Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Regulation of sirtuins in...
SIRT1 in energy metabolism
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(5):575-587. doi:10.7150/ijbs.7.575 This issue Cite
Review
Mammalian Sirtuins and Energy Metabolism
Xiaoling Li ![]() , Nevzat Kazgan
, Nevzat Kazgan
Laboratory of Signal Transduction, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA
Received 2011-2-28; Accepted 2011-3-30; Published 2011-5-9
Abstract
Sirtuins are highly conserved NAD+-dependent protein deacetylases and/or ADP-ribosyltransferases that can extend the lifespan of several lower model organisms including yeast, worms and flies. The seven mammalian sirtuins, SIRT1 to SIRT7, have emerged as key metabolic sensors that directly link environmental signals to mammalian metabolic homeostasis and stress response. Recent studies have shed light on the critical roles of sirtuins in mammalian energy metabolism in response to nutrient signals. This review focuses on the involvement of two nuclear sirtuins, SIRT1 and SIRT6, and three mitochondrial sirtuins, SIRT3, SIRT4, and SIRT5, in regulation of diverse metabolic processes.
Keywords: sirtuins, energy metabolism, aging, nutrients
Introduction
Yeast silent information regulator 2 (Sir2) protein and its homologues in other prokaryotes and eukaryotes, also known as sirtuins, are highly conserved NAD+-dependant protein deacetylases and/or ADP-ribosyltransferases (1-3). In yeast, Sir2 is of great importance in the maintenance of the silent chromatin at the mating-type loci, telomeres, and rRNA-encoding DNA repeats (reviewed in (4)). However, sirtuins in other model organisms have been increasingly recognized as crucial regulators for a variety of cellular processes, ranging from energy metabolism and stress response to tumorigenesis and aging (5).
About a decade ago, Sir2 was first implicated as a limiting component of yeast longevity (6, 7). Since then, many members of this family have been identified as key longevity regulators in species ranging from yeast to fly (reviewed in (8, 9)). For example, in S. cerevisiae, deletion of the Sir2 gene dramatically shortens lifespan, whereas an extra copy of the Sir2 extends lifespan by 40% (7). In C. elegans, overexpression of Sir2.1, a Sir2 homologue, extends lifespan by up to 50% (10). Similar results are seen in Drosophila (11). The mammalian genome encodes seven sirtuins, named SIRT1 to SIRT7 (12). Currently, the role of mammalian sirtuins in the regulation of mammalian aging is an area of intense research.
The seven mammalian sirtuin proteins share a highly conserved NAD+-binding and catalytic core domain, but have distinct flanking N- and C-terminal extensions (12) (Figure 1). Many mammalian sirtuins bears a NAD+-dependant protein deacetylase activity. A variety of acetyl-proteins have been identified as their substrates at different physiological and pathological conditions (Table 1), although whether p53 is a true substrate for SIRT7 is still not clear. The only reported activity of SIRT4, on the other hand, is the NAD+-dependant ADP-ribosyltransferase activity (13, 14). The divergent N and C-termini of sirtuins are responsible for their variation binding partners, substrates, and subcellular localization ((9), Figure 1). Three of the mammalian sirtuins, SIRT1, SIRT6, and SIRT7, are nuclear proteins (Figure 1). SIRT1, while predominately a nuclear enzyme, can also shuttle between cytosol and nucleoplasm in various tissues in response to different environmental signals (15). SIRT6 is a nuclear, chromatin-bound protein (16), whereas SIRT7 is highly enriched in nucleolus (17). Three additional mammalian sirtuins, SIRT3, SIRT4, and SIRT5, are found in mitochondria. They participate in a number of metabolic and survival processes associated with the mitochondrial activity (reviewed in (18)). The other mammalian sirtuin, SIRT2, is primarily cytosolic.
Schematic representation of seven mammalian sirtuins.
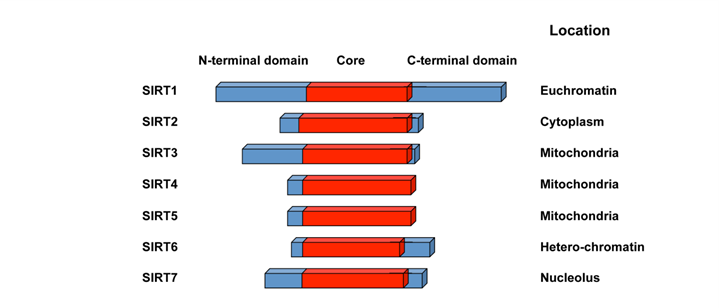
Mammalian sirtuins. Abbreviations: p53, tumor suppressor protein 53; Foxo, forkhead box O; Bax, Bcl2 associated X protein; Hif, hypoxia-inducible factor; HSF1, heat shock factor 1; STAT3, signal transducer and activator of transcription 3; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; NF-κB, nuclear factor-kappa B; TORC2/CRTC2, a transcriptional coactivator for the transcription factor CREB; LXR, liver X receptor; FXR, farnesoid X receptor; SREBP, sterol regulatory element binding protein; PER2, period circadian protein homolog 2; H4, histone 4; AceCS2, acetyl-CoA synthetase 2; LCAD, long-chain acyl CoA dehydrogenase; HMG-CoA synthase 2, 3-hydroxy-3-methylglutaryl CoA synthase 2; IDH2, isocitrate dehydrogenase 2; MnSOD, Mn-superoxide dismutase; SOD2, superoxide dismutase 2; GDH, glutamate dehydrogenase; CPS1, carbamoyl phosphate synthetase 1; CtIP, C-terminal binding protein interacting protein; H3K9, histone 3 acetyl-lysine 9; H3K56, histone 3 acetyl-lysine 56; CtIP, C-terminal binding protein interacting protein.
