Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Centrosome amplification in...
Centrosome amplification in...
Diagnostic, predictive and...
The centrosome as a cancer...
Conclusion and future...
Review criteria
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(8):1122-1144. doi:10.7150/ijbs.7.1122 This issue Cite
Review
A Clinical Overview of Centrosome Amplification in Human Cancers
Jason Yongsheng Chan ![]()
Medical Resident, Ministry of Health Holdings, Singapore
Received 2011-7-15; Accepted 2011-10-6; Published 2011-10-16
Abstract
The turn of the 21st century had witnessed a surge of interest in the centrosome and its causal relation to human cancer development - a postulate that has existed for almost a century. Centrosome amplification (CA) is frequently detected in a growing list of human cancers, both solid and haematological, and is a candidate "hallmark" of cancer cells. Several lines of evidence support the progressive involvement of CA in the transition from early to advanced stages of carcinogenesis, being also found in pre-neoplastic lesions and even in histopathologically-normal tissue. CA constitutes the major mechanism leading to chromosomal instability and aneuploidy, via the formation of multipolar spindles and chromosomal missegregation. Clinically, CA may translate to a greater risk for initiation of malignant transformation, tumour progression, chemoresistance and ultimately, poor patient prognosis. As mechanisms underlying CA are progressively being unravelled, the centrosome has emerged as a novel candidate target for cancer treatment. This Review summarizes mainly the clinical studies performed to date focusing on the mechanisms underlying CA in human neoplasia, and highlights the potential utility of centrosomes in the diagnosis, prognosis and treatment of human cancers.
Keywords: Boveri hypothesis, chromosomal instability, aneuploidy, centrosome, aurora kinase, p53
Introduction
Genomic instability is a hallmark of cancer cells, and may be broadly classified based on origin into chromosomal instability (CIN) and the less common microsatellite instability (MIN). CIN, describing the accelerated rate of change in chromosome number, gives rise to states of aneuploidy [1]. The mechanisms underlying CIN remain largely unknown, but may include defects in chromosome cohesion, mitotic checkpoint function, centrosome copy number, kinetochore-microtubule attachment dynamics, and cell-cycle regulation [2]. A causal association between centrosome amplification (CA) and human cancer development has long been hypothesized [3], yet evidence for this proposal has not been firmly established. CA is detected in a broad range of tumours, both solid and haematological, and is implicated as the major mechanism underlying the generation of multipolar mitoses, CIN, and aneuploidy. Several oncogenic and tumour suppressor proteins are known to localize to the centrosomes, deregulation of which may evoke centrosome abnormalities [4]. Emerging data are demonstrating the detection of centrosome defects in several pre-neoplasia, as well as the correlation of more extensive centrosome alterations in the continuum of advancing disease. Intuitively, this supports a non-bystander role in both tumourigenesis and disease progression. Moreover, the frequent association with extensive karyotypic aberrations and poor patient outcome supports its clinical significance in human cancer [Tables 1-3].
Centrosome amplification in human solid tumours.
| Cancer site | n | CA (%) | CIN | Association | Ref | |
|---|---|---|---|---|---|---|
| Samples | Intratumoural | |||||
| Breast | ||||||
| NOS | 19 | 95 | - | - | - | [10] |
| NOS | 19 | 95 | 10 | - | Nuclear AhR expression | [65] |
| AC | 30 | - | - | - | Cytoplasmic LMW Cyclin E | [41] |
| AC | 35 | 97 | - | - | - | [25] |
| AC | 31 | 77 | - | - | - | [26] |
| IDC | 8 | 75 | - | - | - | [153] |
| IDC | 20 | 100 | - | Y | p53 mutation | [27] |
| AC | 103 | 89 | 46 | - | HER2 positive ER/PR negative ˟p53, BCL2 | [34] |
| AC | 56 | 100 | - | N | Pin1 expression | [61] |
| AC | 75 | 100 | 7 | HER2 positive ER/PR negative | [35] | |
| AC | 50 | 100 | 1-87 | N | BRCA1 mutation ˟Aurora-A, p53, ER/PR, HER2 | [37] |
| AC - Diploid - Aneuploid | 3 13 | 100 100 | - | Y | ˟p53, ER | [33] |
| AC - Diploid - Aneuploid-stable - Aneuploid-unstable | 3 3 4 | 100 100 100 | 3 2 10 | Y | Cyclin E/A expression | [32] |
| AC - Diploid - Aneuploid-stable - Aneuploid-unstable | 12 7 14 | 100 100 100 | 2 2 10 | Y | - | [29] |
| DCIS + IDC | 21 | 100 | 30-100 | Y | Aurora-A expression | [52] |
| DCIS | 7 | Most | - | - | - | [27] |
| DCIS | 42 | 75 | - | Y | - | [28] |
| DCIS | 7 | 100 | 8 | Y | - | [29] |
| Ovarian | ||||||
| Mainly endometroid and serous | 63 | 78 | - | - | - | [102] |
| Mainly serous | 18 | 100 | - | - | Aurora-A expression | [105] |
| NOS | 10 | 100 | 17 | Y | - | [103] |
| Anogenital | ||||||
| AC | 5 | 100 | 8 | Y | High-risk HPV | [84] |
| CIN* | 14 | 71 | 2 | Y | High-risk HPV | [84] |
| CIN* | 48 | 62 | - | Y | - | [28] |
| Anal SCC Anal LSIL Anal HSIL | 14 13 6 | 100 100 100 | 5.8 2.5 4.7 | - | High-risk HPV | [205] |
| Prostate | ||||||
| AC | 18 | 89 | - | - | - | [10] |
| AC | 140 | 94 | 15-100 | Y | - | [66] |
| AC | 63 | - | - | Y | - | [67] |
| PIN | 45 | 28 | - | Y | - | [28] |
| Testicular | ||||||
| Infantile yolk sac tumour Infantile teratoma Seminoma Mixed non-seminoma Embryonal carcinoma Teratoma Spermatocytic seminoma | 1 6 11 12 2 1 3 | 100 33 100 100 100 100 100 | - 20 - - - - 60 | Y | ˟Aurora-A expression | [104] |
| Urothelial | ||||||
| Bladder TCC | 22 | 82 | - | Y | - | [71] |
| Bladder TCC | 45 | 89 | 9-100 | Y | - | [70] |
| Bladder TCC | 50 | 60 | - | Y | 20q gain | [72] |
| Bladder TCC | 65 | 52 | - | - | Cyclin E expression p53 mutation | [76] |
| Renal + Ureteral TCC | 90 | 50 | - | Y | 20q gain | [74] |
| Bladder TCC | 100 | 64 | - | Y | 20q gain Aurora-A expression | [73] |
| Bladder TCC | 101 | 101 | 65 | - | PLK1/Aurora-A expression p53 mutation | [79] |
| Bladder TCC | 104 | 65 | - | - | BUBR1 expression | [80] |
| Bladder TCC | 21 | 60 | - | Y | Cyclin D1 amplification | [75] |
| Renal | ||||||
| RCC | 8 | 25 | 10 | Y | Telomere dysfunction | [81] |
| Adrenal | ||||||
| Carcinoma Adenoma | 10 4 | 100 100 | 1-7 1-19 | - | - | [83] |
| Neural | ||||||
| NOS | 20 | 95 | - | - | - | [10] |
| Cerebral PNET | 2 | 100 | - | Y | p53 mutation | [140] |
| Pituitary adenoma | 12 | 100 | - | Y | Securin expression | [142] |
| Diffuse astrocytic glioma | 46 | 100 | - | - | [141] | |
| Glioma | 34 | - | - | - | Aurora-A mRNA expression | [144] |
| Neuroblastoma | 20 | - | - | - | MYCN amplification | [153] |
| Neuroblastoma | 27 | 89 | 23 | Y | ˟MYCN | [149] |
| Medulloblastoma | 20 | - | - | - | - | [143] |
| Lung | ||||||
| NOS | 15 | 100 | - | - | - | [10] |
| AC SCC | 19 40 | 53 58 | - | Y | Cyclin E/E2F1 expression ˟p53 | [128] |
| AC SCC | 88 87 | 33 24 | - | Y | p16 expression pRb loss ˟p53 | [129] |
| Colorectal | ||||||
| AC | 15 | 87 | - | - | - | [10] |
| AC | 33 | - | - | - | - | [108] |
| AC Adenoma | 30 54 | 100 65 | - | - | TEIF expression | [109] |
| Pancreatic | ||||||
| Ductal Endocrine | 13 3 | 85 0 | - | - | - | [122] |
| Adenoma | 3 | 67 | - | - | - | [122] |
| Hepatobiliary | ||||||
| GC CCC BDC Chronic cholecystitis Adenomatous polyp Hepatolithiasis Choledochal cyst | 13 19 8 15 1 16 5 | 69 58 50 7 0 6 0 | - | - | - | [116] |
| HCC | 33 | 91 | 8.9 | Y | p53 mutation | [117] |
| Head & Neck | ||||||
| Thymic carcinoid tumour | 1 | 100 | - | - | - | [160] |
| SCC | 12 | 83 | - | - | p53 mutation MDM2 expression | [153] |
| Oral SCC | 18 | 94 | - | - | - | [154] |
| SCC PA | 5 3 | 100 100 | - | Y | Telomere dysfunction | [155] |
| SCC | 29 | 62 | - | - | Aurora-A expression | [156] |
| Oral SCC Dysplasia | 15 25 | 100 100 | <1-5 <1-4 | - | - | [158] |
| SCC | 50 | - | 41 | Y | - | [157] |
| Laryngeal SCC | 35 | 94 | - | - | - | [159] |
| BSTT | ||||||
| Leiomyosarcoma | 1 | 100 | - | - | - | [162] |
| Osteosarcoma | 3 | 67 | 25 | - | - | [163] |
| Liposarcoma Chondrosarcoma MFH Haemangiopericytoma Atypical lipomatous tumour Giant cell tumour Benign or tumour-like lesions† | 7 5 5 1 2 1 7 | 87 60 100 100 50 100 29 | - | Y | - | [164] |
| Liposarcoma - Type D near-diploid - Type H near-tetraploid Lipoma | 3 3 9 | 100 100 Rare | 29-50 53-71 - | N | - | [165] |
| Peripheral PNET Synovial sarcoma Rhabdomyosarcoma MFH MPNST Liposarcomma Leiomyosarcoma Benign or tumour-like lesions‡ | 9 8 9 35 18 14 11 14 | 89 75 67 63 39 21 18 29 | >15% | - | TEIF expression | [166] |
| Giant cell tumour - Nonrecurrent - Recurrent - Malignant | 57 35 8 | 66 95 100 | 5 17 37 | N | - | [167] |
n, sample size; CA, centrosome amplification; CIN, chromosomal instability/aneuploidy; AC, adenocarcinoma; IDC, invasive ductal carcinoma; DCIS, ductal carcinoma in-situ; CIN*, cervical intraepithelial neoplasia; LSIL, low-grade squamous intraepithelial lesion; HSIL, high-grade squamous intraepithelial lesion; PIN, prostate intraepithelial neoplasia; TCC, transitional cell carcinoma; PNET; primitive neuroectodermal tumour; SCC, squamous cell carcinoma; GC, gallbladder cancer; CCC, cholangiocellular carcinoma; BDC, bile duct cancer; HCC, hepatocellular carcinoma; PA, pleomorphic adenoma; BSTT, bone and soft tissue tumours; MFH, malignant fibrous histiocytoma; MPNST, malignant peripheral nerve sheath tumour; NOS, not otherwise specified; ˟, no association.
†Includes melorheostosis, lipoma, myxoma, granular cell tumour, non-ossifying fibroma, schwannoma, pigmented villonodular synovitis.
‡Includes leiomyoma, neurilemoma, lipoma, and fibrous histiocytoma.
