Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(6):805-814. doi:10.7150/ijbs.7.805 This issue Cite
Research Paper
MicroRNA-7 Inhibits the Growth of Human Non-Small Cell Lung Cancer A549 Cells through Targeting BCL-2
Shudao Xiong*, Yijie Zheng*, Pei Jiang, Ronghua Liu, Xiaoming Liu, Yiwei Chu ![]()
Department of Immunology, Shanghai Medical College, Key Laboratory of Molecular Medicine of Ministry of Education, Fudan University, Shanghai, China.
* Shudao Xiong and Yijie Zheng contributed equally to this work and should be considered as co-first authors.
Received 2011-3-28; Accepted 2011-6-26; Published 2011-7-4
Abstract
MicroRNAs(miRNAs) are emerging as important regulators in tumorigenesis. Increasing evidences have indicated microRNA-7(miR-7) to be a potential tumor suppressor in several human cancers. However, only a limited number of target genes have been identified so far and its biological function in Non-Small Cell Lung Cancer (NSCLC) remains to be further elucidated. In the present study, we observed a reduction of miR-7 level in NSCLC cell lines. Overexpression of miR-7 not only suppressed NSCLC A549 cells proliferation, induced cell apoptosis and inhibited cell migration in vitro, but also reduced tumorigenicity in vivo. Bioinformatics predictions revealed a potential binding site of miR-7 on 3'UTR of BCL-2 and it was further confirmed by luciferase assay. Moreover, subsequent experiments showed that BCL-2 was downregulated by miR-7 at both transcriptional and translational levels. These results suggest that miR-7 regulates the expression of BCL-2 through direct 3'UTR interactions. Therefore, we postulate BCL-2 to be a novel target possibly involved in miR-7-mediated growth suppression and apoptosis of A549 cells. These findings may provide a basic rationale for the use of miR-7 in the treatment of NSCLC.
Keywords: miR-7, BCL-2, non-small cell lung cancer, A549 cells, apoptosis.
Introduction
MiRNAs are a group of non-coding small RNAs produced from actual genes within the genomic loci of cells[1, 2]. The primary transcripts of miRNAs are processed by Drosha to form 70-80nt precursor miRNAs, which are then exported to the cytoplasm and processed by Dicer into mature microRNAs. Mature miRNAs regulate their target genes through partial sequence complementarity to the 3' untranslated region (UTR) of target genes, thereby resulting in mRNA degradation or/and translational repression[3, 4].
As a new layer of gene-regulation mechanism, miRNAs are emerging as key regulators in pathogenesis of cancer[5]. By regulating the expression of their target genes, miRNAs profoundly influence a wide variety of pathways, thereby acting as oncogenes or tumor suppressors[6, 7]. Indeed, a number of differentially regulated miRNAs, such as miR-155[8, 9], let-7a[10], miR-21[8], miR-34a[11] and miR-7[12-16], have been identified to be functionally associated with cancer cell proliferation, invasion and metastasis. Among them, miR-7 is suggested to be a putative tumor suppressor in breast and glioblastoma[12, 13, 15]. However, the biological function and mechanisms of miR-7 in lung cancer, especially in NSCLC, remain to be further elucidated.
In the present study, we demonstrated a reduction of miR-7 level in NSCLC cell lines. Overexpression of miR-7 suppressed NSCLC A549 cells proliferation, induced A549 cells apoptosis, inhibited cancer cell migration in vitro, and reduced tumorigenicity in vivo. Subsequent experiments confirmed that miR-7 downregulated the expression of BCL-2 at both transcriptional and translational levels through direct 3'UTR interactions. These results suggested BCL-2 to be a novel target through which miR-7 inhibits A549 cells proliferation.
Materials and methods
Cell lines and cell culture
A549, H1299, H1355, H460, MRC5 and 293T cell lines were provided by Institute of Biochemistry and Cell Biology of Chinese Academy of Science (China) and originated from ATCC. All cells were grown in DMEM supplemented with 10% fetal bovine serum, 2 μM glutamine, 100 IU/ml penicillin, and 100μg/ml streptomycin sulfate.
RNA extraction
Total RNA of cultured cells was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. RNAs were then stored at -80℃ before RT-PCR analysis.
