed total of 2891 homologous miRNAs in other species according to 324 miRNA out of total 415 newly sequenced miRNAs (Fig.2B). Generally, the miRNA population of urchin is more conserved to that of vertebrates than Ciona and Xenopus, in line with the high similarity of urchin genome sequences to human [37]. Moreover, the miRNAs homology to the oldest conserved miRNAs termed miR-100, let-7, miR-125 and miR-375, which shared by cnidarians and bilaterians [11], were found in this research. Among these four oldest miRNAs, miR-100, let-7 and miR-375 have never been found in urchin previously. We also identified six miRNAs that previously were thought to be vertebrate specific, including mir-15, -27, -126, -132, -183, and -196. A recent study on amphioxus and Ciona also identified mir-96, mir-183 and mir-454 [10,23], suggesting at least some of these miRNAs might be present throughout the deuterostomia lineages.
The most conserved region was mature sequence followed by complementary sequence, whereas the sequence far from the mature region was highly varied. Regions at 2-8 nucleotides in mature sequences were highly conserved. In terms of the spatial location of substitutions within the mature gene product, most changes occurred at the 3' end of the mature sequence. However, other regions, especially nucleotide 1 and nucleotide 11, showed a relatively high percentage of substitutions, such like U to C and C to U substitutions (Fig.4). This result was in line with Wheeler's observations that nucleotide 2-8 and 12-15 were highly conserved, whereas 3' end showed sequence diversity, especially for the last three nucleotides [42].
Sequences alignment of Snu-mir-375 and its homologous miRNAs

In addition the identification of conserved miRNAs, a group of 70 miRNAs was identified in this research, which termed urchin specific miRNAs and prefixed with “PC” in their names since they could be mapped to S.purpuratus genome without homology to any known deuterostomia miRNAs. These findings underscore the power of sequencing approaches in discovering novel miRNAs. To find homologous miRNAs of this group in genomes of other deuterostomia lineages, the mature sequencess were mapped to the genomes of Ciona intestinalis, Ciona savignyi, Caenorhabditis elegans, Danio rerio and Homo sapiens. The results showed 12 genomic cognates were predicted as homologous miRNAs, of which 11 cognates in Homo sapiens, 2 in Caenorhabditis elegans, 2 in Ciona intestinalis and 2 in Danio rerio, respectively. In these 12 unique “PC” series miRNAs, PC-184-3P-1209 was detected among four lineages, while others existed in a species-specific manner, suggesting the majority of “PC” series miRNAs are urchin specific.
Interestingly, the frequency of urchin specific miRNAs was lower than the average of all miRNAs, account for 38.5 copies per miRNA in average, while the corresponding value of all miRNAs is about 300. This is consistent with previous conclusion indicating that non-conserved miRNAs are usually expressed at lower levels and with a tissue- or developmental-specific pattern [46]. Another significant feature of this group of miRNAs was that their length of mature sequences were much shorter than the average of overall, approximately at 18.3 nt in average (21.3 nt for all miRNAs). Among 70 sequences, 49 were of 18 nt in length, 8 of 19 nt, 12 of 20 nt and 1 of 22 nt. Accordingly, the average length of pre-miRNAs sequences of this group was 85.18 nt, whereas 94.53 nt for all pre-microRNAs, revealing a significant shorter mature and pre-microRNA length of S.nudus specific miRNA than conserved miRNAs. The shorter of sequences of Ciona specific miRNAs were observed (out of 200 Ciona miRNAs short than 60bp in length, 189 are Ciona specific, such as cin-mir-4037 and cin-mir-4044), implying a common features of specific miRNAs in basal deuterostomia lineages. The mechnaism of generating such like miRNAs was remained to be understand. As stated by Starega-Roslan et al (2010) [38], asymmetrical structural motifs present in precursor hairpins are primarily responsible for the length diversity of miRNAs. In other words, this means that the structure of pre-miRNA provide influence on mature sequence length diversity. As mentioned above, it was found that the precursor sequences of “PC” series miRNAs was shorter than conserved miRNAs, accordingly, the asymmetrical structural motifs was lower frequently appeared in these precursors (Fig.3C), generating shorter mature sequences.