Centrosome amplification in human haematological cancers and pre-neoplasia
| Cancer type | n | CA (%) | CIN | Association | Ref | |
|---|---|---|---|---|---|---|
| Samples | Intratumoural | |||||
| cHL | 2 | 100 | - | - | ˟Aurora-A, PLK1 | [177] |
| cHL | 31 | 90 | - | - | - | [178] |
| Total NHL DLBCL MCL - Tetraploid - Diploid FL - High grade - Low grade MZBCL | 92 18 12 19 8 16 17 | 100 100 100 100 100 100 100 | 32 42 56 31 30 23 29 | - - Y - - | ˟p53 | [175] |
| MCL - Tetraploid - Diploid | 9 24 | 100 100 | 56 36 | Y | - | [179] |
| BL | 1 | 100 | 30-50 | N | - | [182] |
| B-CLL | 64 | 100 | 18 | N | - | [180] |
| B-CLL MBL | 70 4 | 100 100 | 26 12 | N | - | [176] |
| ALCL | 17 | 41 | - | - | ˟p53 | [181] |
| CML - CP - BC | 18 16 | 100 100 | 29 54 | Y | - | [183] |
| CML - CP - BC | 5 5 | 100 | 35 59 | - | - | [184] |
| CML (CP) SM | 29 2 | 100 100 | 13-26 16 | - | - | [188] |
| AML | 51 | 100 | 36 | Y | - | [190] |
| Marrow failure - AA - MDS - PNH | 15 9 1 | 27 11 0 | 10-24 | Y | - | [192] |
| ATL | 8 | 88 | 19 | - | - | [194] |
| MM (Plasma cells) MGUS | 41 8 | 100 - | 26 14 | - | RHAMM expression | [197] |
| MM (Plasma cells) SMM MGUS | 14 7 3 | 64 71 67 | 26 20 12 | N | - | [12] |
| MM - Plasma cells - B cells | 50 38 | 17 37 | 9 10 | - | - | [198] |
| GIST | 1 | 100 | 17 | - | - | [188] |
n, sample size; CA, centrosome amplification; CIN, chromosomal instability/aneuploidy; cHL, classic Hodgkin's lymphoma; NHL, non-Hodgkin's lymphoma; DLBCL, diffuse large B-cell lymphoma; MCL, mantle cell lymphoma; FL, follicular lymphoma; MZBCL, marginal zone B-cell lymphoma; BL, Burkitt's lymphoma; B-CLL, B-cell chronic lymphocytic leukaemia; MBL, monoclonal B-cell lymphocytosis; ALCL, ALK-positive anaplastic large cell lymphoma; ATL, adult T-cell leukaemia; AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CP, chronic phase; BC, blast crisis; SM, systemic mastocytosis; AA, aplastic anemia; MDS, myelodysplastic syndrome; PNH, paroxysmal nocturnal hemoglobinuria; MM, multiple myeloma; SMM, smouldering multiple myeloma; MGUS, monoclonal gammopathy of undetermined significance; ˟, no association; Y, positive association; N, no association
Clinical correlates of centrosome aberration in human tumours
| Cancer type | Patient cohort | Correlation | Ref |
|---|---|---|---|
| Breast | AC, surgical resection, n = 16 | Nodal metastasis | [33] |
| IDC, surgically treated, n = 20 | Tumour grade | [27] | |
| DCIS, surgically treated, n = 42 | Tumour grade | [28] | |
| AC, surgically treated (6 received neoadjuvant chemotherapy), n = 103 | Axillary nodal metastasis Not with tumour size, nuclear grade, DNA index, S-phase fraction, or proliferation index | [34] | |
| Mainly IDC, surgically treated, n = 10 | Tumour grade | [32] | |
| AC, surgically treated, n = 73 | Not with tumour size, grade, or nodal metastasis | [35] | |
| Mainly IDC, surgically treated, n = 50 | Not with tumour size, tumour grade, or nodal metastasis | [37] | |
| NOS, surgically treated, n = 30 | Tumour grade | [41] | |
| Prostate | AC, surgically treated, n = 140 | Tumour grade and distant metastasis | [66] |
| AC, surgically treated, n = 63 | Tumour grade and stage | [67] | |
| PIN, surgically treated, n = 45 | Tumour grade | [28] | |
| Bladder | TCC, surgically treated, n = 45 TCC, surgically treated, n = 22 TCC, surgically treated, n = 65 | Tumour grade | [70] [71] [76] |
| TCC, surgically treated, n = 50 | Tumour number, grade, poor recurrence-free survival, and PFS | [72] | |
| TCC, surgically treated, n = 100 | Disease progression | [73] | |
| Renal/ Ureteral | TCC, surgically treated, n = 90 | Tumour grade Not with intravesical or local recurrence, distant metastasis, or cancer-specific death | [74] |
| Cervix | CIN, surgically treated, n = 14 | Tumour grade | [84] |
| CIN, surgically treated, n = 48 | Tumour grade | [28] | |
| Ovary | Mainly endometroid and serous Surgically treated, n = 63 | Stage, tumour grade | [102] |
| Colorectum | AC, surgically treated, n = 30 Adenoma, surgically treated, n = 54 | Tumour grade of CRC and adenoma | [109] |
| Hepatobiliary | GC + CCC + BDC, surgically treated, n = 40 | Stage | [116] |
| HCC, surgically treated, n = 33 | Not with tumour size, stage, or proliferative activity | [117] | |
| Lung | AC + SCC, surgically treated, n = 59 | Not with stage, tumour grade | [128] |
| AC + SCC, surgically treated, n = 175 | Not with tumour type, age, gender, size, stage, or cancer-specific death | [129] | |
| Neural | Diffuse astrocytic glioma, surgically treated, n = 46 | Tumour grade | [141] |
| Glioma, surgically treated, n=34 | Tumour grade | [144] | |
| Head & neck | Oral SCC, surgically treated +/- RT, n = 18 | Tumour recurrence | [154] |
| Oral SCC, surgically treated, n = 15 | Tumour grade | [158] | |
| SCC, surgically treated, n = 50 | Tumour size, stage, distant metastasis, poor DFS, and OS | [157] | |
| Laryngeal SCC, surgically treated, n = 35 | Tumour recurrence | [159] | |
| BSTT | Soft tissue sarcoma, surgically treated, n = 104 | Tumour grade | [166] |
| Giant cell tumour, surgically treated, n = 100 | Tumour recurrence and malignancy | [167] | |
| Blood | NHL, n = 92 | Tumour grade (FL), mitotic and proliferation indices (FL, DLBCL, MCL) | [175] |
| CML (chronic phase), n = 34 | Not with Hasford score | [183] | |
| AML, n = 51 | Cytogenetically-defined risk groups | [190] | |
| B-CLL, untreated, n = 70 | Lymphocyte doubling time, time to 1st treatment Not with IgVH genes mutation status or cytogenetically-defined risk groups | [176] | |
| MM, multiple modalities†, n = 97 | High CI with Chr 13 deletion, t(4;14), t(14;16), high plasma cell labelling index, and poor OS | [12] | |
| MM, multiple modalities‡, n = 539 | High CI with poor PFS and OS | [199] |
n, sample size; AC, adenocarcinoma; IDC, invasive ductal carcinoma; PIN, prostate intraepithelial neoplasia; HCC, hepatocellular carcinoma; GC, gallbladder cancer; CCC, cholangiocellular carcinoma; BDC, bile duct cancer; SCC, squamous cell carcinoma; TCC, transitional cell carcinoma; CIN, cervical intraepithelial neoplasia; NHL, non-Hodgkin's lymphoma; FL, follicular lymphoma; DLBCL, diffuse large B-cell lymphoma; MCL, mantle cell lymphoma; CML, chronic myeloid leukaemia; B-CLL, B-cell chronic lymphocytic leukaemia; AML, acute myeloid leukaemia; MM, multiple myeloma; CI, centrosome index; PFS, progression-free survival; DFS, disease-free survival; OS, overall survival.
†Includes single autologous stem cell transplantation, dexamethasone-based, melphalan and prednisolone, novel agents, thalidomide-based
‡Includes chemotherapy, high-dose therapy with stem cell transplantation, total therapy II, and novel agents such as bortezomib
The centrosome constitutes the major microtubule organizing centre of the cell (MTOC) in mammalian cells, and plays a dominant role in the maintenance of cell polarity and cytoplasmic architecture through the nucleation and spatial organization of microtubules [5], as well as in coordinating efficient cell cycle progression [6]. Structurally, the centrosome is a tiny cytoplasmic organelle consisting of a core formed by a pair of orthogonally-arranged centrioles, embedded in a structurally complex amorphous protein matrix termed the pericentriolar material (PCM). Centrioles are small barrel-shaped organelles consisting of a cylindrical array of nine triplet microtubules, whilst the PCM is composed of a lattice of coiled-coil proteins including gamma-tubulin ring complexes, pericentrin [7], and calcium-sensitive fibres such as Sfi1p and centrin [8]. During a normal cell cycle, centrosome duplication occurs strictly once to yield two centrosomes that function as the spindle poles of the dividing cell [9]. The centrosome duplication cycle is exactly coordinated with cell cycle progression, and is tightly coupled to DNA replication, mitosis, and cytokinesis through parallel pathways of regulation. The presence of only two centrosomes in the cell as it enters mitosis favours the formation of a bipolar spindle and the equipartition of chromosomes to each daughter cell.
Centrosome defects observed in human cancers may be classified into 2 broad categories based on structural or functional alterations. Structural defects include changes in centrosome shape (string-like elongated linear arrays, ring-like, amorphous, atypical filaments, corkscrew), size (usually large patchy aggregates, but may range from tiny flecks), number (more than 2 per cell), position (diffuse patchy cytoplasmic staining, scattered, clustered) and/or composition (higher protein levels, inappropriate phosphorylation, absent centrioles). Functionally, defective centrosomes are capable of abnormal microtubule nucleation, and formation of disorganized mitotic spindles, leading to chromosomal missegregation and aneuploidy [10-12]. These defects, collective referred to as CA, are major contributors to mechanisms underlying loss of cell cycle fidelity, genomic instability and loss of tissue architecture in human cancers.
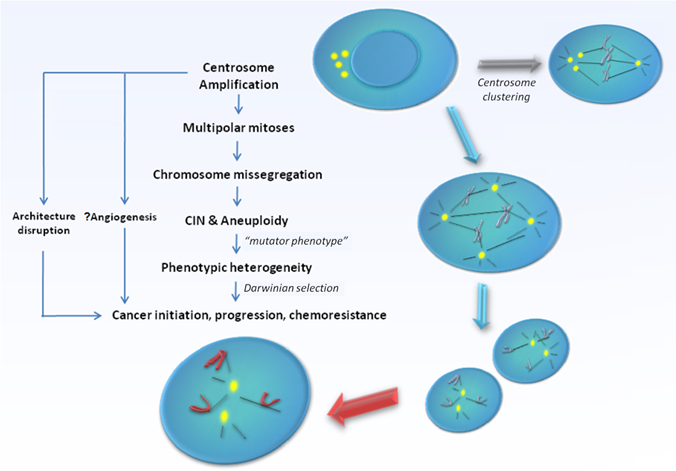
CA can arise via several fundamentally distinct but yet not mutually exclusive mechanisms. Firstly, deregulation of the centrosome duplication cycle may occur, leading to centriole overduplication (loss of cell-cycle control) or excessive centriole multiplication (loss of copy number control) during a single S-phase. The former involves successive rounds of centriole reproduction, whilst the latter entails the rapid simultaneous formation of multiple daughter centrioles at single maternal centriole templates. Both would result in the formation of supernumerary centrosomes in near-diploid cells [13]. Secondly, failure of cytokinesis may generate polyploid cells with supernumerary centrosomes [14]. In such cells, centrosome number accumulates in parallel with nuclear abnormalities and do not primarily originate from enhanced duplication. The potential for such cells to re-enter S-phase and give rise to viable progeny seems to depend at least partially on the fidelity of the tetraploidy checkpoint governed by p53 function, which in such cells triggers cell cycle arrest and apoptosis. Thirdly, centrosome number can increase via cell fusion, for example, when cells are under the influence of fusogenic viruses [15, 16]. Fourthly, functional centrioles may also form de novo [17, 18]. Lastly, the disintegration of centrosomes via fragmentation of PCM may lead to an appearance of CA which otherwise may not necessarily be functional [19, 20]. The exact mechanisms triggering CA and the relative importance of each of their contribution towards the development of specific human cancers remain under intensive research. The issue gets further complicated since the presence of extra centrosomes does not necessarily lead to major cell division errors as extra centrosomes have been demonstrated to undergo clustering [21], or even gradual inactivation [22] during mitosis, thereby preserving bipolarity of the mitotic spindle. At the same time, a limited level of CIN that permits continued bipolar cell division may be maintained consequent to segregation errors via merotelic kinetochore-microtubule attachment possibly during a transient 'multipolar spindle intermediate' prior to centrosome clustering and anaphase [23]. Still, cells with amplified centrosomes are often too unstable to survive, and are eliminated through cell death mechanisms such as apoptosis, mitotic catastrophe, or replicative senescence [23, 24]. It is likely in order for tumourigenesis to occur, these selection forces ultimately must result in the generation of a rare centrosome-amplified, genomically-unstable but yet viable cellular subclone. In possession of a sustainable mutator phenotype, cells that eventually harbour the rare genomic complement that confer survival advantage through a Darwinian selection process would promote cancer development and progression [Figure 1].