| Sirtuin | Activity | Substrates | Functions | References |
|---|---|---|---|---|
| SIRT1 | Deacetylase | p53, Foxo1, Foxo3, Bax, Hif-1α, Hif-2a, HSF1, Ku70, b-catenin, E2F1, Myc, STAT3, PGC-1a, NF-κB, TORC2, LXR, FXR, SREBP, PER2, CLOCK | Energy metabolism, stress response | Reviewed in (9) |
| SIRT2 | Deacetylase | Tubulin, H4, Foxo3a | Cell cycle | (123-125) |
| SIRT3 | Deacetylase | Oxidative phosphorylation complex I, AceCS2, LCAD, HMG-CoA synthase 2, IDH2, MnSOD, SOD2 | ATP production, anti-oxidative stress, thermogenesis | (35, 36, 104-109) |
| SIRT4 | ADP-ribosyl-transferase | GDH | Insulin secretion, fatty acid oxidation | (13) |
| SIRT5 | Deacetylase | CPS1 | Urea cycle | (117) |
| SIRT6 | Deacetylase/ADP-ribosyl-transferase | H3K9, H3K56, CtIP, SIRT6 | DNA repair, metabolism, inflammation | (89-92) |
| SIRT7 | Deacetylase? | p53? | rDNA transcription | (17, 126) |
Regulation of sirtuins in Calorie Restriction
Despite diverse subcellular localizations and a broad range of substrate specificities, the activity of all sirtuins is directly controlled by cellular NAD+ levels, which is an indicator of cellular metabolic status. The activity of these enzymes is also inhibited by their common enzymatic product, nicotinamide (19), and possibly by NADH (20). It is therefore not surprising that the activity of sirtuins changes in response to environmental cues that impact cellular metabolic state. A prominent example of this is Caloric Restriction (CR), a 20-40% reduction in calories consumed below ad libitum intake without malnutrition. CR has been shown to extend the median and maximum life span of numerous organisms including yeast, flies, worms, fish, as well as rodents and other higher mammals (21). In mammals, CR has been seen to ameliorate many of the pathologies associated with obesity and metabolic syndrome (22, 23).
CR is thought to promote lifespan extension through multiple signaling pathways. On the one hand, CR decreases the activity of pro-aging pathways such as insulin and growth hormone signaling and oxidative stress. On the other hand, CR stimulates the activity of cellular stress-resistance pathways including DNA repair and autophagy (24, 25), promoting cell survival in response to environmental stress. It has been shown that in yeast, CR reduces cellular NADH concentrations, thereby increasing the NAD+/NADH ratio and promoting Sir2 activity (20). In a number of lower model organisms, sirtuins are required for the lifespan extension provided by CR (11, 26). Whether sirtuins mediate the life-extending effects of CR in mammals is currently an area of great interest. Recent studies have shown that in mice, SIRT1 protein levels are elevated during CR in the brain, white adipose tissue (WAT), muscles, liver, and kidney (27, 28). CR also induces the expression of SIRT3, particularly in brown adipose tissue (BAT) (29, 30). However, during CR in mice, cellular NAD+ levels and the NAD+/NADH ratio fluctuate significantly depending on tissue type (31), suggesting that sirtuins have tissue-specific responses to CR. Nevertheless, it appears that SIRT1 regulates energy metabolism and physical responses to CR (32-34), while SIRT3 is able to mediate CR-associated reduction of oxidative damage, preventing age-associated hearing loss (35, 36). Moreover, transgenic mice overexpressing SIRT1 display multiple phenotypes resembling those of CR mice, including lower body weight, greater metabolic activity, and reduced serum levels of cholesterol, adipokines, insulin, and glucose (37-39). Taken together, these studies highlight the importance of sirtuins in CR-mediated prevention of age-associated functional decline, suggesting that sirtuins may be important therapeutic targets for a number of age-related diseases.
SIRT1 in energy metabolism
As the best-studied mammalian sirtuin, SIRT1 has been implicated in a variety of metabolic processes including hepatic lipid metabolism and gluconeogenesis, pancreatic insulin secretion, fat cell accumulation and maturation, central nutrient sensing, as well as circadian regulation of metabolism. Further elucidation of the role SIRT1 plays in energy metabolism will likely provide key insights into developing treatments for obesity-induced metabolic disease.
1. SIRT1 in hepatic glucose and lipid metabolism
The liver is a central metabolic organ controlling key aspects of lipid and glucose metabolism in response to nutritional and hormonal signals (40). For example, in fasting conditions, the liver converts lipid and glycogen stores into available energy through the process of fatty acid β-oxidation and glycogenolysis/gluconeogenesis. In the fed condition, metabolic programs in the liver are switched on to store energy in the form of glycogen and lipid droplets.
Recent reports have shown that SIRT1 is an important regulator of hepatic glucose and lipid metabolism (Figure 2). For instance, during short-term fasting phase, SIRT1 inhibits TORC2, a key mediator of early phase gluconeogenesis, leading to decreased gluconeogenesis (41). Prolonged fasting leads to increased SIRT1 deacetylation and activation of PGC-1α, an essential co-activator for a number of transcription factors, resulting in increased fatty acid oxidation and improved glucose homeostasis (42-44). SIRT1 also deacetylates and activates transcriptional factor Foxo1, resulting in increased gluconeogenesis (45). Consistently, adenoviral knockdown of SIRT1 reduces expression of fatty acid β-oxidation genes in the liver of fasted mice (46). Specific deletion of the exon 4 of the hepatic mouse SIRT1 gene, which resulted in a truncated non-functional SIRT1 protein, impairs fatty acid β-oxidation, thereby increasing the susceptibility of mice to high-fat diet induced dyslipidemia, hepatic steatosis, inflammation and endoplasmic reticulum (ER) stress (43). A complete deletion of hepatic SIRT1 by floxing exons 5 and 6 leads to the development of liver steatosis even under normal chow diet (47). Conversely, hepatic overexpression of SIRT1 mediated by adenovirus attenuates hepatic steatosis and ER stress, and restores glucose homeostasis in mice (48), confirming the essential role of SIRT1 in maintaining hepatic metabolic homeostasis.