End variations of microRNAs in S. nudus
It has been frequently found that miRNAs exhibit heterogeneous 5′ and 3′ ends, and post-transcriptional nontemplate 3′ end additions of uridines or adenosines [16,42]. Here, frequent end variation with regard to the reference sequences were found in this research with highly preferential at the 3′-end of sequenced miRNAs, in line with previous reports of miRNA editing [16,42]. Total 239 newly identified miRNAs had multi-isoforms, achieving up to 149 isoforms for miR-184-3p. Importantly, these 239 miRNAs could be divided into three groups, 16 of which prefixed with “miR-”, 22 prefixed with “PC-” and 201 prefixed with “PN-”, indicating that the majority of highly diversitied miRNAs was conserved across species.
Diversity of variations at 5' end could be divided to two types, termed base addition and base substitution, respectively. We found the G addition predominated at 5'end variations, then followed by C and TA addition. Compared with the frequency of base addition, base substitution was occurred with lower frequency. For example, among 258 isoforms of PN-199-5p-1550, 48 isoforms showed replacement of CC with TA at 5' end, and 3 isoforms with CA, wherase remains appeared varieties of base additions. Variations at 3' end were a more frequent phenomenon in this research, most of them were base deletions, then followed by base replacing and base additions, and to more lower frequency, were combinational vriations that comprise of three or two types mentioned above (Table 1). However, patrial of end variations was likely due to miamatches generated in solexa sequencing, and experimantal validations such as 5' and 3' RACE need to be carried out in further research.
End variation pattern of PN-101-3p
| PN-101-3p Isoforms | Sequence | Sequence length | Copy frrequency | Variation type |
|---|---|---|---|---|
| 1 | TACAGTACTGTGATAACTGAAG | 22 | 811 | standard |
| 2 | GTACAGTACTGTGATAACTGA | 21 | 294 | 5'A; 3'D; |
| 3 | GTACAGTACTGTGATAACTGAA | 22 | 168 | 5'A; 3'D; |
| 4 | TACAGTACTGTGATAACTGAA | 21 | 127 | 3'D |
| 5 | TACAGTACTGTGATAACTGAAA | 22 | 111 | 3'C |
| 6 | TACAGTACTGTGATAACTGAAGA | 23 | 62 | 3'D |
| 7 | TACAGTACTGTGATAACTGAAT | 22 | 57 | 3'C |
| 8 | GTACAGTACTGTGATAACTGAAA | 23 | 41 | 5'A; 3'C |
| 9 | TACAGTACTGTGATAACTGAAGT | 23 | 29 | 3'D |
| 10 | GTACAGTACTGTGATAACTGAC | 22 | 10 | 5'A; 3'D; |
| 11 | TACAGTACTGTGATAACTGCAG | 22 | 9 | 3'C |
| 12 | GTACAGTACTGTGATAACTGAT | 22 | 9 | 5'A; 3'D; 3'C |
| 13 | TACAGTACTGTGATAACTGAAC | 22 | 8 | 3;C |
| 14 | GTACAGTACTGGGATAACTGA | 21 | 7 | 5'A; 3'D; |
| 15 | ACAGTACTGTGATAACTGAAGT | 22 | 4 | 5'D; 3;A |
| 16 | ACAGTACTGTGATAACTGAAG | 21 | 4 | 5'D |
| 17 | GTACAGTACTGTGATAACT | 19 | 4 | 5'A; 3'D |
| 18 | GTACAGTACTGTGATAACTGAAT | 23 | 4 | 5'A; 3'C |
| 19 | TACAGTACAGTGATAACTGAA | 21 | 3 | 5'D; 3'D |
| 20 | GTACAGTACTGTGATAACTGAAG | 23 | 3 | 5'A |
| 21 | GTACAGGACTGTGATAACTGA | 21 | 3 | 5'A; 3'D |
| 22 | TACAGTACTGTCATAACTGAAG | 22 | 3 | 5'D |
Note: This table showed typical end variations of S.nudus microRNAs. 5'A means the base addition at 5' end, 5'D means deletion at 5' end, 3'D means 3' end deletion, 3'C means 3' changing, 3'A means 3' addition. Additional nucleotides both at 5' and 3' end were marked with red color. “Seed” sequences which located at 2-7 nt of 5' end were underlined.