Model supporting centrosome amplification as a cause of carcinogenesis. Centrosome amplification has been detected in broad range of tumours, both solid and haematological, and has been implicated in the generation of multipolar mitoses, chromosomal instability (CIN), and aneuploidy. Centrosome amplification also contributes to loss of tissue architecture, and possibly angiogenesis in human cancers. Defective centrosomes are capable of abnormal microtubule nucleation and formation of disorganized mitotic spindles, leading to chromosomal missegregation and aneuploidy. However, the presence of extra centrosomes does not necessarily lead to major cell division errors as extra centrosomes may undergo clustering, thereby preserving bipolarity of the mitotic spindle. Ultimately, the "mutator phenotype" generated as a result raises the possibility of producing cells with the rare genomic complement that may confer survival advantage through a Darwinian selection process, thereby promoting cancer development and progression.
Despite the vastly expanding amount of studies demonstrating the clinical relevance of CA in human cancers, there is as yet no comprehensive review of the existing literature. This Review will summarize studies performed to date implicating CA on human neoplasia, with a focus on those based on human cancer tissue samples. Relevant in vitro studies that shed light on the underlying mechanisms leading to CA in specific tumour types are also included. Finally, the potential utility of centrosomes in the diagnosis, prognosis and treatment of human cancers are discussed.
Centrosome amplification in solid tumours
Breast cancer
In primary human cancers, structural alterations of centrosomes have been first systematically studied in specimens of high grade metastatic breast adenocarcinoma. Several centrosome defects, including an increase in centrosome number and volume, with chaotic subcellular locations, accumulation of excessive PCM, supernumerary centrioles, as well as inappropriate accumulation of phosphocentrin during non-mitotic phases are frequently observed. In addition, breast tumour cells display functional centrosome abnormalities, as characterized by unusually large arrays of microtubules nucleated by an increased number of MTOCs [25]. Ultrastructural analysis of centrosomes via electron microscopy revealed that breast tumour cells contain numerous large centrioles surrounded by abundant densely-stained PCM, disrupted centriole barrel structure (open-ring and missing triplet microtubules), unincorporated microtubule complexes, centrioles of unusual length, centrioles functioning as ciliary basal bodies, and mispositioned centrioles [25, 26]. Interestingly, tumours with excess PCM are highly anaplastic, lack normal cell polarity, and tend to have a higher median frequency of abnormal mitoses compared to tumours with other centrosome abnormalities [26].
Supporting CA as a causal and early event in breast tumourigenesis, CA is also present in ductal carcinoma in-situ (DCIS), and correlates with CIN in these lesions [27-29]. Gamma-tubulin gene amplification and the concomitant protein overexpression are also seen in atypical ductal hyperplasia of the breast, albeit significantly lower than in lesions of DCIS and invasive ductal carcinoma (IDC) [30, 31]. Moreover, within DCIS lesions, centrosome defects increases with advanced histological grade, which suggests a higher propensity of evolution into invasive carcinoma [28].
Kronenwett et al. classified aneuploid human breast cancers into genomically-stable and -unstable subtypes using a surrogate marker based upon the proportion of cells with non-modal DNA content values referred to as the Stemline Scatter Index (SSI) [32]. In keeping with earlier studies [27], greater CA, along with aberrant mitotic spindles are observed in genomically-unstable and clinically more aggressive aneuploid breast cancers with high SSI, compared to genomically-stable aneuploid and diploid counterparts [29, 32]. Furthermore, CA correlates with high histological grade and lymph node metastasis in most studies, supporting it as a possible marker for aggressive disease and poor prognosis in breast cancer [27, 32-35]. The implication of CA upon breast cancer prognosis is further supported by its association with HER2/neu overexpression, negative estrogen receptor (ER) status, and negative progesterone receptor (PR) status in some studies [34-36]. One study however, did not find any association of CA with CIN, nor with hormonal receptor status, HER2/neu status, tumour size, grade or nodal status [37].
Numerous studies have been performed attempting to uncover the underlying mechanisms and significance of CA in breast cancer development. Available evidence suggests a cooperative model of different mechanisms for the development of CA during breast carcinogenesis. In human breast carcinoma, both centrosome size and number, but not microtubule nucleation capacity, shows a positive correlation with aneuploidy and CIN that is independent of p53 mutation [27, 33, 37]. Centrosomal microtubule nucleation capacity is enhanced with p53 mutation, and in addition, correlates with loss of tissue differentiation (high histological grade) and poor patient prognosis. This suggests that in breast cancer, the mechanisms driving CIN and loss of tissue differentiation are determined by independent centrosome defects (structural and functional, respectively), which may not be necessarily dependent on the absence of p53 function [38]. p53 loss may play a critical role in the generation of supernumerary centrosomes in breast cancer cells only following genotoxic stress or mitogenic stimulation through its abrogation of the G1/S cell cycle checkpoint, involving a CDK2/cyclin-dependent pathway [39, 40]. In relation, CA and multinucleation are present in MCF7 cells as a result of cytokinesis failure, due to the expression of the low molecular isoform of cyclin E. This phenotype is exacerbated in the absence of p53 and occurs via premature activation of cdc25C [41, 42].
BRCA1 is a breast and ovary-specific tumour suppressor that in association with BRCA1-associated RING Domain 1 (BARD1), acts as a powerful E3 ubiquitin ligase. The BRCA1-BARD1 complex, through the ubiquitination of gamma-tubulin, maintains centrosome homeostasis by prevention of its aberrant reduplication [43]. Inhibition of BRCA1 causes rapid centrosome overduplication and possibly fragmentation in human breast cell lines [43-45], as well as stimulates centrosome microtubule nucleation function [46]. Consistent with these reports, disruption of the BRCA1 gene in mice leads to CA and aneuploidy [47, 48]. Numerical centrosome aberration is also associated with negative BRCA1 expression and its germline mutations in human breast cancer specimens [37]. Overexpression of Ninein-like protein (Nlp), a BRCA1-associated centrosomal protein overexpressed in human breast cancers, is associated with CA in rodent fibroblasts as well as spontaneous breast tumourigenesis in transgenic mice, suggesting that Nlp overexpression mimics BRCA1 loss [49]. NPM is a BRCA2-associated protein, which together with ROCK2 may form a complex that maintains the numerical integrity of centrosomes and accurate cell division. Dysfunction of this regulation might be involved in the tumourigenesis of breast cancer through both centrosome overduplication and fragmentation [50].
Overexpresson of Aurora-A kinase, a centrosome-associated serine/threonine kinase, and gain of its associated gene locus on 20q13.2, leads to aneuploidy, cell transformation, and CA in mammalian cells [51]. Aurora-A overexpression is associated with CA in DCIS [52] but not in IDC [37], suggesting that it may be more relevant in tumour initiation than progression. Correspondingly, Aurora-A overexpression and CA are found to be early events in mammary tumour development in animal models [53, 54], which may be mediated by estrogen exposure [55]. The mechanism of CA triggered by Aurora-A may be in part due to cytokinesis failure, which is facilitated by its effects on promoting p53 degradation [56]. Aurora-A-overexpressed mice display CA, tetraploidization and premature sister chromatid segregation prior to tumour formation, which is aggravated by haploinsufficiency of p53. The severe chromosomal abnormality did not cause cell death due to the concomitant activation of the pro-survival AKT pathway, allowing continuous proliferation of the tetraploid cells and accumulation of centrosomes [54]. On the other hand, Aurora-A kinase is also known to interfere with BRCA1 ubiquitin ligase function though inhibitory phosphorylation [57], thereby also possibly promoting centrosome overduplication and fragmentation. MDC1 and BRIT1, both negative regulators of Aurora-A and PLK1, are observed to be inversely correlated with CA in human breast cancer. MDC1-depleted cells exhibit centrosome overduplication, whilst BRIT1 depletion leads to defective cytokinesis and centrosome accumulation [58]. Similar to Aurora-A kinase is another centrosomal kinase Nek2 that is frequently elevated in DCIS and IDC, which promotes the accumulation of supernumerary centrosomes via cytokinesis failure in breast epithelial cells [59]. Oncogenic k-ras but not c-myc initiates CA in mammary precursor lesions. Instead, c-myc induces CA in tumours, pointing to a role for c-myc in tumour progression and k-ras in initiation. Abolishing the expression of CDK4/cyclin D1 or Nek2 abrogates Ras-induced CA in human mammary epithelial cells, whereas silencing cyclin E1 or B2 has no such effect [60], suggesting that Ras-evoked CA may be an early breast oncogenic event that occurs through a Nek2 and CDK4/cyclin D1-dependent pathway.
Pin1, a prolyl isomerase implicated in the activation of multiple oncogenic pathways, is overexpressed and positively correlates with CA in human breast cancer tissue. Additionally, Pin1 localizes to centrosomes in interphase but not mitotic cells in vitro. Overexpression of Pin1 induces multiple rounds of centrosome duplication in S-phase arrested cell lines, resulting in formation of multipolar mitoses, chromosomal missegregation, aneuploidy, and malignant transformation [61]. Pin1 overexpression in mouse mammary glands induces CA, leading to mammary hyperplasia and malignant tumours. This suggests that a mechanism of Pin1 overexpression and uncontrolled centrosome duplication cycles may contribute to CA in breast cancer [61]. Y-box binding protein YB-1, which is overexpressed in 75% of human breast carcinomas, has been implicated in the generation of CA in breast cancers as well. In transgenic mice, YB-1 expression in mammary epithelial cells provokes breast carcinomas of different histologic types through induction of CIN that emerges from mitotic failure and CA. Moreover, YB-1-evoked CA occurs in premalignant lesions and proceeds during breast cancer development [62]. The mechanism underlying CA in YB-1-dependent breast cancer development probably occurrs through cytokinesis failure induced by LIMK mislocalization, and is independent of p53 and pRb deregulation [63]. Several other mechanisms implicated in the generation of CA in breast cancer include the aberrant expression of LMO4 [64], centrosome overduplication via nuclear expression of endogenous aryl hydrocarbon receptor (AhR) and cyclin E [65], as well as cell fusion following ectopic expression of the RAD6 ubiquitin conjugating enzyme [15].
Urogenital cancer
Prostate cancer
Support for CA contributing to genomic instability during prostate cancer development has been derived from data showing the increased frequency of centrosome abnormalities progressing from prostate intraepithelial neoplasia (PIN) to non-metastatic and metastatic cancers, with the extent of defects correlating with the degree of CIN. Intra-tumoural centrosome abnormalities are also more severe and less variable in metastatic tumours than those in localized prostatic carcinoma. Centrosome size and number, as well as pericentrin levels are greater in high Gleason grade tumours, which correlates with the more advanced extent of CIN in these lesions [10, 28, 66, 67]. In support of these findings, artificial induction of centrosome abnormalities in cultured prostate cells by overexpression of the centrosome protein pericentrin reproduces many features of aggressive prostate cancer. Overexpression of pericentrin induces severe centrosome and spindle defects, cellular disorganization, genomic instability and enhances growth in prostate epithelial cell lines [66]. There is evidence to support a role for cytokinesis defects in the generation of CA in prostate cancer. Overexpression of oncogenic kinase PIM-1 in prostatic epithelial cells leads to the development of mitotic spindle defects, multinucleation, and supernumerary centrosomes [68]. Non-functional p53 also promotes the formation of increased centrosome number and size in prostate cancer cell lines by abrogating the G2/M checkpoint control [69].
Urothelial cancer
In transitional cell carcinoma (TCC) of the urinary tract, CA is associated with CIN and aneuploidy [70-75], but not with MIN - tumours with low expression of MLH1 or MSH2 mismatch repair proteins are not linked with CA [73]. Interestingly, coalesence of multiple centrosomes into two functional spindle poles occurs in 80% of chromosomally-unstable bladder tumours, suggesting that this may be an important mechanism limiting the deleterious effects of multipolar mitoses on the cancer cells [75]. Several studies have found an association of centrosome aberration with clinical prognostic indicators, including high histological grade [70-72, 74, 76]. CA correlates with recurrence-free survival and progression-free survival, and is the strongest predictor for tumour recurrence in non-muscle invasive urothelial cancers of the bladder [72, 73]. However, no association with recurrent disease, distant metastasis or cause-specific death was demonstrated in another study on renal and ureteric urothelial cancers [74]. Collectively, this may suggest the presence of two distinct types of urothelial cancers classified according to genomic instability: i) MIN cancers that show relatively favourable prognosis and ii) CIN cancers with CA that display highly malignant behaviour.