Quantitative RT-PCR (qRT-PCR) for miRNA
For mature miRNA expression analysis, approximately 10 ng of RNA was converted to cDNA using the ABI miRNA reverse transcription kit (Applied Biosystems, Foster City, CA) along with miR-7-specific primers (Applied Biosystems, Foster City, CA). After reverse transcription, quantitative polymerase chain reaction (PCR) was performed on the ABI 7500 thermocycler (Applied Biosystems, Foster City, CA) according to the manufacture's protocol. U6 gene was used as a normalization control for all samples.
qRT-PCR for mRNA expression
Synthesis of cDNA was performed on 1μg of total RNA per sample with the primerScript RT reagent Kit (TaKaRa, Dalian, China) according to the manufacturer's manual. Quantitative reverse transcription-PCR was performed in triplicate for each sample by FastStart Universal SYBR Green Master kit (Roche, Switzerland) according to the manufacturer's instructions. Oligonucleotides were designed by the PrimerExpress software. GAPDH was used as a housekeeping gene for normalization. The sequences of primers in this section are the followings: (1) BCL-2: 5'-ATGTGTGTGGAGAGCGTCAACC-3' (forward) and 5'-TGAGCAGAGTCTTCAGAGACAGCC-3' (reverse); (2) GAPDH: 5'-TGCACCACCAACTGCTTAGC-3' (forward) and 5'-GCATGGACTGTGGTCATGAG-3' (reverse); (3) MMP-2: 5'-CCAACACTGGGACCTGTCACTC-3' (forward) and 5'-TGTCACTGTCCGCCAAATAAACC-3' (reverse); (4) MMP-9: 5'-TGGGCTACGTGACCTATGACAT-3' (forward) and 5'-GCCCAGCCCACCTCCACTCCTC-3' (reverse).
MiRNAs mimic and transfection
The human miR-7 duplex mimic (miR-7) and negative control oligonucleotide duplex mimic (miR-NC) were designed and provided by Ribobio (Guangzhou, Guangdong, China). 30-50% confluent cells were transfected with miRNAs by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Total RNA was extracted 24 hours after transfection, and total cell protein were extracted 48 or 72 hours after transfection.
Cell proliferation assay
Cell proliferation assay was determined by Cell Counting Kit-8 assay (Dojindo, Japan), a redox assay similar to 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) according to the manufacture's protocol. Cell proliferation assay was carried out in hexakis.
Apoptotic morphology by DAPI staining
Cells were stained with 4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) and those with fragmented or condensed nuclei were defined as apoptotic cells. At least five visual fields were observed under fluorescence microscope for each sample.
Caspase 3/7 assay
Caspase 3/7 activity was performed by manufacture's protocol of Caspase 3/7 assay (Pierce, USA). Briefly, cells cultured in a white-walled 96-well plate were added with 100μL of Caspase-Glo® 3/7 Reagent. Mix gently and incubate the cells at room temperature for 30 minutes to 3 hours, then measure the luminescence of each sample in a plate-reading luminometer of Tecan´s Infinite M200. Caspase 3/7 assay was carried out in triplicate.
Vector constructs
To construct BCL-2-3'UTR plasmid, a wild-type 3'-UTR fragment of human BCL-2 mRNA (1384-1390 nt, Genbank accession no.NM_000633.2) containing the putative miR-7 binding sequence was amplified by PCR and cloned into the site between XbaI and FseI of the pGL3-control vector (Promega, USA) which is the downstream of the luciferase reporter gene. A mutant of the single miR-7 binding site (5'- AGUCUUC-3' to 5'- AccCggC-3') in the 3'-UTR of BCL-2 was included by Site-Directed Mutagenesis Kit (SBS Genetech, Beijing, China). Wild and mutant type of pGL3-con-BCL-2-UTR vectors were validated by DNA sequencing. The nucleotide sequences of primers for BCL-2-3'UTR clone were: 5'-gctctagagcGCCACAAGTGAAGTCAACA-3' (forward) and 5'-tataggccggcctaACAGGCACAGAACATCCAG-3' (reverse) and the two pairs of mutagenesis primers were: (1) 5'-GAGTCAGGATGCTGTCTgCgCACGAATAGTACTCAGT-3'(forward) and 5' ACTGAGTACTATTCGTGcGcAGACAGCATCCTGACTC-3'(reverse); (2) 5'-GAGTCAGGATGCTGTCcGcGCACGAATAGTACTCAGT-3' (forward) and 5' ACTGAGTACTATTCGTGCgCgGACAGCATCCTGACTC-3' (reverse).
Lentiviral vector production, titration and transduction
The pre-hsa-miR-7-1 sequence was obtained from human genomic DNA by PCR with the following primers: 5'-GGGCCCGCTCTAGACTCGAGATATTTGCATGTCGCTATGTG-3' (forward) and 5'-CGCGGCCGCCTAATGGATCCAAAAAAGGCACAGTCGAGGCTGATC-3' (reverse). Then, the sequence was inserted into pGCSIL-GFP lentivirus vector with T4 DNA ligase. The ligated vector was transformed into competent Escherichia coli DH5α cells. To produce lentiviral vectors, 293T cells were co-transfected with the three vector plasmids: pGC-LV、pHelper 1.0 and pHelper 2.0. For lentiviral transduction, A549 cells were plated at a concentration of 1×106 cells in 25 mL flask to reach 60-80% confluence after overnight culture and were then infected at MOI of 20 in the presence of polybrene (5μg/ml). Positively infected cells were sorted by FACS to be continued to culture. The level of miR-7 was detected by realtime PCR.