Importantly, several types of 5' end variation resulted in shifting of “seed” sequences. For PN-101-3p, 5' addition of G or deletion of T genetated a shifting in the “seed sequences” from “ACAGTA” to “GACAGT” and “CAGTAC”, respectively (Table1). These shifting of “seed” sequence maybe cause off-targeting, thus we predicted the targets of PN-101-3p and the two isoforms using RNAhybrid in a selected UTR sets that belong to genes included in GO:0019953. Results showed significant difference in the predicted targets between reference miRNAs and two isoforms. For refence sequence, 21 targets were found, of which 4 targets differ with that of isoform1, 7 targets differ with isoform 2, while 14 differ between isoform1 and isoform2, yielding 12 common targets among these three miRNAs. Considering that 5' base addition and deletion were occurred frequently, the “seed shift” would generate more complex regulation network. Further investigation need to be conducted to elucidate the biological relevance of these miRNA isoforms and whether the composition of isoforms of urchin miRNAs is dynamitically regulated.
Target Prediction
With the specific value at 0.85 and signal-noise value at 6.28, total 5396 genes were predicted as targets of 311 miRNAs, suggesting approx. 17 targets per miRNA. The corresponding value for 70 urchin specific miRNAs were 2209 targets with a ratio of 33 targets per miRNA, indicating that the urchin specific miRNAs were targets enriched than conserved miRNAs. These targets were then sent to GO, KOG and KEGG annotation, and all of the results showed a diversity of regulation roles of these miRNAs. Given 1e-10 as cutoff, 4367 targets were functional annotated in KOG database, which could be divided to 25 subgroups (Fig.1A), signal transduction mechanisms of which was the subgroup comprising the maximum targets, then followed by the categories of post-translation modification. One target (XM_001179984) of PN-222a-3p-9101 is similar to leucine zipper bearing kinase (LZK) and was mapped onto MAPK signaling pathway (http://www.genome.jp/kegg-bin/highlight_pathway?scale=1.0&map=map04010&keyword=MAPK2), through which participate in a complex signal transduction system. XM_001179984 located upstream of p38 and JNK pathways, suggesting its critical role in regulation on biological process involving in cell proliferation, differentiation and apoptosis [40]. Except for regulator genes, many targets were annotated to enzymes in metabolism pathways. It is well known that urchin gonad is enriched in polyunsaturated lipids, and the identification miRNAs in urchin as well as targets prediction provided the first clue for understanding of regulation of urchin polyunsaturated lipids synthesis. For example, out of 81 targets were predicted to be associate with function of lipid transport and metabolism, one target of PC-130c-3p-1672 encode a protein with hydrolase activity and was thought to be an ortholog of bifunctional leukotriene A4 hydrolase/aminopeptidase (LTA4H), which an enzyme participates in arachidonic acid metabolism. Arachidonic acid is an important biological molecular and the precursor to generate other essential substances; therefore, it is likely that the metabolism of polyunsaturated lipids in urchin gonad is regulated by miRNAs.
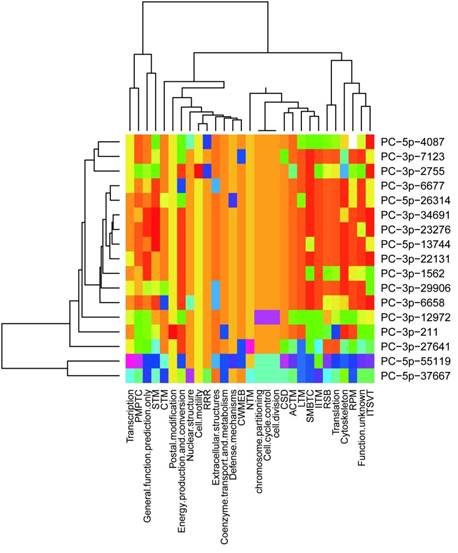
It was concluded previously that clustered miRNAs were usually functional coordinated [26], thus it was stand to reason to investigate whether the miRNAs clustered in scaffold78427 have functions related to each other. We analyzed the targets of 21 miRNAs in scaffold78427 (Fig.3A). As mentioned above, miRNAs clustered in scaffold78427 have no consensus mature sequences, however, the targets of them showed similarities to somewhat, focusing on several common KOG categories. It was more obvious for miRNAs from PC-3p-6677 to PC-3p-6688 (Fig.5). Nucleotide transport and metabolism, replication, recombination and repair, chromatin structure and dynamics, as well as cell divison and cell cycle were the common KOG classes they regulated, suggesting their incooperating functions on regulation. In addition, in the categories of posttranslational modification, protein turnover and chaperones, targets with chaperones functions were intensively regulated by miRNAs in this cluster. Among the targets, TCP-1 alpha subunit (CCT1) of chaperonin complex, Cpn60/Hsp60p (mitochondrial chaperonin), AAA+-type ATPase, chaperones of DnaJ superfamily, subunit 2 and 3 of molecular chaperone prefoldin, molecular co-chaperone STI1, thiol-disulfide isomerase and thioredoxin, mitochondrial chaperonin, proprotein convertase (PC) 2 chaperone, as well as chaperones GRP78/BiP/KAR2 and chaperones mortalin/PBP74/GRP75 of HSP 70 superfamily, were functional with unfold protein binding (GO:0051082). These genes were targeted by PC-5p-4087, PC-3p-29066, PC-5p-55119, PC-5p-6321, PC-3p-6677, PC-3p-13592, PC-3p-1562, respectively, which all clustered in scaffold78427 (Fig.3A). This results revealed the functional associations between clustered miRNAs in urchin.