Like in breast cancers, the association of CA with 20q13.2 copy number gain and Aurora-A kinase overexpression has been reported in urothelial carcinoma, specifically in those harbouring CIN and aneuploidy rather than MIN [72-74, 77]. Forced overexpression of Aurora-A in urothelial cells induced CA, chromosome missegregation and aneuploidy [77]. Bladder cancer cell lines harbouring Aurora-A and p53 overexpression display supernumerary centrosomes with an even number of chromosome copy number [78]. In clinical studies, CA in bladder TCC specimens strongly correlates with concomitant occurrence of cyclin E overexpression and p53 inactivation [76], as well as cyclin D1 gene amplification [75]. Other proteins like PLK-1 and BUBR1 have been associated with CA, CIN and aneuploidy in bladder cancer as well, but their significance remains to be explored [79, 80]. Overall, a cooperative mechanism for CA in bladder cancers is suggested through both disinhibition of centrosome duplication (as a result of cyclin E and/or cyclin D overactivity) and cytokinesis failure (as a result of p53 mutation and/or Aurora-A overexpression).
Renal and adrenal tumours
In renal cell carcinoma (RCC), centrosome abnormalities and mitotic multipolarity are present in the subgroup displaying complex karyotypes. Interestingly, telomeric dysfunction and anaphase bridges are concomitantly observed in this subgroup of cancers, indicating the presence of two possibly related mechanisms i.e. CA and telomeric dysfunction, in the generation of genetic instability of RCC [81]. There is also a possibility that CA is a result of cytokinetic failure from repeated chromosomal breakage-fusion-bridge cycles, due to mechanical blocking of cytokinesis by anaphase bridges. Overexpression of miR-210, a downstream target of HIF1α, has recently been reported to evoke CA and multipolar spindle formation in renal carcinoma cells [82]. CA has also been reported in adrenocortical adenomas and carcinoma [83].
Cervical cancer
The mechanisms underlying CA in high-risk human papillomavirus (HPV)-associated cervical carcinogenesis has been extensively studied over the past decade. Centrosome aberration has been demonstrated in cervical dysplasia and invasive cervical carcinoma, most of which are aneuploid and positive for high-risk HPV infection [10]. CA is also associated with spindle defects and correlates positively with increasing grade of dysplasia, and is highest in lesions of invasive carcinomas [84]. Furthermore, CA is detected in organotypic raft cultures of human keratinocytes containing episomal HPV-16 DNA, even in basal cells with low copy numbers of HPV-16 genome [85], supporting its involvement in tumour initiation and progression.
In high-risk HPV-associated cervical carcinoma, a collaborative effort by the HPV-encoded oncoproteins E6 and E7 have been implicated in the generation of supernumerary centrosomes and multipolar mitoses, predisposing to CIN and carcinogenesis. Whilst stable expression of E6 or E7 in primary human keratinocytes results in abnormal centrosome numbers [13], co-expression results in an even higher number of cells with supernumerary centrosomes [86]. In addition, although acute expression of E7 alone rapidly induces abnormal centrosome numbers, that of E6 alone has no such effect [13]. In a transgenic mouse model of estrogen-induced carcinogenesis, E7 alone is sufficient to evoke CA, producing both high-grade cervical dysplasia and invasive cervical malignancies. E6 alone elevates centrosome copy number but do not produce cancer in the mice. However, E6 plus E7 additionally elevate centrosome copy number and create large, extensively invasive cancers, supporting the cooperative mechanism [87].
E7 drives the generation of abnormal centrosome numbers by directly interfering with centrosome duplication control. E7 expression rapidly increases daughter centriole formation in otherwise normal diploid cells within a single cell division cycle prior to the development of extensive genomic instability [13, 88]. This rapid mode of centriole overduplication has been demonstrated to proceed through a pathway characterized by a single maternal centriole initiating the simultaneous synthesis of two or more daughter centrioles, necessitating CDK2/cyclin activity and PLK4 upregulation [89-91] as well as RNA polymerase II transcription [92]. The absolute requirement of CDK2 dysregulation is consistent with the known effects of E7 on pRB inactivation [93] and the resultant release of E2F transcription factors as well as upregulation of CDK2/cyclin E activity needed for S phase entry. Interestingly, although a E7 deletion mutant lacking pRB binding/degradation properties is unable to induce centriole overduplication [13], wild-type E7 is able to induce centriole abnormalities in pRB/p107/p130-deficient cells [94], implying that pRB degradation is not the only mechanism by which oncogenic E7 induces centriole overduplication. This pRB/p107/p130-independent mechanism has been suggested to be contributed by the association of E7 with gamma-tubulin [95].
On the other hand, unlike E7, E6 probably does not affect centrosome duplication, but instead allows for centrosome accumulation over a prolonged period of time in genomically-unstable cells. This centrosome accumulation occurs in parallel with nuclear atypia (e.g. multi-nucleation, micronuclei). Whilst many of these cells display features of replicative senescence which are unlikely to proliferate further [88], there remains the likelihood of some polypoidal cells escaping senescence and successfully completing mitosis, leading to the propagation of genomically-unstable progeny. Such abnormal cells would have avoided mitotic arrest or apoptosis since E6 interferes with p53-dependent checkpoints by triggering the proteasomal degradation of p53 [96]. The involvement of E6 in the early stages of cervical carcinogenesis is supported by studies demonstrating the presence of centrosome abnormalities in raft cultures harbouring genomes mutated in E7 expression [85]. Moreover, the HPV-16 E2 protein that is required for early viral replication inhibits E7 but not E6-triggered CA [97]. Intriguingly, E2 promotes G2/M arrest independent of E6 and E7, characterized by the accumulation of active CDK/cyclin B1 complexes and histone H3 phosphorylation, followed by metaphase-specific apoptosis. E2-expressing cells which spontaneously escape arrest and apoptosis display CA and CIN [98]. Taken together, it may be speculated that in the early stages of HPV-induced cervical carcinogenesis, centrosome accumulation triggered by E6 and/or E2 may be the predominant mechanisms for the generation of supernumerary centrosomes; in the later stages, it is possible that the loss of E2 may then permit E7 to take on a predominant role through centrosome overduplication, whilst E6 takes on a cooperative role by creating a permissive milieu for genomic disintegrity and centrosome accumulation, through the relaxation of p53 checkpoint control.
Interestingly, recent evidence suggests that genital infection by Chlamydia trachomatis may contribute to HPV-induced cervical cancer formation through its disruptive effects on centrosome homeostasis. Similar to high-risk HPV, chlamydial infection triggers the production of supernumerary centrosomes and chromosome segregation defects which persist even after cells are cured with antibiotics [99]. The underlying mechanism possibly involves abortive cytokinesis, but centriole overduplication through CDK2 and PLK4-dependent pathways appear to be predominant [100]. Additionally, CA is exacerbated by chlamydial infection via an increase in centrosome spread and inhibition of the spindle assembly checkpoint delay to disrupt centrosome clustering [101]. This latter phenomenon may interfere with centrosome clustering in HPV-infected cells, thereby facilitating multipolar mitosis and CIN.
Ovarian and testicular cancers
In ovarian adenocarcinoma, near-tetraploid tumours, which display greater numerical CIN, possess a higher degree of intratumoural CA; whereas near-diploid tumours show fewer abnormal centrosomes and comparatively lower numerical but higher degrees of structural CIN [102, 103]. This implicates upon two distinct but possibly inter-related mechanisms for the generation of chromosomal aberration in ovarian carcinoma - one via copy-number instability associated with mitotic segregation abnormalities, cytokinesis errors and CA, and the other through structural change possibly as a result of impaired DNA repair [103]. Of clinical relevance, the prognostic significance of CA in ovarian cancer is demonstrated by its association with higher stage, histological grade, and the more aggressive serous-type compared to endometroid-type tumours [102]. In testicular germ cell tumours, those with aneuploidy are frequently associated with CA. Aneuploid seminomas and non-seminomas (including carcinoma-in-situ and infantile yolk sac tumours) show increased numbers of centrosomes. In contrast, the occurrence of CA in diploid infantile teratomas is less frequent [104].
High Aurora-A kinase expression strongly associates with supernumerary centrosome count in primary ovarian tumour cells but not in testicular germ cell tumours [104], and is an independent predictor of decreased survival in ovarian cancer patients [105]. Interestingly, HeyA8-MDR cells are more aggressive and chemoresistant compared to their parental HeyA8 cells, and their Aurora-A expression and centrosome count are significantly higher as well [105]. In relation, Aurora-A overexpression in ovarian cancer cell lines promotes CA, malformation of mitotic spindles and chromosome aberration. At the same time, Aurora-A suppresses the expression of p21, pRb, and BRCA2 to advance cell cycle progression and to abolish cellular apoptosis and DNA damage response [106]. Ectopic expression of Aurora-A kinase renders ovarian cancer cells resistant to drug-evoked apoptosis by activating the AKT survival pathway in a p53-dependent manner [107]. Taken together, this may suggest that poor outcome in ovarian cancer patients with Aurora-A kinase overexpression and CA may be a result of concomitant chemoresistance.
Gastrointestinal cancer
Colorectal carcinoma
CA is detected in as early as low-grade dysplastic lesions of the colorectal adenoma-adenocarcinoma sequence [10, 108, 109], is more frequent in carcinoma compared to adenoma, and is associated with higher histological grade of both dysplastic as well as invasive lesions [109], supporting its roles in the initiation and progression of colorectal cancer (CRC) development. In CRC cell lines, the occurrence of CA lies exclusively in aneuploid but not diploid tumours, correlating with severe impairment of microtubule nucleation ability and chromosome segregation errors in the aneuploid cell lines [110]. These in vitro observations correspond with findings in primary human tissue that defects in centrosome organization, spindle structure, and chromosome segregation often occur within the same tumour cells, correlating with CIN and presence of aberrant nuclei [10].
In addition to the association with aneuploidy, a proportion of CRC cell lines with CA also contains p53 mutations or overexpression, gains or genomic amplification of 20q, as well as increased copies of 16p [110]. Ectopic overexpression of Aurora-A kinase in HCT116 cells leads to supernumerary centrosomes and aneuploidy regardless of p53 status [111]. In the same cell line, oncogenic beta-catenin is also required for S-phase arrest and centrosome overduplication [112], whilst insufficient PLK4 is associated with aberrant centrioles [113]. Loss of hSgo1, a protein regulating chromosome segregation that is downregulated in CRC, results in G2/M arrest and apoptosis, while promoting CIN and CA, cytokinesis defects and mitotic catastrophe [114]. Telomerase transcriptional elements-interacting factor (TEIF), a centrosome-associated protein and transactivator of human telomerase reverse transcriptase subunit (hTERT), is overexpressed in colorectal adenoma and CRC compared to normal tissue, and positively correlates with CA and tumour grade [109], suggesting that telomere dysfunction may be responsible for generation of CA in CRC. At the same time, it is also possible that both telomere dysfunction and CA contribute separately to genomic instability in CRC.
Esophageal cancer
In cases of chronic esophagitis followed by Barrett's metaplasia-derived adenocarcinoma, CA is frequently observed in both Barrett's metaplasia and adjacent histopathologically-normal native squamous epithelium. Interestingly, centrosome aberrancies are less frequent in frank adenocarcinoma compared to Barrett's metaplasia, perhaps suggesting its relative importance in earlier stages of carcinogenesis [115].
Hepatobiliary cancer
CA may play a role in the initiation and progression of biliary cancers. Biliary tumours including gallbladder cancers (GC), intrahepatic cholangiocellular carcinomas (CCC), and extrahepatic bile duct cancers (BDC) display CA. CA also occurs in chronic cholecystitis and hepatolithiasis, both potentially neoplastic conditions. In addition, advanced stage cancers possess a higher frequency and greater levels of CA than in the early stages [116].