Migration assay
Migration of the cells infected with miR-7-GFP-LV lentivirus or its control were measured by counting the number of cells that migrated through Transwell inserts with 3μm pores, as described previously. Cells were trypsinized, and 200μl of cell suspension (1×106 cells/ml) from each treatment were added in triplicate wells. After 24h incubation, the cells that had migrated through the filter into the lower wells were quantitated by gentian violet assay and expressed as the total cell numbers in the lower wells.
Tumorigenicity assay in nude mice
Nude mice (4-6 weeks old) were used for xenograft studies. MiR-7 and miR-NC transfected A549 cells (3×106 in matrigel) were injected s.c. into either side of the posterior flank of the same female nude mouse. After 7 days, the tumor surface areas were measured with a vernier caliper at weekly intervals. The formula (L/2)×(W/2)×π (where L is maximum diameter of each tumor, and W is the length at right angles to L) was used to calculated the tumor surface areas as previously described[17, 18].
MiRNA target prediction
Putative miRNA target genes were searched by application of miRanda (http://cbio.mskcc.org/microrna [19], then calculated the binding free energy and analyzed biding sites on the website of http://bibiserv.techfak.uni-bielefeld.de/rnahybrid [20]).
Luciferase assay
A549 cells were transfected with BCL-2 3'UTR plasmid, Renilla luciferase pRL-TK vector (Promega USA), and miR-7 mimic or miR-NC mimic using lipofectamine 2000 reagent (Invitrogen, USA) according to the manufacturer's protocol. Luminescence was assayed 48 hours later using the Dual-Luciferase Reporter Assay System (Promega USA) according to the manufacturer's instructions. Results were normalized to the Renilla luminescence from the same vector and shown as the ratio between the various treatments and cells transfected with control vector.
Western blot analysis
Proteins extracted from cells were immunoblotted with different antibodies following a published protocol[17]. The primary antibodies used were BCL-2 (1:5000 dilutions) and GAPDH (1:5000 dilutions) (Cell Signaling, Danvers, MA, USA).
Statistical analysis
Data were expressed as Mean±SEM of three independent experiments. For all statistical tests, PRISM 5.0 (GraphPad Software Inc., San Diego, CA, USA) was used. P values less than 0.05 were considered statistically significant.
Results
miR-7 was frequently downregulated in NSCLC cell lines
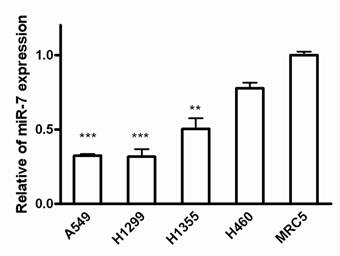
To assess the biological role of miR-7 in lung cancer cells, we first examined the expression of miR-7 in lung cancer cell lines including A549, H1299, H1355 and H460 cells. Since most of the miRNA-related studies on cancers are based on the different expression of miRNAs in cancer cells versus normal cells[21], we compared miR-7 expression between these lung cancer cell lines and normal human fetal lung fibroblast cell line MRC5 using realtime RT-PCR. As a result, miR-7 showed significantly lower expression in human non-small cell lung cancer A549, H1299 and H1355 cells than in MRC5 cells (Fig. 1). However, expression of miR-7 exhibited no significant difference in large cell cancer H460 cells compared to MRC5 cells. These data suggest that expression of miR-7 may vary in different lung cancer cell types and is frequently down-regulated in NSCLC cell lines. Therefore, it indicated that the downregulation of miR-7 might be involved in lung carcinogenesis.
Expressions of miR-7 are downregulated in NSCLC cell lines. The miR-7 expression levels of A549, H1299, H1355, H460 and MRC5 cells were detected by realtime RT-PCR. The relative expression of miR-7 was normalized to the endogenous control U6. Each sample was analyzed in triplicate. (**p<0.01, ***p<0.001).
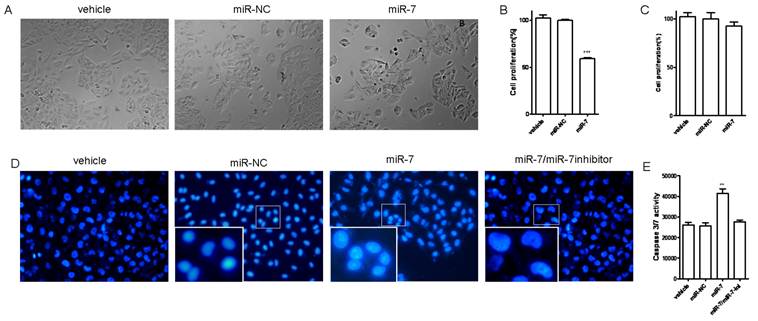
Overexpression of miR-7 suppressed proliferation and induced apoptosis of A549 cells in vitro
We next investigated the influence of miR-7 on phenotypes of A549 cells. We transiently transfected A549 cells with mature miR-7 mimic (miR-7). As a control, cancer cells were transfected with negative control mimic (miR-NC) without specifically targeting any human gene products. After 48h, cells transfected with miR-7 were found to grow more slowly than the negative control and vehicle group (Fig. 2A). In CCK-8 proliferation assay, A549 cells also exhibited reduced cell proliferation in the presence of miR-7 (Fig. 2B) with a dose-dependent manner (Fig.S1). However, miR-7 showed no significant influence on cell growth of MRC5 cells (Fig. 2C).