Distribution of predicted targets of miRNAs clustered scaffold78427. ACTM: Amino acid transport and metabolism; CTM: Carbohydrate transport and metabolism; CWMEB: Cell wall/ membrane/ envelope biogenesis; PMPTC: Post-translational modification, protein turnover, chaperones; CSD: Chromatin structure and dynamics; IITM: Inorganic ion transport and metabolism; ITSVT: Intracellular trafficking, secretion and vesicular transport; LTM: Lipid transport and metabolism; NTM: Nucleotide transport and metabolism; RRR: Replication, recombination and repair; RSB: ribosomal structure and biogenesis; RPM: RNA processing and modification; SMBTC: Secondary metabolites biosynthesis, transport and catabolism; STM: Signal transduction mechanisms.
Discussion
It was well established that miRNAs possess important effects on normal biological regulation through which bind to target mRNAs and repress the translation or induce deadenylation of targets in animal cells [4]. Identifying the total number of miRNA genes in a species, especially those low-abundance and species-specific ones, is helpful for appreciating the breadth of miRNA functions [6]. It was estimated that miRNAs comprise 1-5% of animal genes [17], making them become one of the most abundant classes of gene regulators. NGS is an ideal method to identify small ncRNAs in an unbiased and unselected fashion. It does not require a priori knowledge of the sequence of the RNA species to be detected, but provides exact sequence information quantitatively, enabling it as a ideal approach for identification of miRNA and other smanll non-coding RNA in a diverse species, especially in those less studied. Therefore, researches on deep sequencing of the miRNAs and other small ncRNAs have been performed intensively [6,10,13,22,29,30,42].
Sea urchins in the genus Strongylocentrotus are important research models in many areas of bioscience including developmental and cell biology, reproductive biology and evolutionary biology. The sea urchin S.nudus is widely distributed in the cold seas along the coasts of northern China, the Korean Peninsula, the Russian Far East and northern Japan [45]. S. nudus has been studied in ecology and reproductive biology [28]. As the gonads of S. nudus are extensively eaten as a delicacy in China, Korea and Japan, the sea urchin is an important resource for coastal fisheries. In China, however, natural stocks of S. nudus have declined dramatically due to overfishing and damage of natural habitat. At present, S. nudus has been listed as endangered in China Species Red List [28]. The economic and biomedical importance of the urchin lead to significant efforts to decode the urchin genome and its genetics. However, only 45 records were currently repoisted in MiRBase 16.0 version, laging behind other deuterostomia species. The results of this study will help us identify the miRNA-based regulatory system not only of this urchin species but also of basel deuterostomia lineages.
In the present research, we have sequenced and characteried miRNAs from from a pooled RNA samples from five organs of S.nudus, based on the Solexa high-throughput sequencing system. In total, we identified 415 novel miRNA expressed in very different frequence, 70 of which were urchin specific, 21 has been reported in S.purpuratus and 5 were recognized as miRNA*s. The results showed that the method is effective for identifying low-abundance and species-specific small RNAs. It should be note that the filter used in this research is >=3 copy, thus the miRNAs identified in this study might represent only portion of novel miRNA components found in S.nudus due to the fact that the small RNA library that can not cover all tissues and development stages. Further experiments would provide more insight into the function of these miRNAs.