In hepatocellular carcinomas (HCC), intratumoural CA is significantly higher in non-diploid tumours than in diploid tumours. CA is associated with p53 mutation but is not related to tumour stage, size or proliferative activity [117]. The upstream mechanisms leading to CA in HCC have been investigated in in vitro models of hepatitis B virus (HBV)-associated hepatocarcinogenesis. Specifically, the HBV-encoded hepatitis B virus X (HBX) oncoprotein promotes centrosome overduplication and cytokinesis defects through its interaction with cellular protein HBXIP [118, 119]. Additionally, HBX evokes centrosome overduplication, multipolar spindles and aneuploidy through cytoplasmic sequestration and inactivation of Crm1, a Ran-GTP binding nuclear export receptor [120], as well as by activating the Ras-MEK-MAP kinase pathway [121].
Pancreatic cancer
CA is detected in primary pancreatic exocrine tumours, including both ductal carcinomas and adenomas [122], as well as in pancreatic cancer cell lines, particularly those with multipolar spindles, defective mitoses and CIN [123]. Interestingly, although CA is prevalent in exocrine tumours, endocrine malignancies show normal centrosome patterns and thus may arise through different genomic instability pathways [122]. Using orthotopic transplantation of Suit-2 human pancreatic carcinoma cells into nude mice as a model, supernumerary centrosome numbers are found at higher frequencies in metastatic foci than in pancreatic xenografts, correlating with multipolar mitotic spindles and enhanced degree of CIN [124]. Aurora-A kinase is amplified in pancreatic cancer cell lines and primary pancreatic cancer [125], and is associated with CA, giant nuclei formation and CIN in vitro [126]. Taken together, this supports cytokinesis failure as a major mechanism underlying CA in pancreatic cancer. Cytokinesis failure and the tendency of p53-proficient binuclear and tetraploid cells to evade the tetraploidy checkpoint are commonly observed in an acinar-ductal transdifferentiating culture model of pancreatic carcinogenesis, predisposing to pleiotropic mitotic defects. In the context of p53 deficiency, uncontrolled polyploid progression ensues due to evasion of the tetraploidy checkpoint during cytokinesis failure, leading to a rapid exacerbation of CA and aneuplody [127].
Lung cancer
CA has been demonstrated in a proportion of lung cancers, including squamous cell carcinoma and adenocarcinoma [10, 128, 129], and correlates with aneuploidy. However, there is no significant relationship with clinical characteristics such as tumour size, disease stage, or patient survival [129]. In human lung cancer tissue, CA positively correlates with loss of pRb expression as well as with E2F1 and cyclin E overexpression [128, 129]. At least some cells with CA ars also positive for p53 mutation, although correlative significance is absent [128-130]. The possible association of p53 loss with centrosome anomalies is supported by studies on p53-knockout murine bronchiolar epithelial “Clara” cells, presumably the origin of lung adenocarcinoma, which possess increased centrosome number and multinucleation, as compared to their wild-type p53 counterparts [131]. In addition, several studies have demonstrated a relationship between exposure to carcinogens implicated in lung cancer and the development of centrosome abnormalities in vitro. Specifically, exposure to chrysotile asbestos fibres [132], chromate particles [133, 134], arsenite [135, 136] and benzo[a]pyrene diol epoxide [137] have been shown to induce CA, multipolar spindles, aneuploidy, as well as malignant transformation in human lung cell lines, which may be conditional on p53 dysfunction. Expression of Skp2 in lung cancer cells is associated with CA as well [138]. Recently, NORE1A, a gene commonly downregulated in non-small cell lung cancer, has been shown to suppress CA induced by hydroxyurea in lung cancer cell lines, implying a preventive role of NORE1A against carcinogenesis of non-small cell lung cancer [139].
Neural cancer
CA has been demonstrated in several neural cancers [10], including aneuploid cerebral primitive neuroectodermal tumours (PNET) with p53 mutation [140], diffuse astrocytic gliomas of various histological grades [141], pituitary adenomas (PA) [142], as well as in medulloblastoma [143]. Gamma-tubulin staining is greater in adult high grade anaplastic astrocytomas and glioblastomas as compared to low-grade diffuse astrocytomas [141]. Similarly, supernumerary centrosomes as well as higher Aurora-A mRNA expression levels are observed in high grade but not low grade glioma [144]. Comparing with normal brain tissue, mRNA expression for centrosomal structural proteins, such as centrin 3, gamma-tubulin, hNinein isoforms 1/2/5/6, Aurora-A and Aurora-B are elevated in glioma tissue [144]. Survivin suppression leads to CA in glioma cell lines, especially in the absence of p53 [145]. Loss of PTEN and expression of EGFRvIII transform neural precursor cells into tumours resembling glioblastomas, displaying CA, Aurora-A/B upregulation, and activation of Ras/Erk/AKT pathways [146]. Interestingly, variously pronounced gamma-tubulin localization is present in the cytoplasm of vascular endothelial cells in areas of tumour angiogenesis (microvascular proliferation) in glioblastomas [141]. This indicates that CA may underlie neoplastic neovascularization in glioblastomas. In support of this notion, endothelial cells in solid tumours are aneuploid and are curiously associated with multiple centrosomes as well [147].
In PA, CA is observed in somatotroph and lactotroph adenomas, which are characterized by aneuploidy and securin overexpression, in comparison to non-functioning adenomas or normal pituitary tissue [142]. In a mouse study, forced transgenic expression of cyclin E in the pituitary intermediate lobe results in CA [148]. In medulloblastoma, overexpression of gamma-tubulin is widespread in poorly differentiated, proliferating tumour cells but is significantly diminished in quiescent differentiating tumour cells undergoing neuritogenesis [143]. In primary neuroblastoma tumours, childhood and infant diploid tumours display greater CA than infant triploid tumours. Ploidy divergence accompanying tetraploid cells, implying cytokinesis failure, occurs very frequently in infant diploid but not infant triploid tumours. However, although CA is found in the majority of childhood diploid tumours, none of these show ploidy divergence, suggesting centrosome overduplication as the mechanism of CA in this group of tumours [149]. CA may be associated with MYCN locus amplification in primary neuroblastoma [149, 150]. This is supported by in vitro observations, whereby in neuroblastoma cell lines, MYCN overexpression induces CA in response to a DNA damage stimulus such as ionizing radiation [150, 151]. Suppression of p27 expression through increased expression of Skp2 [152], as well as MDM2-mediated negative regulation of p53 activity [150] may underlie this DNA damage-evoked CA.
Head and neck cancer
CA has been observed in squamous cell carcinomas of the head and neck (HNSCC) [153-159], benign salivary gland pleomorphic adenomas [155], as well as in thymic carcinoid tumour [160]. Strong prognostic implications have been demonstrated in HNSCC. High intratumoural CA is associated with local tumour recurrence of surgically-resected oral SCC despite histologically-negative margins. Interestingly, analysis of the tumour margins for CA found a trend toward local tumour recurrence in patients whose margins showed high degree of CA [154]. CA is also observed in oral epithelial dysplasia. The percentage of cells containing CA is more frequent in oral SCC than dysplasia, as well as in poorly-differentiated oral SCC relative to moderate- and well-differentiated subtypes [158]. Collectively, this strongly implicates upon CA as a major mechanism driving the initiation and progression HNSCC. In laryngeal SCC, presence of cells with large centrosomes is a better predictor of tumour recurrence compared to T stage, and even predicts recurrence in node-negative tumours. Moreover, this group of patients shows a near-significant trend for shorter survival and thus poor prognosis [159]. In another study, CA is associated with tumour size, stage, distant metastasis, disease-free survival and overall survival [157].
There is evidence to support cytokinesis failure as the major mechanism underlying CA in HNSCC, which may involve Aurora-A kinase upregulation [156], MDM2-p53 dysregulation [153], and decreased myosin light chain phosphorylation [161]. Aurora-A kinase mRNA and protein upregulation are frequently occurring in HNSCC and are associated with CA and poor patient prognosis [156]. The underlying mechanism linking Aurora-A kinase overexpression and CA remains unclear, but its interaction with important cell cycle regulators such as p53 may be contributory. Notably, Aurora-A kinase is known to phosphorylate and promote the MDM2-mediated degradation of p53 [56]. In earlier studies, CA have been correlated with the occurrence of either mutation/deletion of p53. Interestingly, in tumours that retained wild-type p53, CA is associated with MDM2 overexpression instead [153]. In corroboration with these findings, Swiss 3T3 cells overexpressing stably-transfected MDM2 show extensive CA and CIN, despite retaining wild-type p53 [153]. The actual relationship between Aurora-A kinase and the MDM2-p53 pathway in generating CA in HNSCC remains to be further elucidated.
Dysfunctional telomeres have been suggested to play a possible role in the generation of CA in HNSCC. HNSCC and benign salivary gland pleomorphic adenomas displaying abnormal karyotypes showed widespread centrosome aberration and multipolar division, together with anaphase bridges. The frequency of anaphase bridges correlates with number of chromosome ends lacking TTAGGG signals, indicating the presence of telomere shortening. This repeated chromosomal breakage-fusion-bridge cycles has been proposed to prevent normal cytokinesis, leading to centrosome accumulation along with multinucleation, multipolar cell division, and aneuploidy [155]. These cells however, may be at an evolutionary dead-end with limited contribution towards tumour development, since mitoses with five or more poles are very rarely observed [155].
Bone and soft tissue tumour
Bone and soft tissue sarcoma
CA has been detected in various types of malignant and borderline malignant sarcomas, including osteosarcoma, chondrosarcoma, malignant fibrous histiocytoma (MFH), haemangiopericytoma, atypical lipomatous tumour, liposarcoma, giant cell tumour, peripheral PNET, synovial sarcoma, rhabdomyosarcoma, leiomyosarcoma as well as malignant peripheral nerve sheath tumour (MPNST) [162-167]. In several types of sarcomas, aneuploid karyotypes are strongly associated with supernumerary centrosomes and multipolar mitoses, as well as anaphase bridges. Furthermore, supernumerary centrosomes and multipolar mitoses are positively correlated with anaphase bridges, supporting both types of abnormal chromosome segregation in the evolution of CIN in these soft tissue sarcomas [164]. A small study of six well-differentiated liposarcomas of both near-diploid (Type D) and near-tetraploid (Type H) cytogenetic subtypes display CA, independent of their ploidy status, although Type H tumours demonstrate higher intratumoural degrees of centrosome number short of statistically significance [165].
Numerical CA is more frequently observed in highly aggressive types of sarcomas, including PNET, synovial sarcoma, rhabdomyosarcoma, MFH, as compared to moderate grade sarcomas such as liposarcoma and leiomyosarcoma [166]. The degree of CA is also significantly higher in recurrent and malignant cases than in non-recurrent cases of giant cell tumour of bone [167], in which a positive correlation exists between aneuploidy and CA. Interestingly, benign or tumour-like lesions of soft tissue may also feature CA, albeit at lower frequencies [164-166].
Analysis of p53-mutant osteosarcoma cell lines reveals a wide range of aneusomy, high levels of atypical mitotic figures, and high frequencies of abnormal centrosome numbers, as compared to p53 wild-type cell lines [163]. Furthermore, treatment with colcemid, a spindle-disrupting polyploidizing agent, induces multipolar cell division and supernumerary centrosomes in an MDM2-amplified osteosarcoma cell line but not in normal fibroblasts [164]. This may implicate a defective p53 pathway in the generation of centrosome aberration and CIN in osteosarcoma.
Similar to earlier reports in colorectal adenoma and CRC [109], TEIF positively correlates with CA in soft tissue sarcomas, and is related to tumour grade [166]. In addition, in vitro overexpression of TEIF levels either exogenously introduced, or evoked by DNA-damaging agents or telomeric dysfunction promotes multinucleation, CA, mitotic defects, and chromosome missegregation [166]. Collectively, this suggests that DNA damage evokes TEIF centrosomal expression, resulting in CA and genomic instability that contributes to tumour progression.