Overexpression of miR-7 suppressed cell proliferation and induced apoptosis of A549 cells. (A) A549 cells were transfected with 50nM of miR-7 mimic (miR-7) or negative control mimic (miR-NC), and incubated for 48h. Cell morphology was detected by a microscope. (B) Cell proliferation was measured by CCK-8 assay. (C) MRC5 cells were transfected with 50nM of miR-7 or miR-NC. CCK-8 assay was performed at 48h after transfection. (D) A549 cells were transfected with 50nM of miR-7, miR-NC or along with miR-7 inhibitor (200nM). Cells were then incubated with low-serum (3%) DMEM mediums for 48h. Morphology of apoptotic cell nuclei was observed by DAPI staining using a fluorescence microscope. (E) Subsequently, activities of caspase-3 and caspase-7 were examined by caspase 3/7 assay. Data are representative of three independent experiments. (**p<0.01, ***p<0.001).
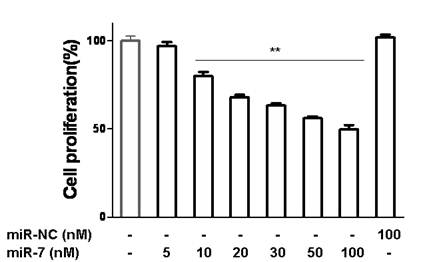
miR-7 suppressed the growth of A549 cells in a dose-dependent manner. A549 cells were transfected with various doses of miR-7(from 0 to 100nM) in low-serum (3%) DMEM for 48h and then cell growth was detected by CCK-8.
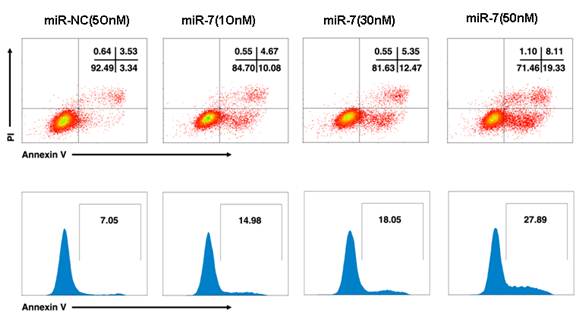
To further understand whether the reduced cell growth was due to apoptosis, we studied the apoptosis of cells following Annexin V/PI assay, DAPI staining and caspase-3/7 assay. A549 cells were transfected with various doses of miR-7 mimic (from 0 to 50nM) or negative control mimic in low-serum (3%) DMEM for 48h and subsequently analyzed by Annexin V/PI assay. We observed a dose-dependent increase of cell apoptosis after miR-7 transfection (Fig.S2).Similarly, the percentage of cells with apoptotic nuclei significantly increased in miR-7- transfected group compared to the control in DAPI staining (Fig. 2D), suggesting the induction of apoptosis by miR-7. We also found substantial increases in the activities of caspase-3 and caspase-7 after miR-7 transfection (Fig. 2E). Moreover, the effect of miR-7 on activities of caspase-3 and caspase-7 could be rescued by miR-7 inhibitor (Fig. 2E). Together, these results indicated that the inhibition of cell growth by miR-7 was associated with increased apoptosis.
miR-7 induced apoptosis of A549 cells in a dose-dependent manner. A549 cells were transfected with various doses of miR-7(from 0 to 50nM) in low-serum (3%) DMEM for 48h and then cell apoptosis was detected by Annexin V /PI assay.
miR-7 inhibits migration of A549 cells
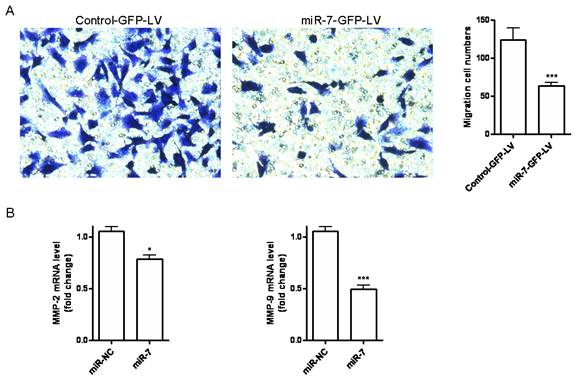
Cell migration is an essential process in cancer metastasis. To assess the effect of miR-7 on migration of A549 cells, we first established a stable miR-7 expressing A549 cell line (miR-7-GFP-LV) by pGCSIL-GFP lentivirus vector through lentiviral transduction. We observed a significant decrease in cell migration in miR-7-GFP-LV cells compared with mock vector control ones (control-GFP-LV) (Fig. 3A). We further analyzed the MMP-2 and MMP-9 expression by realtime RT-PCR. It showed a significant reduction of MMP-2 and MMP-9 expression in miR-7-GFP-LV cells (Fig. 3B).