The diverse functions of SIRT1 in central nutrient sensing and peripheral energy metabolism. The activity of SIRT1 is regulated by the cellular metabolic status, small molecule activators, interacting proteins, as well as post-translational modifications. After activation, SIRT1 modulates a variety of metabolic activities systemically and locally through either direct protein deacetylation or indirect chromatin remodeling.
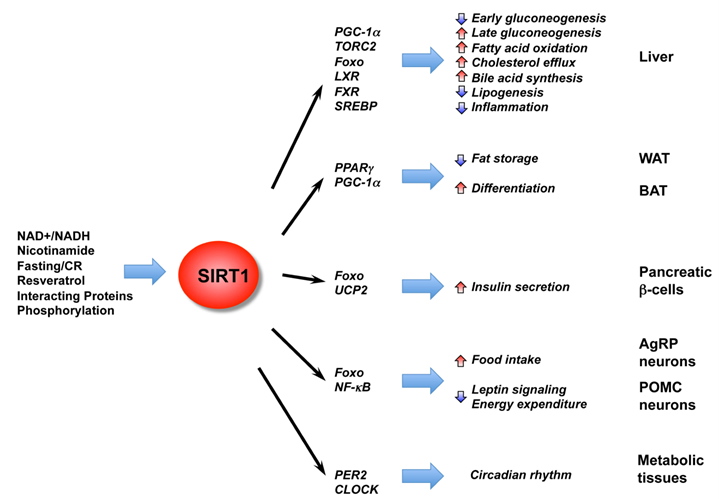
SIRT1 also regulates hepatic cholesterol and bile acid homeostasis through direct modulation of the liver X receptor (LXR), farnesoid X receptor (FXR), and the sterol regulatory element binding protein (SREBP) family of transcription factors (49-52). LXR and FXR are nuclear receptors that function as important cholesterol and bile acid sensors (53). We have previously shown that SIRT1 can directly deacetylate LXRs, resulting in increased LXR turnover and target gene expression (49). Systemic deletion of SIRT1 in mice results in decreased serum HDL levels (49). Recently, Kemper et al. showed that the bile sensor FXR is also a target of hepatic SIRT1 in metabolic regulation (50). Acetylation of FXR inhibits its activity and downregulation of hepatic SIRT1 increases FXR acetylation, causing deleterious metabolic outcomes such as liver steatosis and decreased bile output. Walker et al. and Ponugoti et al. further showed that SIRT1 may also regulate hepatic lipid metabolism through deacetylation of SREBPs (51, 52), critical regulators of lipid metabolism that promote expression of lipogenic and cholesterolgenic genes involved in lipid storage (54). These two reports revealed that SIRT1 can directly deacetylate SREBP, and that SIRT1 activity is important in the fasting-dependent attenuation of SREBP (51, 52). In addition, chemical activators of SIRT1 inhibit SREBP target gene expression in vitro and in vivo, correlating with attenuated liver steatosis in diet-induced and genetically obese mice. In summary, these findings imply that hepatic SIRT1 plays a critical role in metabolic regulation and activation of SIRT1 in the liver may prove beneficial in treating obesity-associated diseases.
2. SIRT1 in fat cell maturation and fat accumulation
Adipose tissue functions both to store fat and as a conduit for hormone signaling. For example, WAT-derived hormones such as leptin and adiponectin control energy balance, glucose regulation, and fatty acid catabolism. Among numerous factors involved in adipose tissue differentiation, the nuclear receptor PPARγ plays an essential role in modulating fatty acid storage and glucose metabolism (55). SIRT1 has been shown to repress PPARγ, thereby suppressing the expression of genes such as the mouse aP2 gene and fat storage ((27), Figure 2). Consistently, treatment of mice on a high fat diet with resveratrol, a polyphenol that activates SIRT1, has been shown to protect against high-fat induced obesity and metabolic derangements (56-58). Although the mechanisms of resveratrol's apparent protective effect on metabolic disorders are not fully understood, questions still remain whether or not resveratrol directly activates SIRT1 or functions through multiple signaling pathways (59, 60), these findings demonstrate that SIRT1 acts in concert with lipid regulating transcription factors to adapt gene transcription to changes in nutrient levels.
As an important non-shivering thermogenesis organ, brown adipose tissue (BAT) plays an essential role in survival of non-shivering animals and neonates. However, the role of SIRT1 in BAT is still not clear. On the one hand, SIRT1 in BAT may directly promote BAT differentiation through repression of the MyoD-mediated myogenic gene expression signature and stimulation of PGC-1α-mediated mitochondrial gene expression (61). On the other hand, SIRT1 also appears to regulate BAT differentiation and functions through non-cell autonomous mechanisms. It has been shown that SIRT1 in propiomelanocortin (POMC) expressing neurons selectively controls perigonadal WAT-to-BAT-like remodeling to increase energy expenditure in female mice (62). Consistent with these observations, activation of SIRT1 by a specific SIRT1 activator, SRT1720, enhances oxidative metabolism in skeletal muscle, liver, and BAT (63). Together, these data indicate that SIRT1 modulates BAT through both cell autonomous and non-cell autonomous mechanisms.
3. SIRT1 in pancreatic insulin secretion
Pancreatic β cells are systemic metabolic sensors that release insulin in response to blood glucose levels. Dysfunction of these cells is the leading cause of type 1 diabetes mellitus, and partially contributes to the pathogenesis of type 2 diabetes. SIRT1 has been shown to be a major positive regulator for pancreatic insulin secretion, which in turn triggers glucose uptake and utilization. For example, it has been reported that increased dosage of SIRT1 in pancreatic β cells improves glucose tolerance and enhances insulin secretion in response to glucose in mice (64), whereas deletion of SIRT1 impairs glucose-stimulated insulin secretion (65). In both studies, SIRT1 has been shown to promote insulin secretion by repressing transcription of the uncoupling protein 2 (UCP2) (Figure 2). Furthermore, it has been shown that treatment with resveratrol potentiates glucose-stimulated insulin secretion in β cells (66). In line with these observations, activation of SIRT1 by its activators in animals protects against high-fat induced obesity and insulin resistance (56-58), and modest overexpression of SIRT1 has a protective effect against high-fat induced glucose intolerance (38, 39). These observations suggest that modulation of SIRT1 activity may regulate whole-body glucose metabolism at both systemically and locally.