Kaposi sarcoma
The Kaposi sarcoma-associated herpes virus (KSHV) has been linked with several human malignancies, including all subtypes of Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Kaposi sarcoma lesions are latently infected by KSHV and express latent genes, such as KSHV cyclin (K cyclin) and latency associated nuclear antigen (LANA). Expression of K cyclin in human endothelial cells induces senescence and strong DNA damage response, leading to CA and S-phase arrest through a CDK6 and ATM-Chk2-dependent pathway [168]. In human and mouse primary fibroblasts, K cyclin expression results in a p53-dependent growth arrest, abortive cytokinesis and centrosome accumulation [169]. In the absence of p53, the K cyclin-induced CA is exacerbated, leading to the acquisition of an aneuploid population of cells which is not only genetically unstable but also resistant to apoptosis [169]. In line with these findings, in vivo studies demonstrated accelerated K cyclin-induced lymphomagenesis in mice without p53 [170]. Notably, KSHV infection of endothelial cells generates supernumerary centrosomes and multinucleation [168, 171], suggesting that the hindrance of tumour progression due to p53-triggered growth arrest and apoptosis may be overcome by other latent viral genes. Indeed, LANA is able to bind p53 and inhibits the ability of p53 to transactivate cellular genes, resulting in abnormal centrosomes, multinuclear cells, and other genomic abnormalities [172]. In KSHV-infected cells, NPM1 is a phosphorylation substrate for CDK6/K cyclin necessary for centrosome duplication, and p53-driven apoptosis occurring downstream of NPM1 phosphorylation is a critical checkpoint mechanism that prevents accumulation of cells with supernumerary centrosomes, which may be overcome by LANA co-expression [173].
Centrosome amplification in haematological malignancies
In addition to solid neoplasms, centrosome aberrations are common in several haematological malignancies including Hodgkin's (HL) as well as non-Hodgkin's lymphomas (NHL), acute (AML) and chronic myeloid leukaemias (CML), HTLV-1-associated adult T-cell leukaemia (ATL) and multiple myeloma (MM). In analogy to solid tumours, a correlation between CA and CIN as well as clinical aggressiveness also exists in certain haematological malignancies [174].
Non-Hodgkin's and Hodgkin's lymphoma
Centrosome defects, predominantly structural in nature, are frequently detected in several subtypes of B-cell lineage NHL including diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), Burkitt's lymphoma (BL), follicular lymphoma (FL), marginal zone B-cell lymphomas (MZBCL), as well as chronic lymphocytic leukaemia (CLL) [175, 176]. Similar abnormalities occur in Hodgkin and Reed-Sternberg (HRS) cells in classic HL [177, 178].
Aggressive lymphomas like DLBCL, MCL and BL harbour more centrosome abnormalities than indolent lymphomas including FL and MZBCL. In accordance, the degree of CA is associated with the proliferation/mitotic index, irrespective of histological subgroup (FL, DLBCL and MCL). In FL, the number of cells with CA correlates with increased histological grading; whilst in MCL, CA occurs at a higher frequency in blastoid variants harbouring near-tetraploid chromosome numbers as compared to the less aggressive diploid subtypes [175]. In contrast, the latter correlation is not seen in FL and DLBCL. The significance of the centrosome in MCL subtyping and prognostication is further supported by a gene expression signature inclusive of centrosome proteins CAMKK2, PCNT2, TUBGCP3 and TUBGCP4, which discriminated between near-tetraploid and diploid MCL [179]. Centrosome abnormalities are observed in B cell CLL and even at the premalignant monoclonal B-cell lymphocytosis (MBL) stage. The extent of CA correlates with the clinical aggressiveness and proliferative activity of the CLL cases as measured by lymphocyte doubling time as well as with time to first treatment [176]. However, there is no correlation with IgVH gene mutation status or cytogenetically-defined risk groups (11q22-23, 17p13 and 13q14 deletions; trisomy 12). This is corroborated with the finding that no difference exists between CLL subgroups with or without the same specific chromosome aberrations with prognostic significance [180].
Apart from B-cell neoplasms, numerical and structural centrosome aberrations are present in ALK-positive anaplastic large cell lymphoma (ALCL) as well, a T-cell derived neoplasm characterized by t(2;5) rearrangements, resulting in the creation of fusion genes involving the ALK locus [181]. No correlation between CA and p53 status is apparent in ALCL. Despite the prevalence of CA, ALK-positive ALCL is known to be relatively chromosomally stable, with only few secondary abnormalities on top of the primary t(2;5) or variant translocations. On a similar note, in a detailed analysis of a single case of BL with extensive CA, there was an absence of ongoing numerical CIN, supporting the notion that CA is not a universal surrogate marker CIN [182].
Chronic myeloid leukaemia
In CML patients, the presence of CA in CD34+ BCR-ABL1-positive cells is an early event in CML development, and precedes CIN that is associated with advanced disease. Cells from chronic phase (CP) specimens display mostly numerical centrosome abnormalities with a uniform morphology, in contrast to those from blast crisis (BC) specimens showing irregularly-shaped amorphous amplification (so-called "centriolar satellite material") on top of a high rate of numerical alteration [183, 184]. Cells with CA are more evident in BC in comparison to CP specimens while the extent of CA strongly correlate with karyotype instability and aneuploidy, giving support to a prognostic value of centrosome status in CML [183].
CML is characterized by the BCR-ABL1 translocation, which produces the p210BCR-ABL1 fusion protein that localizes to the centrosome in K562 cell lines. Cell lines expressing p210BCR-ABL1 exhibit more CA than p210BCR-ABL1 negative cells [184], in keeping with earlier observations that BCR-ABL1 transgene expression induces CA in cell lines in a time-dependent manner [185]. Interestingly, tyrosine kinase inhibitors including imatinib, nilotinib and dasatinib lead to de novo induction of CA, lending a possible explanation to the emergence of BCR-ABL1-negative clones with aberrant karyotypes and secondary malignancies (e.g. AML and myelodysplastic syndrome) with prolonged treatment using these drugs [184-188]. CA has also been detected in disease-unrelated cells/tissues in patients treated with tyrosine-kinase inhibitors bosutinib, dasatinib, imatinib, nilotinib, sorafenib and sunititib [189]. These findings though, remain more of a scientific enigma. It cannot be excluded that these genomically-unstable cells would be incapable of further proliferation, and thus do not pose any significant danger towards neoplastic development.
Acute myeloid leukaemia
Numerical and structural CA has been described as a potential cause of aneuploidy in AML. AML blasts with high degree of numerical and structural CA contain numerical CIN, and possess higher expression levels of genes coding for proteins involved in stimulating G1/S transition, DNA replication, as well as centrosome-associated proteins such as pericentrin and TUBGCP2. Furthermore, higher frequency of CA is associated with cytogenetically-defined adverse risk groups, as defined by the MRC AML 10 trial as favourable (22.5%), intermediate (35.3%), and adverse (50.3%). Interestingly, this is mainly attributed to structural rather than numerical centrosome aberrations [190, 191]. Numerical centrosome abnormalities are observed in a study of 25 patients with hematopoietic bone marrow failure disorders with a risk for evolution into AML, including myelodysplastic syndrome, aplastic anaemia, and paroxysmal nocturnal haemoglobinuria. Increased centrosome copy number positively correlates with aneuploidy, and samples with the highest percentage of CA tends to be aneuploid for the most chromosomes. This suggests that CA may be associated with the development of a clonal population of potentially pre-leukemic aneuploid hematopoietic progenitor cells in bone marrow failure patients [192].
Adult T-cell leukaemia
CA is also observed in ATL, a condition causally linked to human T lymphotropic virus-1 (HTLV-1) infection [193, 194]. Tax, a HTLV-1 transactivator protein, localizes to the centrosome, deregulates CDK activity and induces CA in JPX-9 cell lines, suggesting an association with cellular transformation by HTLV-1 and CIN in HTLV-1-infected human T cells [194]. Tax may be directly responsible for genomic instability and aneuploidy in ATL cells through the interaction with Ran-BP1 and coiled-coil protein TaxIBP2. The interaction of Tax with Ran-GTPase pathway via Ran-BP1 regulates centrosome stability and is necessary for Tax localization to centrosomes as well as its induction of CA. Disruption of TaxIBP2-Tax interaction results in failure of CA triggering by Tax, while TaxIBP2 downregulation leads to CA, suggesting that TaxIBP2 is an intrinsic block to centrosome overduplication [193, 195, 196].
Multiple myeloma
CA is common in MM and is already present in monoclonal gammopathy of undetermined significance (MGUS), suggesting an early role in myelomagenesis. The percentage of plasma cells with CA increases progressively from MGUS to smouldering MM to MM, implicating that CA contributes towards increasing genomic instability and disease progression [12, 197]. Structural centrosome abnormalities are mainly seen in MM rather than MGUS. Interestingly, no difference in CA exists between ploidy categories in myeloma, suggesting that CA contributes to genomic instability in both tumour subgroups. Expanding upon these observations, CA is also evident in B-cells of MM patients, supporting the existence of an abnormal clonal population of B-cells which may be precursors of malignant plasma cells [198].
Chng et al. demonstrated that a gene expression-derived "centrosome index" (CI) composed of the expression of genes encoding for major centrosome components - centrin, gamma-tubulin, and pericentrin, is a strong surrogate of CA in MM. Tumours with high CI overexpressed genes coding for proteins involved in cell cycle, proliferation, DNA damage, and G2/M checkpoints, as well as those associated with the centrosome and kinetochore or microtubules. In MM patients, a high CI is associated with poor prognostic features, including chromosome 13 deletion, t(4;14), t(14;16) and high plasma cell labelling index. Correspondingly, a high CI is a powerful independent poor prognostic factor regardless of disease phase (newly diagnosed or relapsed), therapeutic modality, or disease stage [12, 199, 200].
Some of the plausible mechanisms underlying CA in multiple myeloma have been discussed recently [200]. Of particular significance, elevated expression of RHAMM correlates with centrosome structural abnormalities in multiple myeloma; whilst introduction of exogenous RHAMM, which localizes to the centrosome, triggers an increase in centrosome size as well as gamma-tubulin levels, and induces aberrant mitoses [197]. In addition, Aurora-A kinase has been shown to be significantly overexpressed in multiple myeloma with a high centrosome index [199].
Diagnostic, predictive and prognostic implications
The diagnostic utility of centrosome abnormalities has been repeatedly discussed in previous studies, yet its usefulness as a biomarker for cancer detection has never been thoroughly investigated. Based on earlier observations, CA in normal or benign tissue is a rare phenomenon. In most studies, no CA could be detected in normal human cells including those of the breast, prostate, lung, brain, colon [10], bladder [76], kidney [81], cervix [84], testis [104], ovary [105], liver [117], pancreas [122], head and neck [157], and blood [183] in contrast to their malignant counterparts. However, in studies on benign breast lesions, centrosome abnormalities were not found in mastopathia or fibroadenoma [29], while another study observed structural centrosomal abnormalities occasionally in the benign lesions studied (3 of 25, including mastopathia, intraductal papilloma and fibrocystic mastopathy) [35]. Apart from those of the breast, non-malignant tumours from other sites such as soft tissue [81, 165, 166] and pancreatic adenoma [122] have been shown to harbour CA, suggesting that the presence of CA alone is not a sufficient criterion to diagnose malignancy. In addition, although CA is frequently present to some extent in most cancers, it is not an invariable phenomenon [Table 1]. Thus, the lack of CA may not necessarily exclude malignancy either. Nonetheless, the utility of centrosome status as a differential diagnostic marker may be augmented when employed in adjunct to contemporary methods. CA may also be of clinical utility where tissue availability is limited, as demonstrated in cytological aspirates of breast lesions [29, 35].
In HNSCC, those with high degree of CA in tumour margins recur more frequently than those with less CA. This finding is interesting, given that these margins are histologically negative for malignant cells, the latter of which clinically implies adequate surgical resection [154]. Given such observations, it is tempting to speculate that malignant, or at least potentially malignant cells harbouring CA within these margins that escape histological detection, may be responsible for eventual tumour recurrence and poor prognosis. In parallel, CA occurs in matched adjacent normal or hyperplastic regions of NSCLC as well [128, 129], although their clinical significance remains unknown. In such cases, CA in the tumours and/or their margins may define a subgroup of patients who may benefit from more extensive curative surgery, more aggressive adjuvant treatment, and closer monitoring for disease recurrence.
Finally, can CA be a surrogate for aggressive tumour phenotype? Phenotypic heterogeneity generated as a result of CIN and aneuploidy allows for selection of a superior karyotypic clone that may confer survival advantage through a Darwinian selection process, which in turn promotes tumour aggressiveness, progression, metastasis, and chemoresistance [Figure 1]. As summarized in Table 3, CA is not only characteristic of tumours in general, but is also more pronounced in advanced stage and tumour grade, as well as correlates with early disease recurrence, disease progression and poor survival in many cases. Together with other established prognostic factors, CA may prove to be helpful in predicting outcomes and survival of patients with cancer.