Among the reported 45 urchin miRNAs (all of S.purpuratus), 21 were found in our research. These 24 failures are probably due to several chance events including incompleteness of the sampling (missing some stage or tissue-specific miRNAs), the delicate RNA processing procedure, and uneven sequencing depth [15]. Another potential reason contributed to the failure of detecting of 24 remained miRNAs lies in that the genomic sequence of urchin is more variable than vertebrate, account for about 4-5% [37]. In addition, miRNAs with PN as prefix in their names denote that they were high homology to miRNAs of deuterostoma lineages without genomic mapping information on S.purpuratus. Both of these two results showed that the stringent filter for cross-genome alignment used in this study hamper the finding of more conserved miRNAs. We cannot estimate how many miRNAs were missed in this research, since we only used five tissues to construct small RNA library for sequencing. In addition, the real population of miRNA is dynamic changed in different tissue and at different development stages. Tthe miRNAs identified here maybe not represente all miRNAs existing in urchin and more sequencing researches were need to achieve full set of urchin miRNAs.
The sequencing frequency of the miRNAs generally reflected their relative abundance and was used to establish miRNA expression profiles [10]. For the 20 conserved miRNAs between S.nudus and S.purpurtus, their sequencing frequency was not evenly distributed. The most abundance is snu-mir-184, followed by snu-mir-10, whereas others below 100 copies. The relationship of copy frequency to miRNA functional roles is unclear yet in the present case, although it is believed that high-throughput sequencing not only is capable of identifying miRNAs but also provides information on gene expression albeit with variable results [24]. The human has-mir-184 and mir-10 both regulate more than 1000 targets. By contrast, only 30 targets were found for snu-mir-184, this value was below than that of PC series of miRNAs, which expressed in low frequency. As speculated by Cai et al (2010) [6], the higher expressed bmo-miR-1 and bmo-miR-263a alludes to their functional roles in regulating silkworm development. In the present case, it is far away to underline the relationship of miRNA expression level with its function in urchin, which need investigate further.
Several recent reports have demonstrated that microRNAs (miRNAs) can exhibit heterogeneous ends and post-transcriptional nontemplate 3' end additions of uridines or adenosines [16]. Experimental reslts evidented that the composition of isoforms including size of mature sequences and end modification was dynamically regulated and biologically meaningful, variated according to developmental stage and differrent tissues [16]. In the presented research, frequent end heterogeneous and size variation of mature sequences was observed, both at 3' and 5' ends, and in some cases resulted in “seed shift” through which alert its targets, suggesting a complex population of urchin miRNAs and a complicated regulaton networks. The nontemplate addition of A or U at 3' end was found for several miRNAs, such as mir-184, demostrating a similar end modification pattern in urchin miRNAs. On the other hand, there is evidence in A. thaliana that terminal nucleotide additions occur after Dicer processing of the miRNA precursor, and that some variants are differentially loaded into Argonautes [16 and references therein]. Likewise, in P. trichocarpa, miRNAs that possess adenosine additions are less prone to degradation [37]. Further investigation need to be conducted to elucidate the biological relevance of these miRNA isoforms and whether the composition of isoforms of urchin miRNAs is dynamitically regulated.
In the present research, miRNAs conserved in deuterostomia lineages were identified, including those that thought to be only existed in verbrated. It is interested that the higher level of miRNA repertoire homology between echinodermata and vertebrates, such as Danio rerio and Homo sapiens, whereas largely different with Ciona intestinalis and Ciona savignyi, which would due to the fact that urchin has a more higher morphological complex than species of urochordata. Fu et al. (2008) predicted 14 homologs of vertebrate miRNAs in C. intestinalis. Similarity, out of the total 49 miRNA families of O. dioica, a species in the urochordate group, only eight are homologs of the vertebrates (Fu et al. 2008) [18]. Therefore, the comparisons of the miRNA repertoires among the three chordate groups in this study indicated that echinodermata possess twenty-five more miRNAs homologous to vertebrates than the urochordates do. Given the fact that urochordates are morphologically simplified, the higher level of miRNA overlapping between the echinodermata and vertebrates provided a clear evidence that miRNA was continuous added following morphologically complex in deuterostomia lineages
Our study showed majority of the potential targets of urchin miRNAs is highly enriched in signal transduction and posttranslation modification, as well as transcription and translation. Out of targets hit signalling transduction system, several members mapped onto MAPK pathway which vital for p38, JNK, NFκB and p53 to regulate cell proliferation, differentation and apoptosis [40]. All of these proteins plus many other targets binding DNA and RNA showed urchin miRNAs are regulators for maintain cell process under normal or stressed condions. It should be note that many atrgets were failed in KOG and GO classification, due to their relative low homology to orthologs and uncomplete annotation, thus the miRNA-targets network is likely more complex than the results presented here. Beside, more efforts and experiments shoule be conducted to provide detailed informations on how urchin miRNAs regulation.