4. SIRT1 in central control of metabolic homeostasis
The brain is essential in controlling whole-body metabolism through both neurologic and endocrine functions. The hypothalamus/pituitary axis is the primary brain structure that interprets adiposity and nutrient-related inputs to regulate energy homeostasis. It has been shown that both CR and fasting enhance SIRT1 expression and activity in the hypothalamus (67, 68). Mice lacking SIRT1 in the brain show specific anterior pituitary cell defects and fail to mediate changes in pituitary signaling and physical activity in response to CR (34), while brain-specific SIRT1 transgenic mice display enhanced neural activity in the hypothalamus (68). These findings suggest that SIRT1 in the brain may function as a link between the hypothalamus/pituitary hormones and CR longevity pathways in mammals.
However, the role of SIRT1 in the central control of whole body energy homeostasis is complex (Figure 2). In the hypothalamus, the anorexigenic POMC expressing neurons and the orexigenic agouti-related protein (AgRP) expressing neurons are the major regulators of feeding and energy expenditure (69). The POMC neurons produce satiety peptides thereby inhibiting food intake after feeding, while the AgRP neurons promote feeding in response to fasting and CR. It appears that SIRT1 plays distinct roles in these two neuron populations. A recent study has shown that inhibition of hypothalamic SIRT1 activity reverses the fasting induced decrease of FOXO1 acetylation, resulting in increased POMC and decreased AgRP expressions, thereby decreasing food intake and body weight gain (67). This hypothalamic activity of SIRT1, which is in contrast to its anti-obesity functions in the peripheral tissues, appears to act through AgRP neurons, as AgRP neuron-specific deletion of SIRT1 decreases AgRP neuronal activity, thereby alleviating the inhibitory tone on the POMC neurons, resulting in decreased food intake and body weight (70). Specific deletion of SIRT1 in POMC neurons in mice, on the other hand, causes a blunted response to leptin signaling and reduced energy expenditure, leading to hypersensitivity to diet-induced obesity (62). Nevertheless, these studies confirm that SIRT1 is an essential element in the periphery-central feedback circuits that mediate normal responses to nutrient deprivation. Consistent with these notions, central administration of small molecule SIRT1 activator has shown promise in controlling of diet-induced obesity. For example, long-term intracerebroventricular infusion of resveratrol normalizes hyperglycemia and greatly improves hyperinsulinemia in mice with diet-induced obesity and diabetes (71), although resveratrol may elicit its functions through both SIRT1 dependent and independent pathways. In summary, SIRT1 activity appears to be an important player in the central regulation of nutrient sensing. As relatively little is known at this stage, this area of sirtuin biology may yield important discoveries in coming years.
5. SIRT1 in the regulation of circadian rhythm
All living entities on Earth have endogenously driven 24-hour cycles in many biological processes. This circadian rhythm depends on internal clocks that work in part through chromatin modification and epigenetic control of gene expression (72). In mammals, the circadian clock is largely controlled by negative-feedback loops mediated by the heterodimeric transcription factors CLOCK-BMAL1 and their transcriptional targets, including the PER and CRY proteins that in turn directly repress CLOCK-BMAL1 activity, as well as REV-ERB and ROR nuclear receptors that control BMAL1 expression (73). The CLOCK protein itself is a transcriptional activator that functions as a histone acetyltransferase (HAT) (74), highlighting the importance of chromatin modification in the regulation of circadian gene expression.
SIRT1 has recently been linked to the regulation of the circadian rhythm (Figure 2). Two studies have shown that SIRT1 interacts with CLOCK-BMAL1 to directly regulate the amplitude of circadian clock-controlled gene expression through deacetylation of PER2 and/or BMAL1 (75, 76). However, whether it is the amount of SIRT1 or its activity that is cyclically regulated by the circadian clock remains unclear. Two recent studies appear to favor the circadian regulation of SIRT1 activity (77, 78). The expression of NAMPT, an enzyme that controls a rate-limiting step in NAD+ biosynthesis, is directly controlled by CLOCK-BMAL1. It has been proposed that this regulation leads to circadian oscillation of cellular NAD+ levels, resulting in a cyclical regulation of SIRT1 activity (77, 78). Together, these studies add a new feedback loop in the circadian clock that involves CLOCK-BMAL1, NAMPT, NAD+, and SIRT1, and provide an important link between the circadian clock and cellular metabolism.
It remains to be determined whether the observed oscillation of cellular NAD+ levels (77, 78) is indeed responsible for the oscillation of SIRT1 activity observed in earlier studies (76). An earlier study had shown that the activity of immuno-purified endogenous SIRT1 proteins display a circadian oscillating pattern when measured in vitro with fixed amount of exogenous NAD+ (Figure 1D in (76)), suggesting that post-translational modifications, or protein-protein interactions also play a role in the circadian regulation of SIRT1 activity. SIRT1 has several interacting partners that can directly inhibit or activate its activity (79-82). Its activity can also be modulated by phosphorylation (83-85), particularly by DYRK1A (86), an essential clock component that governs the rhythmic phosphorylation and degradation of CRY2 protein (87). Therefore, exploring the possible role of these factors in the circadian regulation of SIRT1 activity may provide novel insights into its function in circadian rhythm.