The centrosome as a cancer therapeutic target
Potentially, the centrosome can be a therapeutic target for the treatment of cancers. The selective killing of cells with CA has been investigated via targeting specific altered proteins/pathways peculiar to these cells. Centrosome duplication in normal non-neoplastic cells differs mechanistically from pathways leading to CA in cancer cells, and these differences may be exploited to selectively kill target cancer cells while leaving normal cells relatively unaffected [89-91]. Of particular significance, small molecule centrosome-associated protein kinase inhibitors, such as Aurora-A kinase and Polo-like kinase inhibitors have recently shown promising results in clinical trials [201]. Also, AhR agonists such as indirubins reduce centriole overduplication and may imply a novel role for chemoprevention in breast cancer [65]. Likewise, targeting the compensatory mechanisms of bipolar spindle formation, such as inhibition of centrosome clustering, may be a potential mode of promoting cancer cell death [21]. Conversely, it may also be plausible that agents that promote centrosome clustering may delay tumour progression by limiting CIN.
Another attractive approach to discriminatory cancer cell eradication invokes exploiting the possible additional burden of excess centrosomes on the cell cycle [202]. In these cells with CA, the threshold to apoptosis induced by drugs [203] or radiation [145] may be much lower. For example, the partial inhibition of PARP1 in BRCA1-deficient cells has been suggested to represent a possible chemopreventive or therapeutic approach for BRCA1-deficient breast cancers, via its induction of severe chromosome aberrations, CA, telomere dysfunction and apoptosis [204]. Survivin inhibition in glioma cells has also been shown to enhance CIN and radiosensitivity via induction of CA [145]. The induction of CA using agents that delay S-phase but at insufficient doses to trigger apoptosis, such as low dose 5-FU, has been suggested as a novel mode of chemosensitization approach to cancer therapy [203]. However in all these approaches, one should be mindful of evoking CA in normal cells and risk of secondary carcinogenesis as a consequence of amplified genomic instability. In addition, cancer cells which evade apoptosis despite being induced with a greater degree of centrosome aberration and genomic instability may eventually develop an even more aggressive phenotype. These theoretical concerns warrant further investigations before this therapeutic approach may be considered for clinical utility.
Conclusion and future perspectives
CA is a common phenomenon in various human malignancies and may play a dominant role in tumour initiation and progression. Given the present evidence, it is most likely that CA represents, but not necessarily always, one of the major mechanisms underlying CIN and aneuploidy in cancer cells. Although the list of clinical studies on centrosomes and human cancers is growing rapidly, methodological issues place upon a significant limitation on data interpretation, as many studies use only super-numeration as the sole marker for CA rather than considering structural and functional aberrations concurrently. Although these types of aberrations often occur together, their biological and clinical consequences may be different. In addition, studies based on single section histological specimens will discount centrosomes unseen at that particular cut, therefore underestimating the true incidence of CA, as well as their association with concurrent molecular events. Future studies should take on a holistic approach to understanding the contribution of numerical, structural as well as functional centrosome abnormalities to specific human cancers throughout the course of tumour development, with particular respect to the generation of CIN and aneuploidy. Finally, a cell-to-cell based examination for specific molecular alterations associated with centrosome defects may reveal unique mechanistic correlations that would have been missed on simple linear correlative analysis. A better delineation of the detailed molecular circuitry regulating centrosome biology will be indispensable for a more intricate manipulation of these pathways for clinical application.
Review criteria
Information for this Review was compiled by searching the PubMed database for articles published until June 2011. Only articles published in English were considered. Search terms included “centrosome” and "cancer", in association with the terms "breast", "prostate", "bladder", kidney", "cervix", "ovary", “gastric”, “colorectal”, hepatobiliary”, “prostate”, “bladder”, “lung”, ”ovary”, “testicular”, "colorectal", "hepatobiliary", "pancreas", "lung", “cerebral”, "head and neck", "sarcoma", "lymphoma", "leukaemia", "myeloma", "carcinogenesis", “prognosis”, “progression”, “survival”, “biomarker”, and “chemotherapy”. Full articles were retrieved, and further information was obtained from relevant references. The focus was on relevant primary literature rather than review papers to compile this Review. A specific emphasis was placed on literature focusing on the frequency of CA, as well as its clinical relevance in solid and haematological cancers in human. Relevant papers on cell line or animal models were also discussed if deemed related to current evidence from clinical studies.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623-627
2. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285-295
3. Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121:1-84
4. Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911-924
5. Kirschner M, Mitchison T. Beyond self-assembly: from microtubules to morphogenesis. Cell. 1986;45:329-342
6. Hinchcliffe EH, Miller FJ, Cham M. et al. Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science. 2001;291:1547-1550
7. Doxsey SJ, Stein P, Evans L. et al. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell. 1994;76:639-650
8. Salisbury JL. Centrosomes: Sfi1p and centrin unravel a structural riddle. Curr Biol. 2004;14:R27-29
9. Kellogg DR. Centrosomes. Organizing cytoplasmic events. Nature. 1989;340:99-100
10. Pihan GA, Purohit A, Wallace J. et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58:3974-3985
11. Giehl M, Fabarius A, Frank O. et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19:1192-1197
12. Chng WJ, Ahmann GJ, Henderson K. et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood. 2006;107:3669-3675
13. Duensing S, Lee LY, Duensing A. et al. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002-10007
14. Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21:483-492
15. Shekhar MP, Lyakhovich A, Visscher DW. et al. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002;62:2115-2124
16. Duelli DM, Hearn S, Myers MP. et al. A primate virus generates transformed human cells by fusion. J Cell Biol. 2005;171:493-503
17. Khodjakov A, Rieder CL, Sluder G. et al. De novo formation of centrosomes in vertebrate cells arrested during S phase. J Cell Biol. 2002;158:1171-1181
18. La Terra S, English CN, Hergert P. et al. The de novo centriole assembly pathway in HeLa cells: cell cycle progression and centriole assembly/maturation. J Cell Biol. 2005;168:713-722
19. Mikule K, Delaval B, Kaldis P. et al. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol. 2007;9:160-170
20. Difilippantonio MJ, Ghadimi BM, Howard T. et al. Nucleation capacity and presence of centrioles define a distinct category of centrosome abnormalities that induces multipolar mitoses in cancer cells. Environ Mol Mutagen. 2009;50:672-696
21. Quintyne NJ, Reing JE, Hoffelder DR. et al. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127-129
22. Basto R, Brunk K, Vinadogrova T. et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032-1042
23. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278-282
24. Duensing S, Duensing A, Crum CP. et al. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356-2360
25. Lingle WL, Lutz WH, Ingle JN. et al. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U S A. 1998;95:2950-2955
26. Lingle WL, Salisbury JL. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am J Pathol. 1999;155:1941-1951
27. Lingle WL, Barrett SL, Negron VC. et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002;99:1978-1983
28. Pihan GA, Wallace J, Zhou Y. et al. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003;63:1398-1404
29. Kronenwett U, Huwendiek S, Castro J. et al. Characterisation of breast fine-needle aspiration biopsies by centrosome aberrations and genomic instability. Br J Cancer. 2005;92:389-395
30. Liu T, Niu Y, Yu Y. et al. Increased gamma-tubulin expression and P16INK4A promoter methylation occur together in preinvasive lesions and carcinomas of the breast. Ann Oncol. 2009;20:441-448
31. Niu Y, Liu T, Tse GM. et al. Increased expression of centrosomal alpha, gamma-tubulin in atypical ductal hyperplasia and carcinoma of the breast. Cancer Sci. 2009;100:580-587
32. Kronenwett U, Huwendiek S, Ostring C. et al. Improved grading of breast adenocarcinomas based on genomic instability. Cancer Res. 2004;64:904-909
33. D'Assoro AB, Barrett SL, Folk C. et al. Amplified centrosomes in breast cancer: a potential indicator of tumor aggressiveness. Breast Cancer Res Treat. 2002;75:25-34
34. Schneeweiss A, Sinn HP, Ehemann V. et al. Centrosomal aberrations in primary invasive breast cancer are associated with nodal status and hormone receptor expression. Int J Cancer. 2003;107:346-352
35. Guo HQ, Gao M, Ma J. et al. Analysis of the cellular centrosome in fine-needle aspirations of the breast. Breast Cancer Res. 2007;9:R48
36. Montagna C, Andrechek ER, Padilla-Nash H. et al. Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene. 2002;21:890-898
37. Shimomura A, Miyoshi Y, Taguchi T. et al. Association of loss of BRCA1 expression with centrosome aberration in human breast cancer. J Cancer Res Clin Oncol. 2009;135:421-430
38. Salisbury JL, D'Assoro AB, Lingle WL. Centrosome amplification and the origin of chromosomal instability in breast cancer. J Mammary Gland Biol Neoplasia. 2004;9:275-283
39. D'Assoro AB, Busby R, Suino K. et al. Genotoxic stress leads to centrosome amplification in breast cancer cell lines that have an inactive G1/S cell cycle checkpoint. Oncogene. 2004;23:4068-4075
40. D'Assoro AB, Busby R, Acu ID. et al. Impaired p53 function leads to centrosome amplification, acquired ERalpha phenotypic heterogeneity and distant metastases in breast cancer MCF-7 xenografts. Oncogene. 2008;27:3901-3911
41. Bagheri-Yarmand R, Biernacka A, Hunt KK. et al. Low molecular weight cyclin E overexpression shortens mitosis, leading to chromosome missegregation and centrosome amplification. Cancer Res. 2010;70:5074-5084
42. Bagheri-Yarmand R, Nanos-Webb A, Biernacka A. et al. Cyclin E deregulation impairs mitotic progression through premature activation of Cdc25C. Cancer Res. 2010;70:5085-5095
43. Starita LM, Machida Y, Sankaran S. et al. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004;24:8457-8466
44. Schlegel BP, Starita LM, Parvin JD. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene. 2003;22:983-991
45. Ko MJ, Murata K, Hwang DS. et al. Inhibition of BRCA1 in breast cell lines causes the centrosome duplication cycle to be disconnected from the cell cycle. Oncogene. 2006;25:298-303
46. Sankaran S, Starita LM, Groen AC. et al. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol Cell Biol. 2005;25:8656-8668
47. Xu X, Weaver Z, Linke SP. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389-395
48. Deng CX. Tumorigenesis as a consequence of genetic instability in Brca1 mutant mice. Mutat Res. 2001;477:183-189
49. Shao S, Liu R, Wang Y. et al. Centrosomal Nlp is an oncogenic protein that is gene-amplified in human tumors and causes spontaneous tumorigenesis in transgenic mice. J Clin Invest. 2010;120:498-507
50. Wang HF, Takenaka K, Nakanishi A. et al. BRCA2 and nucleophosmin coregulate centrosome amplification and form a complex with the Rho effector kinase ROCK2. Cancer Res. 2011;71:68-77
51. Zhou H, Kuang J, Zhong L. et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189-193
52. Hoque A, Carter J, Xia W. et al. Loss of aurora A/STK15/BTAK overexpression correlates with transition of in situ to invasive ductal carcinoma of the breast. Cancer Epidemiol Biomarkers Prev. 2003;12:1518-1522
53. Goepfert TM, Adigun YE, Zhong L. et al. Centrosome amplification and overexpression of aurora A are early events in rat mammary carcinogenesis. Cancer Res. 2002;62:4115-4122
54. Wang X, Zhou YX, Qiao W. et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148-7158
55. Li JJ, Weroha SJ, Lingle WL. et al. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci U S A. 2004;101:18123-18128
56. Katayama H, Sasai K, Kawai H. et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55-62