Conclussion
This study provides the first large scale identification and characterization of S.nudus miRNAs and of their potential targets, as well as confirmed the expression of 100 miRNAs in female gonad. These miRNAs comprise of 345 deuterostoma conserved and 70 S.nudus specific miRNAs, thereby enriched database of new miRNA and give insights into urchin miRNA populations. Meanwhile, this information provided the foundation for further characterized for their roles in the regulation of diverse physiological pathways in deuterostoma animals.
Acknowledgements
We thank Prof.Yonghua Wang, Mr.Junxiao Xu and Mr.Mingtai Liu for their technical assistance.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Alves-Junior L, Niemeier S, Hauenschild A, Rehmsmeier M, Merkle T. Comprehensive prediction of novel microRNA targets in Arabidopsis thaliana. Nucl Acids Res. 2009;37(12):4010-4021
2. Ambros SV. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;114:673-676
3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297
4. Beilharz TH, Humphreys DT, Clancy JL, Thermann R, Martin DIK, Hentze MW, Preiss T. microRNA-mediated messenger RNA deadenylation contributes to translational repression in mammalian Cells. PLoS One. 2009;4(8):e6783
5. Brennecke J, Hipfner DR, Stark A, Russell PB, Cohen SM. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25-36
6. Cai Y, Yu X, Zhou Q, Yu C, Hu H, Liu J, Lin H, Yang J, Zhang B, Cui P, Hu S, Yu J. Novel microRNAs in silkworm (Bombyx mori). Funct Integr Genomics. 2010;10:405-415
7. Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Na Struct Mol Biol. 2004;11:214-218
8. Chang S, Jr Johnson RJ, Frokjaer-Jensen C, Lockery S, Hobert O. MicroRNAs act sequentially and asymmetrically to control chemosensory laterality in the nematode. Nature. 2004;430:785-789
9. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83-86
10. Chen X, Li QB, Wang J, Guo X, Jiang XR, Ren ZJ, Weng CY, Sun GX, Wang XQ, Liu YP, Ma LJ, Chen JY, Wang J, Zen K, Zhang JF, Zhang CY. Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biology. 2009;10(7):R78
11. Christodoulou F, Raible F, Tomer R, Simakov O, Trachana K, Klaus S, Snyman H, Hannon GJ, Bork P, Arendt D. Ancient animal microRNAs and the evolution of tissue identity. Nature. 2010;463(7284):1084-8
12. Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell. 2004;16:861-865
13. Dai ZH, Chen ZZ, Ye H, Zhou LH, Cao LX, Wang YQ, Peng SH, Chen LB. Characterization of microRNAs in cephalochordates reveals a correlation between microRNA repertoire homology and morphological similarity in chordate evolution. Evolution & Development. 2009;11:41-49
14. Davidson EH. The sea urchin genome: where will it lead us? Science. 2006;314:939-40
15. Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC. High-throughput sequencing of Arabidopsis micro-RNAs: evidence for frequent birth and death of miRNA genes. PLoS One. 2007;2:e219
16. Fernandez-Valverde SL, Taft RJ, Mattick JS. Dynamic isomiR regulation in Drosophila development. RNA. 2010;16(10):1881-1888
17. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92-105
18. Fu XH, Marcin Adamski, Eric M. Thompson. Altered miRNA repertoire in the simplified chordate, Oikopleura dioica. Molecular Biology and Evolution. 2008;25(6):1067-1080
19. Gao X, Gulari E, Zhou X. In situ synthesis of oligonucleotide microarrays. Biopolymers. 2004;73:579-596
20. Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP, Tizard ML. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008;18:957-964
21. Guerra-Assunção JA, Enright AJ. MapMi: automated mapping of microRNA loci. BMC Bioinformatics. 2010;11:133
22. Hao L, Cai P, Jiang N, Wang H, Chen Q. Identification and characterization of microRNAs and endogenous siRNAs in Schistosoma japonicum. BMC Genomics. 2010;11:55
23. Hendrix D, Levine M, Shi W. miRTRAP, a computational method for the systematic identification of miRNAs from high throughput sequencing data. Genome Biology. 2010;11(4):R39
24. Kato M, de Lencastre A, Pincus Z, Slack FJ. Dynamic expression of small non-coding RNAs, including novel micro-RNAs and piRNAs/21U-RNAs, during C. elegans development. Genome Biol. 2009;10:R54
25. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mo Cell Biol. 2005;6:376-385
26. Kim YK, Yu J, Han TS, Park SY, Namkoong B, Kim DH, Hur K, Yoo MW, Lee HJ, Yang HK, Kim VN. Functional links between clustered microRNAs: suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009;37(5):1672-1681
27. Legeai F, Rizk G, Walsh T, Edwards O, Gordon K, Lavenier D, Leterme N, Méreau A, Nicolas J, Tagu D, Jaubert-Possamai S. Bioinformatic prediction, deep sequencing of microRNAs and expression analysis during phenotypic plasticity in the pea aphid, Acyrthosiphon pisum. BMC Genomics. 2010;11:281
28. Li JJ, Li Q. A set of microsatellite markers for use in the endangered sea urchin Strongylocentrotus nudus developed from S. purpuratus ESTs. Conserv Genet. 2008;9:743-745
29. Li M, Xia Y, Gu Y, Zhang K, Lang Q, Chen L, Guan J, Luo Z, Chen H, Li Y, Li Q, Li X, Jiang AA, Shuai S, Wang J, Zhu Q, Zhou X, Gao X, Li X. MicroRNAome of porcine pre- and postnatal development. PLoS One. 2010;5(7):e11541
30. Liang C, Zhang X, Zou J, Xu D, Su F, Ye N. Identification of miRNA from Porphyra yezoensis by High-Throughput Sequencing and Bioinformatics Analysis. PLoS One. 2010;5(5):e10698
31. Lu S, Sun Y-H, Chiang VL. Adenylation of plant miRNAs. Nucleic Acids Res. 2009;37:1878-1885
32. Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343-349
33. Nacci D, Jackim E, Walsh R. Comparative evaluation of three rapid marine toxicity tests: Sea urchin early embryo growth test, sea urchin sperm cell toxicity test and microtox. Environmental Toxicology and Chemistry. 1986;6:521-525
34. Okamura K, Phillips MD, Tyler DM, Duan H, Chou Y, Lai E. The regulatory activity of microRNA* species has substantial influence on microRNA and 3'UTR evolution. Nat Struct Mol Biol. 2008;15:354-363
35. Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226-230
36. Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199-208
37. Sea Urchin Genome Sequencing Consortium, Sodergren E, Weinstock GM, Davidson EH, Cameron RA, Gibbs RA, Angerer RC, Angerer LM, Arnone MI, Burgess DR, Burke RD, Coffman JA, Dean M, Elphick MR, Ettensohn CA, Foltz KR, Hamdoun A, Hynes RO, Klein WH, Marzluff W, McClay DR, Morris RL, Mushegian A, Rast JP, Smith LC, Thorndyke MC, Vacquier VD, Wessel GM, Wray G, Zhang L, Elsik CG, Ermolaeva O, Hlavina W, Hofmann G, Kitts P, Landrum MJ, Mackey AJ, Maglott D, Panopoulou G, Poustka AJ, Pruitt K, Sapojnikov V, Song X, Souvorov A, Solovyev V, Wei Z, Whittaker CA, Worley K, Durbin KJ, Shen Y, Fedrigo O, Garfield D, Haygood R, Primus A, Satija R, Severson T, Gonzalez-Garay ML, Jackson AR, Milosavljevic A, Tong M, Killian CE, Livingston BT, Wilt FH, Adams N, Bellé