SIRT6 in metabolic homeostasis and inflammation
SIRT6, another nuclear sirtuin, plays an essential role in animal development. Previous studies have shown that among the seven mammalian sirtuins, SIRT6 deficiency causes the most striking phenotypes. The SIRT6 null mice appear to have normal embryonic development and intrauterine growth and were born with no obvious abnormalities. However, they suffer a severe metabolic imbalance, acute onset of hypoglycemia, postnatal growth retardation, and premature death at one month of age (16). Unlike the 110 kDa SIRT1 protein, SIRT6 is relatively small (around 36 kDa) with short N- and C-terminal extensions (12). Although it is a nuclear protein, it is predominately chromatin-bound and is enriched in telomeric chromatin in S-phase (16, 89). Moreover, despite possessing a robust NAD+-dependent auto-ADP-ribosyltransferase activity (88), SIRT6 is a highly specific histone 3 deacetylase that targets acetyl-H3K9 and acetyl-H3K56, playing an important role in DNA repair, telomere function, genomic stability, and cellular senescence (89-91). Human SIRT6 also deacetylates C-terminal binding protein interacting protein (CtIP) and promotes DNA end resection (92).
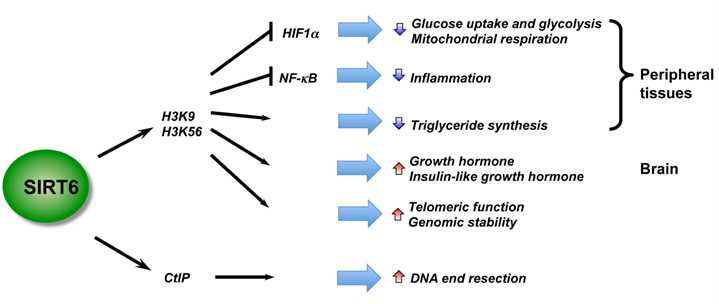
Recent studies using mouse models have revealed essential roles of SIRT6 in metabolic homeostasis and inflammation, two heavily inter-locked biological processes (Figure 3). Kawahara et al. showed that SIRT6 functions at chromatin to attenuate NF-κB signaling (93). SIRT6 interacts with the RelA subunit of NF-κB and deacetylates H3K9 at target promoters. Deficiency of SIRT6 in mice results in increased NF-κB-driven gene expression programs. However, SIRT6 also appears to positively regulate Tumor necrosis factor (TNF) production at a post-transcriptional step in response to an increase in intracellular NAD+ concentrations (94), raising the possibility that SIRT6 may play discrete roles in acute and chronic inflammatory responses. SIRT6 is also a master regulator of systemic glucose metabolism. Using a whole-body knockout mouse model, Zhong et al. reported that SIRT6 functions as a corepressor of the transcription factor Hif1α to suppress glucose uptake and glycolysis (95). SIRT6 knockout mice are hypoglycemic and SIRT6-deficient cells exhibit increased Hif1α activity with augmented glucose uptake, along with upregulation of glycolysis and diminished mitochondrial respiration. Xiao et al. also reported that systemic deletion of SIRT6 in mice results in severe hypoglycemia, but it appears that SIRT6-deficiency enhances both basal and insulin-stimulated glucose uptake through increased insulin signaling and activation of AKT (96). Further, studies using tissue-specific SIRT6 knockout mouse models have indicated that SIRT6 regulates metabolic homeostasis both systemically and locally. For instance, using a liver-specific SIRT6 knockout mouse model, Kim et al. demonstrated that liver-specific deletion of SIRT6 leads to increased glycolysis and triglyceride synthesis, as well as reduced fatty acid oxidation, resulting in liver steatosis (97). Neural-specific deletion of SIRT6 does not lead to hypoglycemia (98). Instead, neural SIRT6-null mice exhibit postnatal growth retardation due to somatotropic attenuation through low growth hormone (GH) and insulin-like growth factor 1 (IGF1) levels. They gradually reach normal size over time and ultimately become obese. These observations indicate that SIRT6 regulates animal body growth through central control, while modulating glucose and lipid metabolism at both systemic and local levels (Figure 3).
SIRT6 is a critical regulator in genome stability, metabolism, and inflammatory response. By deacetylation of H3, SIRT6 regulates metabolic homeostasis and inflammatory response in peripheral tissues, while functioning as a central regulator of somatic growth.
Mitochondrial sirtuins in energy metabolism
In line with the notion that sirtuins are important cellular metabolic sensors coupling energy status to cellular functions, three of the mammalian sirtuins, SIRT3, SIRT4, and SIRT5, are directly localized to mitochondria. These double membrane-bound organelles are the “cellular power plants”, consuming 85-95% of the oxygen used by cells to generate ATP, and account for over 90% of reactive oxygen species (ROS) generated in cells (99). Mitochondria are also at the crossroads between numerous intermediary metabolic pathways that function in nutrient adaptation in response to environmental nutrient cues. Therefore it is not surprising that defects in mitochondrial functions are associated with degenerative diseases, cancer, and aging (100).
Despite the important localization of SIRT3-5, mice with deletions of these mitochondrial sirtuins lack obvious phenotypes (101), explaining why the biochemical and biological functions of these proteins have just started to be elucidated (Figure 4). SIRT3, the most studied mitochondrial sirtuin, is highly expressed in metabolically active tissues such as brown adipose, muscle, liver, kidney, heart, and brain (102, 103). Consistent with this expression pattern and its mitochondrial localization, deletion of SIRT3 in mice leads to striking mitochondrial protein hyperacetylation (101). These hyperacetylated proteins include subunits of oxidative phosphorylation complexes (104), metabolic enzymes such as acetyl-CoA synthetase 2 (105, 106), long-chain acyl CoA dehydrogenase (LCAD) (107) and 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) synthase 2 (108), as well as oxidative stress reducing enzymes isocitrate dehydrogenase 2 (IDH2) (36), superoxide dismutase 2 (SOD2) (35), and MnSOD (109). A number of recent studies have reported that hyperacetylation of these mitochondrial proteins in SIRT3 deficient mice results in a variety of metabolic abnormalities including reduced ATP production, decreased rates of fatty acid oxidation and ketone body production, and fatty liver (104, 107, 108, 110). In addition, SIRT3 also regulates mitochondrial translation (111), a process that may indirectly impact all mitochondrial pathways.