miR-7 inhibits migration of A549 cells. (A)Stable miR-7 expressing A549 cells miR-7-GFP-LV and mock vector control cells control-GFP-LV were allowed to migrate on transwell inserts for 24h. The cells that had migrated through the filter into the lower wells were quantitated by gentian violet assay and were expressed as the total cell numbers of lower wells. (B)MMP-2 and MMP-9 expressions of miR-7-GFP-LV and control-GFP-LV cells were measured by realtime RT-PCR and normalized with respect to endogenous GAPDH and were expressed as fold change compared with control. Data are representative of two independent experiments. (*p<0.05, ***p<0.001).
miR-7 inhibited tumor growth of A549 cells in vivo
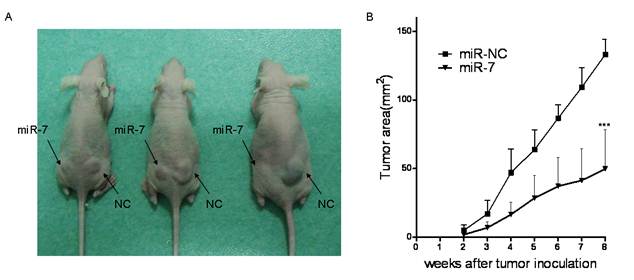
Given the suppression of A549 cell viability by miR-7 in vitro, we further evaluated whether increased level of miR-7 could inhibit tumor growth of A549 cells in vivo. A549 cells were transfected either with miR-7 or miR-NC. Six hours later, they were implanted subcutaneously into either posterior flank of the same nude mice (3 x106 cells per injection site). A549 cells transfected with miR-NC formed tumors 15 days after implantation, while A549 cells transfected with miR-7 partially failed to grow 15 days after injection, and exhibited a marked reduction in tumor size at week 8 post-implantation compared to the control group (Fig4.A/B). These data indicated that elevated miR-7 level in A549 cells markedly reduced their ability to form tumors.
miR-7 inhibited A549 cells tumor growth in vivo. miR-7 and miR-NC mimic transfected A549 cells (3×106) were injected s.c. into either side of the posterior flank of the same female nude mouse respectively (as indicated).(A) A photograph is shown 8 weeks after tumor cell implantation. (B) Tumor sizes are shown as Mean ± SEM. Three mice were used in each time and repeated twice, for a total of six mice. Data are representative of two independent experiments (***p<0.001).
BCL-2 emerged as a novel target for miR-7
According to the previous data, we hypothesized that miR-7 might inhibit the malignant phenotype of A549 cells by regulating genes that control cell proliferation or apoptosis. Thus, we tried to search for the target genes of miR-7 by algorithm of PicTar, Target Scan and miRanda. Among them, BCL-2, a molecule in the anti-apoptotic family, was found to have putative miR-7 binding sites within its 3'UTR in database of miRanda (Fig.5A).
miR-7 downregulated BCL-2 expression by directly targeting its 3'-UTR. (A) BCL-2 3'-UTR and corresponding fragments were inserted into the region immediately downstream of the luciferase gene in pGL3-con vector and validated by DNA sequencing. The sequences of predicted miR-7 binding sites within the BCL-2 3'-UTR, including wild-type UTR or UTR segments containing mutant (dotted lines) binding site are shown. (B) Relative luciferase activity was analyzed after co-transfection of miR-7 or miR-NC with the above reporter plasmids along with an endogenous control Renilla luciferase pRL-TK vector in A549 cells. (C) A549 cells were transfected either with miR-7 or miR-NC for 24 hours. BCL-2 mRNA levels were measured by realtime RT-PCR, normalized with endogenous GAPDH and expressed as fold change compared with control. (D) BCL-2 protein levels were examined by western blots using GAPDH as a loading control (one of two similar blots is shown). Results are shown as Mean ± SEM. Data are representative of three independent experiments (*p<0.05,***p<0.001).