R, Carbonneau S, Cheung R, Cormier P, Cosson B, Croce J, Fernandez-Guerra A, Genevière AM, Goel M, Kelkar H, Morales J, Mulner-Lorillon O, Robertson AJ, Goldstone JV, Cole B, Epel D, Gold B, Hahn ME, Howard-Ashby M, Scally M, Stegeman JJ, Allgood EL, Cool J, Judkins KM, McCafferty SS, Musante AM, Obar RA, Rawson AP, Rossetti BJ, Gibbons IR, Hoffman MP, Leone A, Istrail S, Materna SC, Samanta MP, Stolc V, Tongprasit W, Tu Q, Bergeron KF, Brandhorst BP, Whittle J, Berney K, Bottjer DJ, Calestani C, Peterson K, Chow E, Yuan QA, Elhaik E, Graur D, Reese JT, Bosdet I, Heesun S, Marra MA, Schein J, Anderson MK, Brockton V, Buckley KM, Cohen AH, Fugmann SD, Hibino T, Loza-Coll M, Majeske AJ, Messier C, Nair SV, Pancer Z, Terwilliger DP, Agca C, Arboleda E, Chen N, Churcher AM, Hallböök F, Humphrey GW, Idris MM, Kiyama T, Liang S, Mellott D, Mu X, Murray G, Olinski RP, Raible F, Rowe M, Taylor JS, Tessmar-Raible K, Wang D, Wilson KH, Yaguchi S, Gaasterland T, Galindo BE, Gunaratne HJ, Juliano C, Kinukawa M, Moy GW, Neill AT, Nomura M, Raisch M, Reade A, Roux MM, Song JL, Su YH, Townley IK, Voronina E, Wong JL, Amore G, Branno M, Brown ER, Cavalieri V, Duboc V, Duloquin L, Flytzanis C, Gache C, Lapraz F, Lepage T, Locascio A, Martinez P, Matassi G, Matranga V, Range R, Rizzo F, Röttinger E, Beane W, Bradham C, Byrum C, Glenn T, Hussain S, Manning G, Miranda E, Thomason R, Walton K, Wikramanayke A, Wu SY, Xu R, Brown CT, Chen L, Gray RF, Lee PY, Nam J, Oliveri P, Smith J, Muzny D, Bell S, Chacko J, Cree A, Curry S, Davis C, Dinh H, Dugan-Rocha S, Fowler J, Gill R, Hamilton C, Hernandez J, Hines S, Hume J, Jackson L, Jolivet A, Kovar C, Lee S, Lewis L, Miner G, Morgan M, Nazareth LV, Okwuonu G, Parker D, Pu LL, Thorn R, Wright R. The genome of the sea urchin Strongylocentrotus purpuratus. Science. 2006;314(5801):941-952
38. Starega-Roslan J, Krol J, Koscianska E, Kozlowski P, Szlachcic WJ, Sobczak K, Krzyzosiak WJ. Structural basis of microRNA length variety. Nucleic Acids Res. 2011;39(1):257-68
39. Stark A, Kheradpour P, Parts L, Brennecke J, Hodges E, Hannon GJ, Kellis M. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res. 2007;12:1865-1879
40. Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer. 2009;9:537-549
41. Wei Y, Chen S, Yang P, Ma Z, Kang L. Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biol. 2009;10:R6
42. Wheeler BM, Alysha M. Heimberg, Vanessa N. Moy, Erik A. Sperling, Thomas W. Holstein,Steffen Heber, and Kevin J. Peterson. The deep evolution of metazoan microRNAs. Evolution& Development. 2009;11:50-68
43. Wu H, Ye C, Ramirez D, Manjunath N. Alternative Processing of Primary microRNA transcripts by Drosha generates 5' end variation of mature microRNA. PLoS One. 2009;4(10):e7566
44. Xu P, Vernooy SY, Guo M, Hay BA. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol. 2003;13:790-795
45. Yan J, Peng W, Du H, Li L, Zhou Z, Bao Z, Hu J. Isolation and characterization of 61 microsatellite markers from sea urchin Strongylocentrotus nudus. Conservation Genet Resour. 2010;2:35-38
46. Zhao CZ, Xia H, Frazier TP, Yao YY, Bi YP, Li AQ, Li MJ, Li CS, Zhang BH, Wang XJ. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L). BMC Plant Biology. 2010;10:3
Author contact
![]() Corresponding author: Email: liuxiaolinedu.cn; Tel: +86-29-87092102; Fax: +86-29-87092164
Corresponding author: Email: liuxiaolinedu.cn; Tel: +86-29-87092102; Fax: +86-29-87092164