Mitochondrial sirtuins in the center of mitochondrial energy metabolism and anti-oxidative stress response. Mitochondria are central metabolic organelles for the production of cellular ATP from various nutrients including glucose, fatty acids, and amino acids. However, ROS, such as superoxide, are also produced during the oxidative phosphorylation. Mitochondria also contain numerous enzymatic complexes involved in intermediary metabolism pathways that function for nutrient adaptation and antioxidant defense. Mitochondrial sirtuins are essential for normal mitochondrial functions through interaction and modification of a number of mitochondrial proteins. SIRT3 deacetylates and maintains the normal functions of various mitochondrial proteins (blue) involved in fatty acid oxidation, ketogenesis, oxidative phosphorylation, antioxidant defense, and amino acid metabolism. GDH (orange) can also be ADP-ribosylated and repressed by SIRT4. SIRT5 deacetylates and activates CPS1 (green). This figure is modified from reference 9.
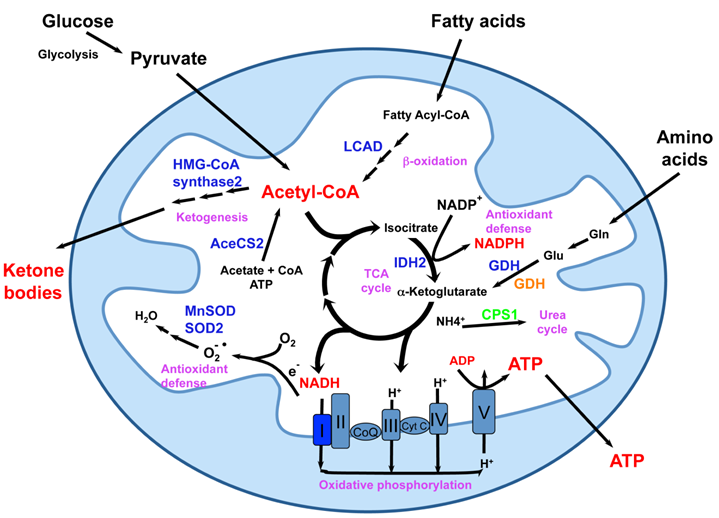
Hyperacetylation of mitochondrial proteins may have biological consequences beyond abnormal metabolic homeostasis. Using SIRT3-deficient and transgenic mouse models, two studies reported that SIRT3 blocks age-related cardiac hypertrophic response (112, 113). Kim et al. showed that SIRT3 is a mitochondria-localized tumor suppressor in response to stress (114). SIRT3 also protects in vitro fertilized mouse preimplantation embryos against oxidative stress (115). Furthermore, two recent studies showed that SIRT3 deficient mice fail to suppress oxidative stress and hearing loss in response to CR (35, 36). These observations demonstrate that SIRT3 can delay the onset of a number of oxidative stress-associated pathologies in multiple tissues and suggest that SIRT3 may be a novel target for these age-associated diseases as well as aging itself. However, since SIRT3 null mice display minimal phenotypes under normal feeding conditions, particularly when they are young (101), much work still needs to be done to determine whether the effects of SIRT3 deletion on these processes are physiologically relevant, and whether they are direct consequences of particular hyperacetylated mitochondrial proteins. Furthermore, it remains to be answered whether SIRT3 is required for CR-mediated extension of lifespan or it is only a part of CR- elicited anti-aging circuits.
Unlike SIRT3, very little is known about the functions of SIRT4 and SIRT5. As a sirtuin that only displays detectable ADP-ribosyltransferase activity, SIRT4 is unique among the mammalian sirtuins (13). For example, although SIRT4 is a mitochondrial enzyme, it is not enriched in mitochondria-dense tissues such as heart and muscle (13). The only known substrate of SIRT4 is glutamate dehydrogenase (GDH), and SIRT4 interacts with GDH and suppresses its activity via ADP-ribosylation (13). It appears that loss of SIRT4 in mice results in increased insulin secretion in response to glucose and amino acids (13), while overexpression of SIRT4 in cultured cells suppresses insulin secretion (14). SIRT4 also plays a role in the regulation of fatty acid oxidation and mitochondrial gene expression in liver and muscle cells, although the mechanisms underlying these phenotypes are still unclear (116). SIRT5 has recently been reported as a specific deacetylase for carbamoyl phosphate synthetase (CPS1), the rate-limiting first step of the urea cycle in mitochondria (117, 118). Deletion of SIRT5 in mice leads to increased acetylation of CPS1 and elevated levels of ammonia after prolonged fasting, whereas SIRT5 transgenic mice display increased CPS1 activity (117). In summary, while it appears likely that SIRT3 is a major mitochondrial deacetylase that helps to prevent non-specific hyperacetylation of mitochondrial proteins by acetyl-CoA over-flood during the process of active energy production, SIRT4 and SIRT5 appear to have specific targets and functions in this organelle. Further work is needed to identify these SIRT4 and SIRT5 specific targets to elucidate any additional roles these sirtuins possess in the regulation of energy metabolism.