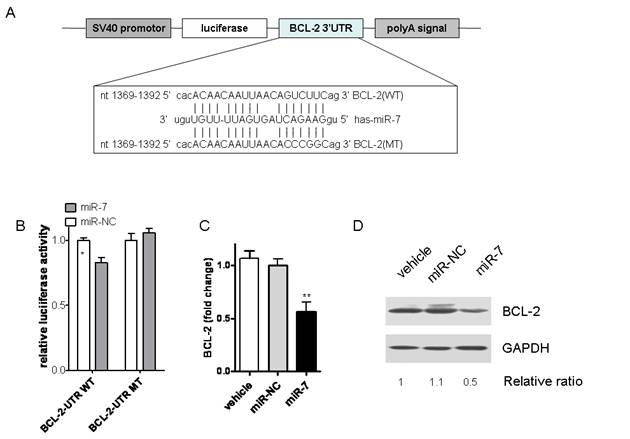
It is well known that miRNAs cause mRNA cleavage or translational repression by forming imperfect base pairing with the 3'UTR of target genes. To directly test whether miR-7 can repress BCL-2 through direct 3'UTR interactions, we cloned the 3'UTR of BCL-2 into a reporter plasmid downstream from luciferase and performed reporter assays using A549 cells. No significant differences of luciferase activities were found between the vehicle control group and negative control one (Data not shown). As shown in Fig.5B, miR-7 repressed the activity of luciferase fused to the WT BCL-2 3'UTR, while it failed to repress the mutated one. These results indicate that miR-7 may suppress BCL-2 expression by targeting the 3'-UTR of BCL-2 mRNA.
To assess whether miR-7 had a functional role in downregulation of endogenous BCL-2 expression, A549 cells were transfected with miR-7 mimic for 24h and then analyzed the BCL-2 expression by realtime PCR. As a result, overexpression of miR-7 significantly reduced the expression of BCL-2 at mRNA level (Fig.5C). Protein level of BCL-2 expression was further analyzed by western blot. Compared with control groups, BCL-2 expression was remarkably reduced in response to miR-7 transfection for 48h (Fig.5D). Taken together, these results suggested that miR-7 could decrease expression of BCL-2 through direct 3'UTR interactions.
Inhibition of BCL-2 induced apoptosis of A549 cells
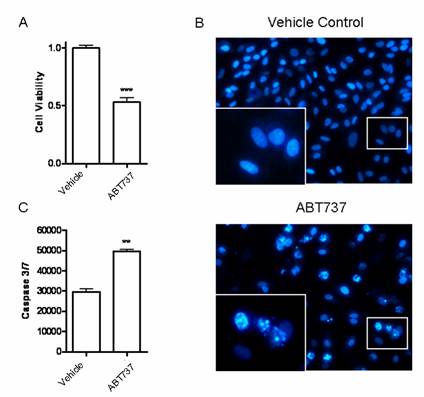
Since miR-7 could decrease expression of BCL-2, we would like to further investigate the influence of such kind of alteration on A549 cells. Blocking BCL-2 with its inhibitor ABT-737(10μM) significantly decreased the cell viability of A549 cells in CCK-8 assay (Fig6A). The percentage of cells with apoptotic nuclei was also increased in DAPI staining (Fig.6B). Meanwhile, significant increases in activities of caspase-3 and caspase-7 were observed after ABT-737 treatment (Fig.6C). These results were consistent with the effect of miR-7 on A549 cells (Fig2.A/B/D), providing further evidence that BCL-2 may be involved in miR-7-mediated growth suppression and apoptosis induction of A549 cells. Accordingly, identification of BCL-2 as a novel miR-7 target gene may explain, at least in part, the molecular mechanism of the tumor suppressor miR-7.
BCL-2 inhibitor ABT737 induced apoptosis of A549 cells. A549 cells were treated with BCL-2 inhibitor ABT737 (10μM) or DMSO (vechicle control) for 24h. (A) Cell viability was detected by CCK-8 assay. (B) Apoptotic nuclei were analyzed by DAPI staining. The inserted enlarged micrograph shows the morphology of the apoptotic cell nuclei. (C) Activities of caspase-3/7 were measured by a Caspase-Glo 3/7 assay kit. Data are representative of three independent experiments (**p<0.01, ***p<0.001).
Discussion
Increasing data have indicated miRNAs to be critical regulators in cancer-related processes[5, 8, 22]. MiRNA expression shows close correlation with various cancers[7, 9, 23, 24]. They are thought to function as either tumor suppressors or oncogenes. During tumorigenesis and development, overexpressed miRNAs may potentially target tumor suppressor genes, whereas downregulated miRNAs would mostly regulate oncogenes[24-26]. For example, miR-155, which is highly expressed in breast cancer, promotes tumor cell growth by downregulating the suppressor of cytokine signaling 1 gene[27]. Conversely, miR-7, which has low expression in brain, breast cancer, and pituitary adenoma, often acts as a tumor suppressor[12-15]. In NSCLC tissues, Duncavage et al also showed that miR-7 expression was significantly decreased in recurrent tumors[28].This result was consistent with the reduction of miR-7 level in NSCLC cell lines in our study. Moreover, we observed a significant inhibition on cell growth by overexpression of miR-7, suggesting miR-7 to be a tumor suppressor in NSCLC cell lines. However, a recent report showed that miR-7 promoted, rather than inhibited, cell growth and tumor formation in another lung cancer line CL1-5[29]. Therefore, the role of miR-7 may be different in various cancer types.
The fundamental function of miRNA is to regulate their targets by direct cleavage of the mRNA or by inhibition of protein synthesis through complementarity with their target 3'-UTRs[3, 6]. Several targets of miR-7 have been identified so far, such as IRS1 (Insulin Receptor Substrate 1), IRS2, EGFR (Epidermal Growth Factor Receptor), RAF1 (v-raf-1 murine leukaemia viral oncogene homologue 1) and PAK1 (p21/CDC42/RAC1-Activated Kinase 1)[12, 14, 15]. On the basis of bioinformatics analysis, we further predicted a number of additional miR-7 targets. Our present experimental results suggested BCL-2 to be a novel regulated molecule of miR-7 in A549 cells. Overexpression of miR-7 significantly reduced the endogenous BCL-2 mRNA and protein levels. MiR-7 also reduced the relative luciferase activity of reporter containing the potential binding sites on 3'-UTR of BCL-2 in reporter assays while it failed to repress the relative luciferase activity of reporter when the potential binding sites were mutated. Therefore, these results suggested that miR-7 decreased the expression of BCL-2 through direct 3'UTR interactions.
BCL-2 family proteins are key regulators of the apoptotic process [30, 31]. The members of this family can be divided into two groups with contradictory biological functions, one with anti-apoptotic (i.e. BCL-2, Bcl-XL, Bcl-w) and the other with pro-apoptotic (i.e.Bax, Bim, Bid) properties[30, 31]. BCL-2 predominantly localized in mitochondria which is the central coordinator of both intracelluar and extracellular signals mediating caspase-dependent and caspase-independent cell death[32-34]. Recently, it has been reported that induction of mitochondrial apoptosis requires the involvement of BCL-2[35, 36]. Thereby, it is emerging with an important role in tumorigenesis by blocking apoptosis[37, 38]. We found that inhibition of BCL-2 by its inhibitor ABT 737 significantly induced apoptosis of A549 cells, which is consistent with the effect of miR-7. Meanwhile, miR-7 could decrease the expression of anti-apoptotic molecule BCL-2. These results indicated that the downregulation of BCL-2 by miR-7 may be involved in the pro-apoptotic function of miR-7. Therefore, identification of BCL-2 as a novel miR-7 target gene may explain, at least in part, the molecular mechanism of the tumor suppressor miR-7.
In summary, this study further extends the biological role of miR-7 in NSCLC A549 cells and for the first time identifies BCL-2 as a novel target possibly involved in miR-7-mediated growth suppression and apoptosis induction of A549 cells. These findings may provide a basic rationale for the use of miR-7 in the treatment of NSCLC.
Acknowledgements
This work was supported by the National Key Technologies R & D Program of China during the eleventh Five-Year Plan Period (2009ZX10004-104), Fudan University Outstanding Doctoral Research Projects, National 973 project (2010CB912603, 2011CB910404), the National Science Foundation of China (No.30872378, 81072408) and the Science and Technology Commission of Shanghai Municipality (10JC1401100) in China.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Zheng ZM. Viral oncogenes, noncoding RNAs, and RNA splicing in human tumor viruses. Int J Biol Sci. 2010;6(7):730-55
2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281-97
3. Maas S. Gene regulation through RNA editing. Discov Med. 2010;10(54):379-86
4. Deng Y, Deng H, Bi F. et al. MicroRNA-137 targets carboxyl-terminal binding protein 1 in melanoma cell lines. Int J Biol Sci. 2011;7(1):133-7
5. Hummel R, Hussey DJ, Haier J. MicroRNAs: predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur J Cancer. 2010;46(2):298-311
6. Garzon R, Marcucci G, Croce CM. Croce. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9(10):775-89
7. Lin PY, Yu SL, Yang PC. MicroRNA in lung cancer. Br J Cancer. 2010;103(8):1144-8
8. Shibuya H, Iinuma H, Shimada R. et al. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology. 2011;79(3-4):313-20
9. Tili E, Michaille JJ, Wernicke D. et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci U S A. 2011;108(12):4908-13
10. Yu CC, Chen YW, Chiou GY. et al. MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol. 2011;47(3):202-10
11. Akao Y, Noguchi S, Iio A. et al. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. 2011;300(2):197-204
12. Kefas B, Godlewski J, Comeau L. et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68(10):3566-72