Cross talks between sirtuins
As one of the most important cellular metabolic sensors in cells, it has been speculated that the seven sirtuins coordinate with each other in various cellular compartments to actively monitor diverse environmental signals, modulating cellular metabolic activity, gene transcription, and genome stability, ultimately affecting aging. Indeed, perturbation of the activity of one sirtuin has been shown to impact the activities of other sirtuin members. For example, we reported that in mouse macrophages, deletion of SIRT1 leads to increased levels of chromatin-associated SIRT6 near the NF-κB binding sites, resulting in reduction of local acetyl-H3K9 levels, compensating for the hyperactivation of NF-κB induced by SIRT1 deficiency (119). Interestingly, this compensatory effect between SIRT1 and SIRT6 does not appear to exist in hepatocytes. Instead, deletion of SIRT1 causes 50% reduction of both SIRT6 mRNA and protein levels in the liver (97). This is a result of SIRT1 binding to Foxo3a and nuclear respiratory factor 1 (NRF-1) on the promoter of SIRT6 in hepatocytes, directly promoting the expression of SIRT6 under both basal and fasting conditions (97). Additional studies will be necessary to address whether the varying coordination patterns between these two nuclear sirtuins are related to the different metabolic profiles of macrophages and hepatocytes.
Intensive crosstalk between nuclear and mitochondrial sirtuins have also been implied in the literature. A recent study reported that SIRT3 is a transcriptional target of PGC-1α via an estrogen-related receptor binding element (ERRE) on its promoter (120). As PGC-1α is a direct deacetylation substrate of SIRT1 (42), this observation suggests that SIRT1 could indirectly modulate the expression of SIRT3 through PGC-1α deacetylation. Additionally, Nasrin et al. recently showed that SIRT4 knockdown in hepatocytes induces an increase in fatty acid oxidation through SIRT1 (116). Although molecular mechanisms underlying these connections remain to be defined, these findings suggest the existence of a sirtuin-network that may be pivotal in the maintenance of systemic metabolic homeostasis.
Conclusions
The discovery that overexpression of sirtuins extends lifespan in lower model organisms has evoked a flood of research on their roles in the mammalian aging process. The disruption of energy metabolism, genome stability, and stress response in sirtuin-deficient mouse models demonstrates that mammalian sirtuins are major contributors to the delicate balance between metabolism and aging. Consistent with this notion, small molecule activators of SIRT1, such as the polyphenol resveratrol and closely related derivatives, have shown promise as therapeutic agents for the treatment of obesity and related metabolic diseases (58, 121, 122), although there were debate on whether these small-molecule drugs are direct activators of SIRT1 that function to prevent obesity and diabetes (59, 60). Further research is needed on the systemic and tissue-specific functions of sirtuins in other aspects of the aging process, such as stem cell renewal and protein quality control, as well as on the identification of novel targets and modulators of these proteins. Ultimately, these studies will enhance our knowledge of sirtuin biology, aiding the development novel small molecule modulators of sirtuins to combat age-associated diseases as well as aging itself.
Acknowledgements
We thank Lawrence J. Forsberg from the UNC-Chapel Hill and members of the Li laboratory for critical reading of the manuscript. The work related to this article was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to X.L. (Z01 ES102205).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000Feb17;403(6771):795-800
2. Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L. et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000May23;97(11):5807-11
3. Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ. et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A. 2000Jun6;97(12):6658-63
4. Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000May1;14(9):1021-6
5. Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet. 2007Nov;8(11):835-44
6. Sinclair D, Guarente L. Extrachromosomal rDNA circles-a cause of aging in yeast. Cell. 1997;91:1033-1042
7. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999Oct1;13(19):2570-80
8. Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417-35
9. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253-95
10. Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001Mar8;410(6825):227-30
11. Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004Nov9;101(45):15998-6003
12. Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000Jul5;273(2):793-8
13. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006Sep8;126(5):941-54
14. Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V. et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007Nov16;282(46):33583-92
15. Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007Mar2;282(9):6823-32
16. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006Jan27;124(2):315-29
17. Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006May1;20(9):1075-80
18. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010Dec;35(12):669-75
19. Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099-107
20. Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004Jan1;18(1):12-6
21. Lomb DJ, Laurent G, Haigis MC. Sirtuins regulate key aspects of lipid metabolism. Biochim Biophys Acta. 2010Aug;1804(8):1652-7
22. Kim C, Park J, Kang E, Ahn C, Cha B, Lim S. et al. Comparison of body fat composition and serum adiponectin levels in diabetic obesity and non-diabetic obesity. Obesity (Silver Spring). 2006Jul;14(7):1164-71
23. Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A. 2004Apr27;101(17):6659-63
24. Kaushik S, Singh R, Cuervo AM. Autophagic pathways and metabolic stress. Diabetes Obes Metab. 2010Oct;12(Suppl 2):4-14
25. Rodriguez-Navarro JA, Cuervo AM. Autophagy and lipids: tightening the knot. Semin Immunopathol. 2010Dec;32(4):343-53
26. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000Sep22;289(5487):2126-8
27. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R. et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004Jun17;429(6993):771-6
28. Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L. et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005Oct14;310(5746):314-7
29. Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005Apr8;280(14):13560-7
30. Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Ward JL3rd. et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany NY). 2009Sep;1(9):771-83
31. Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW. et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008Jul1;22(13):1753-7
32. Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005Dec9;310(5754):1641
33. Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C. et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. 2008;3(3):e1759
34. Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes Dev. 2009Dec15;23(24):2812-7
35. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010Dec1;12(6):662-7
36. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C. et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010Nov24;143(5):802-12
37. Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A. et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007Dec;6(6):759-67
38. Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L. et al. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell metabolism. 2008;8:333-41
39. Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008Jul15;105(28):9793-8
40. van den Berghe G. The role of the liver in metabolic homeostasis: implications for inborn errors of metabolism. J Inherit Metab Dis. 1991;14(4):407-20
41. Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S. et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456(7219):269-73
42. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005Mar3;434(7029):113-8
43. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009Apr;9(4):327-38
44. Dominy JEJr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1alpha's acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta. 2010Aug;1804(8):1676-83
45. Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005May27;280(21):20589-95
46. Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007Jul31;104(31):12861-6
47. Wang RH, Li C, Deng CX. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci. 2010;6(7):682-90
48. Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X. et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011May;25(5):1664-79
49. Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007Oct12;28(1):91-106
50. Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D. et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009Nov;10(5):392-404
51. Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A. et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010Jul1;24(13):1403-17
52. Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M. et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem. 2010Oct29;285(44):33959-70
53. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995Dec15;83(6):841-50
54. Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. 2009Nov15;23(22):2578-91
55. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289-312
56. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A. et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006Nov16;444(7117):337-42
57. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F. et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006Dec15;127(6):1109-22
58. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007Nov29;450(7170):712-6
59. Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L. et al. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des. 2009Dec;74(6):619-24
60. Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS. et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010Mar12;285(11):8340-51
61. Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N. et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci U S A. 2007Mar13;104(11):4401-6
62. Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R. et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab. 2010Jul4;12(1):78-87
63. Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC. et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008Nov;8(5):347-58
64. Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C. et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005Aug;2(2):105-17
65. Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J. et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006Feb;4(2):e31
66. Vetterli L, Brun T, Giovannoni L, Bosco D, Maechler P. Resveratrol potentiates glucose-stimulated insulin secretion in INS-1E beta-cells and human islets through Sirt1 dependent mechanism. J Biol Chem. 2011;286(8):6049-60
67. Cakir I, Perello M, Lansari O, Messier NJ, Vaslet CA, Nillni EA. Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS One. 2009;4(12):e8322
68. Satoh A, Brace CS, Ben-Josef G, West T, Wozniak DF, Holtzman DM. et al. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci. 2010Jul28;30(30):10220-32
69. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006Sep21;443(7109):289-95
70. Dietrich MO, Antunes C, Geliang G, Liu ZW, Borok E, Nie Y. et al. Agrp neurons mediate Sirt1's action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J Neurosci. 2010Sep1;30(35):11815-25
71. Ramadori G, Gautron L, Fujikawa T, Vianna CR, Elmquist JK, Coppari R. Central administration of resveratrol improves diet-induced diabetes. Endocrinology. 2009Dec;150(12):5326-33
72. Wijnen H, Young MW. Interplay of circadian clocks and metabolic rhythms. Annu Rev Genet. 2006;40:409-48
73. Wijnen H. Circadian rhythms. A circadian loop asSIRTs itself. Science. 2009May1;324(5927):598-9
74. Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006May5;125(3):497-508
75. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F. et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008Jul25;134(2):317-28
76. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D. et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008Jul25;134(2):329-40
77. Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009May1;324(5927):654-7
78. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B. et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009May1;324(5927):651-4
79. Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008Jan31;451(7178):587-90
80. Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008Jan31;451(7178):583-6
81. Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007Oct26;28(2):277-90
82. Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV. et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nature cell biology. 2007Nov;9(11):1253-62
83. Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT. et al. Phosphorylation regulates SIRT1 function. PLoS ONE. 2008;3(12):e4020
84. Kang H, Jung JW, Kim MK, Chung JH. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS ONE. 2009;4(8):e6611
85. Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R. et al. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS ONE. 2009;4(12):e8414
86. Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010Apr23;285(17):13223-32
87. Kurabayashi N, Hirota T, Sakai M, Sanada K, Fukada Y. DYRK1A and glycogen synthase kinase 3beta, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol. 2010Apr;30(7):1757-68
88. Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005Jun3;280(22):21313-20
89. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M. et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008Mar27;452(7186):492-6
90. Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O. et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009Aug15;8(16):2664-6
91. Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009Aug15;8(16):2662-3
92. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010Sep10;329(5997):1348-53
93. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M. et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009Jan9;136(1):62-74
94. Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A. et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med. 2009Feb;15(2):206-10
95. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD. et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010Jan22;140(2):280-93
96. Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O. et al. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem. 2010Nov19;285(47):36776-84
97. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010Sep8;12(3):224-36
98. Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J. et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci U S A. 2010;107(50):21790-4
99. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005Feb25;120(4):483-95
100. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359-407
101. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R. et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007Dec;27(24):8807-14
102. Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002Aug19;158(4):647-57
103. Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002Oct15;99(21):13653-8
104. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A. et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008Sep23;105(38):14447-52
105. Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006Jul5;103(27):10224-9
106. Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006Jul5;103(27):10230-5
107. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010Mar4;464(7285):121-5
108. Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB. et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010Dec1;12(6):654-61
109. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H. et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010Dec22;40(6):893-904
110. Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL. et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011Jan14;433(3):505-14
111. Yang Y, Cimen H, Han MJ, Shi T, Deng JH, Koc H. et al. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem. 2010Mar5;285(10):7417-29
112. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009Sep;119(9):2758-71
113. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A. et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY). 2010Dec;2(12):914-23
114. Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD. et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010Jan19;17(1):41-52
115. Kawamura Y, Uchijima Y, Horike N, Tonami K, Nishiyama K, Amano T. et al. Sirt3 protects in vitro-fertilized mouse preimplantation embryos against oxidative stress-induced p53-mediated developmental arrest. J Clin Invest. 2010Aug2;120(8):2817-28
116. Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S. et al. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem. 2010Oct15;285(42):31995-2002
117. Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009May1;137(3):560-70
118. Ogura M, Nakamura Y, Tanaka D, Zhuang X, Fujita Y, Obara A. et al. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem Biophys Res Commun. 2010Feb26;393(1):73-8
119. Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB. et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010Oct;30(19):4712-21
120. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y. et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One. 2010;5(7):e11707
121. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003Sep11;425(6954):191-6
122. Camins A, Sureda FX, Junyent F, Verdaguer E, Folch J, Pelegri C. et al. Sirtuin activators: Designing molecules to extend life span. Biochim Biophys Acta. 2010;1799(10-12):740-9
123. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007Aug;6(4):505-14
124. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Molecular cell. 2003Feb;11(2):437-44
125. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes & development. 2006May15;20(10):1256-61
126. Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T. et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008Mar28;102(6):703-10
Author contact
![]() Corresponding author: lix3nih.gov
Corresponding author: lix3nih.gov