13. Chen H, Shalom-Feuerstein R, Riley J. et al. miR-7 and miR-214 are specifically expressed during neuroblastoma differentiation, cortical development and embryonic stem cells differentiation, and control neurite outgrowth in vitro. Biochem Biophys Res Commun. 2010;394(4):921-7
14. Jiang L, Liu X, Chen Z. et al. MicroRNA-7 targets IGF1R (insulin-like growth factor 1 receptor) in tongue squamous cell carcinoma cells. Biochem J. 2010;432(1):199-205
15. Saydam O, Senol O, Wurdinger T. et al. miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways. Cancer Res. 2011;71(3):852-61
16. Webster RJ, Giles KM, Price KJ. et al. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J Biol Chem. 2009;284(9):5731-41
17. Xiong SD, Yu K, Liu XH. et al. Ribosome-inactivating proteins isolated from dietary bitter melon induce apoptosis and inhibit histone deacetylase-1 selectively in premalignant and malignant prostate cancer cells. Int J Cancer. 2009;125(4):774-82
18. Kern MA, Schöneweiss MM, Sahi D. et al. Cyclooxygenase-2 inhibitors suppress the growth of human hepatocellular carcinoma implants in nude mice. Carcinogenesis. 2004;25(7):1193-9
19. Chaudhuri K, Chatterjee R. MicroRNA detection and target prediction: integration of computational and experimental approaches. DNA Cell Biol. 2007;26(5):321-37
20. Rehmsmeier M, Steffen P, Hochsmann M. et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507-17
21. Guo LM, Pu Y, Han Z. et al. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. Febs J. 2009;276(19):5537-46
22. Bhatt K, Zhou L, Mi QS. et al. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol Med. 2010;16(9-10):409-16
23. Oliveras-Ferraros C, Cufi S, Vazquez-Martin A. et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: Induction of the tumor suppressor miRNA let-7a and suppression of the TGFbeta-induced oncomiR miRNA-181a. Cell Cycle. 2011;10(7):1144-51
24. Shah AA, Leidinger P, Blin N. et al. miRNA: small molecules as potential novel biomarkers in cancer. Curr Med Chem. 2010;17(36):4427-32
25. Farazi TA, Spitzer JI, Morozov P. et al. miRNAs in human cancer. J Pathol. 2011;223(2):102-15
26. Meng F, Glaser SS, Francis H. et al. Functional Analysis of microRNAs in Human Hepatocellular Cancer Stem Cells. J Cell Mol Med. 2011 Epub ahead of print
27. Jiang S, Zhang HW, Lu MH. et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70(8):3119-27
28. Duncavage E, Goodgame B, Sezhiyan A. et al. Use of microRNA expression levels to predict outcomes in resected stage I non-small cell lung cancer. J Thorac Oncol. 2010;5(11):1755-1763
29. Chou YT, Lin HH, Lien YC. et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010;70(21):8822-31
30. Huang Z. Bcl-2 family proteins as targets for anticancer drug design. Oncogene. 2000;19(56):6627-31
31. Antonsson B. Bax and other pro-apoptotic Bcl-2 family "killer-proteins" and their victim the mitochondrion. Cell Tissue Res. 2001;306(3):347-61
32. Mehmeti I, Lenzen S, Lortz S. Modulation of Bcl-2-related protein expression in pancreatic beta cells by pro-inflammatory cytokines and its dependence on the antioxidative defense status. Mol Cell Endocrinol. 2011;332(1-2):88-96
33. Qin LY, Xing WB, Ye WJ. [Effect of combined therapy with Chinese drug and narrow broad ultraviolet B on Bcl-2, caspase-3 and survivin in skin lesion with psoriasis vulgaris]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2010;30(7):706-9
34. Santos AO, Pereira JP, Pedroso DLM. et al. In vitro modulation of Bcl-2 levels in small cell lung cancer cells: effects on cell viability. Braz J Med Biol Res. 2010;43(10):1001-9
35. Hossini AM, Eberle J. Apoptosis induction by Bcl-2 proteins independent of the BH3 domain. Biochem Pharmacol. 2008;76(11):1612-9
36. Stolz C, Hess G, Hahnel PS. et al. Targeting Bcl-2 family proteins modulates the sensitivity of B-cell lymphoma to rituximab-induced apoptosis. Blood. 2008;112(8):3312-21
37. Anagnostou VK, Lowery FJ, Zolota V. et al. High expression of BCL-2 predicts favorable outcome in non-small cell lung cancer patients with non squamous histology. BMC Cancer. 2010;10:186
38. Muilenburg DJ, Coates JM, Virudachalam S. et al. Targeting Bcl-2-mediated cell death as a novel therapy in pancreatic cancer. J Surg Res. 2010;163(2):276-81
Figures
Author contact
![]() Corresponding author: Yiwei Chu, MD, PhD, Department of Immunology, Shanghai Medical College, Fudan University, 138 Yixueyuan Road, mail box 226, Shanghai, 200032, China. Tel/Fax: +86-21-54237324. E-mail: yiwei_chucom
Corresponding author: Yiwei Chu, MD, PhD, Department of Immunology, Shanghai Medical College, Fudan University, 138 Yixueyuan Road, mail box 226, Shanghai, 200032, China. Tel/Fax: +86-21-54237324. E-mail: yiwei_chucom