57. Sankaran S, Crone DE, Palazzo RE. et al. Aurora-A kinase regulates breast cancer associated gene 1 inhibition of centrosome-dependent microtubule nucleation. Cancer Res. 2007;67:11186-11194
58. Rai R, Phadnis A, Haralkar S. et al. Differential regulation of centrosome integrity by DNA damage response proteins. Cell Cycle. 2008;7:2225-2233
59. Hayward DG, Clarke RB, Faragher AJ. et al. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370-7376
60. Zeng X, Shaikh FY, Harrison MK. et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29:5103-5112
61. Suizu F, Ryo A, Wulf G. et al. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol Cell Biol. 2006;26:1463-1479
62. Bergmann S, Royer-Pokora B, Fietze E. et al. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005;65:4078-4087
63. Davies AH, Barrett I, Pambid MR. et al. YB-1 evokes susceptibility to cancer through cytokinesis failure, mitotic dysfunction and HER2 amplification. Oncogene. 2011 in press
64. Montañez-Wiscovich ME, Shelton MD, Seachrist DD. et al. Aberrant expression of LMO4 induces centrosome amplification and mitotic spindle abnormalities in breast cancer cells. J Pathol. 2010;222:271-281
65. Korzeniewski N, Wheeler S, Chatterjee P. et al. A novel role of the aryl hydrocarbon receptor (AhR) in centrosome amplification - implications for chemoprevention. Mol Cancer. 2010;9:153
66. Pihan GA, Purohit A, Wallace J. et al. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61:2212-2219
67. Toma MI, Friedrich K, Meyer W. et al. Correlation of centrosomal aberrations with cell differentiation and DNA ploidy in prostate cancer. Anal Quant Cytol Histol. 2010;32:1-10
68. Roh M, Gary B, Song C. et al. Overexpression of the oncogenic kinase Pim-1 leads to genomic instability. Cancer Res. 2003;63:8079-8084
69. Ouyang X, Wang X, Xu K. et al. Effect of p53 on centrosome amplification in prostate cancer cells. Biochim Biophys Acta. 2001;1541:212-220
70. Jiang F, Caraway NP, Sabichi AL. et al. Centrosomal abnormality is common in and a potential biomarker for bladder cancer. Int J Cancer. 2003;106:661-665
71. Kawamura K, Moriyama M, Shiba N. et al. Centrosome hyperamplification and chromosomal instability in bladder cancer. Eur Urol. 2003;43:505-515
72. Yamamoto Y, Matsuyama H, Furuya T. et al. Centrosome hyperamplification predicts progression and tumor recurrence in bladder cancer. Clin Cancer Res. 2004;10:6449-6455
73. Yamamoto Y, Matsuyama H, Kawauchi S. et al. Biological characteristics in bladder cancer depend on the type of genetic instability. Clin Cancer Res. 2006;12:2752-2758
74. Akao J, Matsuyama H, Yamamoto Y. et al. Chromosome 20q13.2 gain may predict intravesical recurrence after nephroureterectomy in upper urinary tract urothelial tumors. Clin Cancer Res. 2006;12:7004-7008
75. Del Rey J, Prat E, Ponsa I. et al. Centrosome clustering and cyclin D1 gene amplification in double minutes are common events in chromosomal unstable bladder tumors. BMC Cancer. 2010;10:280
76. Kawamura K, Izumi H, Ma Z. et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res. 2004;64:4800-4809
77. Park HS, Park WS, Bondaruk J. et al. Quantitation of Aurora kinase A gene copy number in urine sediments and bladder cancer detection. J Natl Cancer Inst. 2008;100:1401-1411
78. Yamamoto Y, Eguchi S, Junpei A. et al. Intercellular centrosome number is correlated with the copy number of chromosomes in bladder cancer. Cancer Genet Cytogenet. 2009;191:38-42
79. Yamamoto Y, Matsuyama H, Kawauchi S. et al. Overexpression of polo-like kinase 1 (PLK1) and chromosomal instability in bladder cancer. Oncology. 2006;70:231-237
80. Yamamoto Y, Matsuyama H, Chochi Y. et al. Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer. Cancer Genet Cytogenet. 2007;174:42-47
81. Gisselsson D, Gorunova L, Höglund M. et al. Telomere shortening and mitotic dysfunction generate cytogenetic heterogeneity in a subgroup of renal cell carcinomas. Br J Cancer. 2004;91:327-332
82. Nakada C, Tsukamoto Y, Matsuura K. et al. Overexpression of miR-210, a downstream target of HIF1α, causes centrosome amplification in renal carcinoma cells. J Pathol. 2011;224:280-288
83. Roshani L, Fujioka K, Auer G. et al. Aberrations of centrosomes in adrenocortical tumors. Int J Oncol. 2002;20:1161-1165
84. Skyldberg B, Fujioka K, Hellström AC. et al. Human papillomavirus infection, centrosome aberration, and genetic stability in cervical lesions. Mod Pathol. 2001;14:279-284
85. Duensing S, Duensing A, Flores ER. et al. Centrosome abnormalities and genomic instability by episomal expression of human papillomavirus type 16 in raft cultures of human keratinocytes. J Virol. 2001;75:7712-7716
86. Duensing S, Münger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075-7082
87. Riley RR, Duensing S, Brake T. et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862-4871
88. Duensing S, Duensing A, Crum CP. et al. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356-2360
89. Duensing S, Duensing A, Lee DC. et al. Cyclin-dependent kinase inhibitor indirubin-3'-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene. 2004;23:8206-8215
90. Duensing A, Liu Y, Tseng M. et al. Cyclin-dependent kinase 2 is dispensable for normal centrosome duplication but required for oncogene-induced centrosome overduplication. Oncogene. 2006;25:2943-2949
91. Duensing A, Liu Y, Perdreau SA. et al. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280-6288
92. Duensing A, Liu Y, Spardy N. et al. RNA polymerase II transcription is required for human papillomavirus type 16 E7- and hydroxyurea-induced centriole overduplication. Oncogene. 2007;26:215-223
93. Dyson N, Howley PM, Münger K. et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934-937
94. Duensing S, Münger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331-12335
95. Nguyen CL, Eichwald C, Nibert ML. et al. Human papillomavirus type 16 E7 oncoprotein associates with the centrosomal component gamma-tubulin. J Virol. 2007;81:13533-13543
96. Scheffner M, Werness BA, Huibregtse JM. et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129-1136
97. Gammoh N, Isaacson E, Tomaić V. et al. Inhibition of HPV-16 E7 oncogenic activity by HPV-16 E2. Oncogene. 2009;28:2299-2304
98. Bellanger S, Blachon S, Mechali F. et al. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle. 2005;4:1608-1615
99. Grieshaber SS, Grieshaber NA, Miller N. et al. Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic. 2006;7:940-949
100. Johnson KA, Tan M, Sütterlin C. Centrosome abnormalities during a Chlamydia trachomatis infection are caused by dysregulation of the normal duplication pathway. Cell Microbiol. 2009;11:1064-1073
101. Knowlton AE, Brown HM, Richards TS. et al. Chlamydia trachomatis Infection Causes Mitotic Spindle Pole Defects Independently from its Effects on Centrosome Amplification. Traffic. 2011 in press
102. Hsu LC, Kapali M, DeLoia JA. et al. Centrosome abnormalities in ovarian cancer. Int J Cancer. 2005;113:746-751
103. Bayani J, Paderova J, Murphy J. et al. Distinct patterns of structural and numerical chromosomal instability characterize sporadic ovarian cancer. Neoplasia. 2008;10:1057-1065
104. Mayer F, Stoop H, Sen S. et al. Aneuploidy of human testicular germ cell tumors is associated with amplification of centrosomes. Oncogene. 2003;22:3859-3866
105. Landen CN Jr, Lin YG, Immaneni A. et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007;13:4098-4104
106. Yang G, Chang B, Yang F. et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010;16:3171-3181
107. Yang H, He L, Kruk P. et al. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304-2312
108. Kayser G, Gerlach U, Walch A. et al. Numerical and structural centrosome aberrations are an early and stable event in the adenoma-carcinoma sequence of colorectal carcinomas. Virchows Arch. 2005;447:61-65
109. Gao Y, Zhang B. Expression of TEIF protein in colorectal tumors and its correlation with centrosome abnormality. Ai Zheng. 2009;28:1277-1282
110. Ghadimi BM, Sackett DL, Difilippantonio MJ. et al. Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer. 2000;27:183-190
111. Lentini L, Amato A, Schillaci T. et al. Simultaneous Aurora-A/STK15 overexpression and centrosome amplification induce chromosomal instability in tumour cells with a MIN phenotype. BMC Cancer. 2007;7:212
112. Bahmanyar S, Guiney EL, Hatch EM. et al. Formation of extra centrosomal structures is dependent on beta-catenin. J Cell Sci. 2010;123:3125-3135
113. Kuriyama R, Bettencourt-Dias M, Hoffmann I. et al. Gamma-tubulin-containing abnormal centrioles are induced by insufficient Plk4 in human HCT116 colorectal cancer cells. J Cell Sci. 2009;122:2014-2023
114. Iwaizumi M, Shinmura K, Mori H. et al. Human Sgo1 downregulation leads to chromosomal instability in colorectal cancer. Gut. 2009;58:249-260
115. Segat D, Cassaro M, Dazzo E. et al. Pericentriolar material analyses in normal esophageal mucosa, Barrett's metaplasia and adenocarcinoma. Histol Histopathol. 2010;25:551-560
116. Kuo KK, Sato N, Mizumoto K. et al. Centrosome abnormalities in human carcinomas of the gallbladder and intrahepatic and extrahepatic bile ducts. Hepatology. 2000;31:59-64
117. Nakajima T, Moriguchi M, Mitsumoto Y. et al. Centrosome aberration accompanied with p53 mutation can induce genetic instability in hepatocellular carcinoma. Mod Pathol. 2004;17:722-727
118. Fujii R, Zhu C, Wen Y. et al. HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res. 2006;66:9099-9107
119. Wen Y, Golubkov VS, Strongin AY. et al. Interaction of hepatitis B viral oncoprotein with cellular target HBXIP dysregulates centrosome dynamics and mitotic spindle formation. J Biol Chem. 2008;283:2793-2803
120. Forgues M, Difilippantonio MJ, Linke SP. et al. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282-5292
121. Yun C, Cho H, Kim SJ. et al. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol Cancer Res. 2004;2:159-169
122. Sato N, Mizumoto K, Nakamura M. et al. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res. 1999;5:963-970
123. Sato N, Mizumoto K, Nakamura M. et al. Correlation between centrosome abnormalities and chromosomal instability in human pancreatic cancer cells. Cancer Genet Cytogenet. 2001;126:13-19
124. Shono M, Sato N, Mizumoto K. et al. Stepwise progression of centrosome defects associated with local tumor growth and metastatic process of human pancreatic carcinoma cells transplanted orthotopically into nude mice. Lab Invest. 2001;81:945-952
125. Li D, Zhu J, Firozi PF. et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9:991-997
126. Zhu J, Abbruzzese JL, Izzo J. et al. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet Cytogenet. 2005;159:10-17
127. Sphyris N, Harrison DJ. p53 deficiency exacerbates pleiotropic mitotic defects, changes in nuclearity and polyploidy in transdifferentiating pancreatic acinar cells. Oncogene. 2005;24:2184-2194
128. Koutsami MK, Tsantoulis PK, Kouloukoussa M. et al. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol. 2006;209:512-521
129. Jung CK, Jung JH, Lee KY. et al. Centrosome abnormalities in non-small cell lung cancer: correlations with DNA aneuploidy and expression of cell cycle regulatory proteins. Pathol Res Pract. 2007;203:839-847
130. Haruki N, Harano T, Masuda A. et al. Persistent increase in chromosome instability in lung cancer: possible indirect involvement of p53 inactivation. Am J Pathol. 2001;159:1345-1352
131. Armit CJ, O'Dea S, Clarke AR. et al. Absence of p53 in Clara cells favours multinucleation and loss of cell cycle arrest. BMC Cell Biol. 2002;3:27
132. Cortez BA, Machado-Santelli GM. Chrysotile effects on human lung cell carcinoma in culture: 3-D reconstruction and DNA quantification by image analysis. BMC Cancer. 2008;8:181
133. Xie H, Holmes AL, Wise SS. et al. Neoplastic transformation of human bronchial cells by lead chromate particles. Am J Respir Cell Mol Biol. 2007;37:544-552
134. Holmes AL, Wise SS, Pelsue SC. et al. Chronic exposure to zinc chromate induces centrosome amplification and spindle assembly checkpoint bypass in human lung fibroblasts. Chem Res Toxicol. 2010;23:386-395
135. Liao WT, Lin P, Cheng TS. et al. Arsenic promotes centrosome abnormalities and cell colony formation in p53 compromised human lung cells. Toxicol Appl Pharmacol. 2007;225:162-170
136. Liao WT, Yu HS, Lin P. et al. Arsenite promotes centrosome abnormalities under a p53 compromised status induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Toxicol Appl Pharmacol. 2010;243:55-62
137. Shinmura K, Iwaizumi M, Igarashi H. et al. Induction of centrosome amplification and chromosome instability in p53-deficient lung cancer cells exposed to benzo[a]pyrene diol epoxide (B[a]PDE). J Pathol. 2008;216:365-374
138. Jiang F, Caraway NP, Li R. et al. RNA silencing of S-phase kinase-interacting protein 2 inhibits proliferation and centrosome amplification in lung cancer cells. Oncogene. 2005;24:3409-3418
139. Shinmura K, Tao H, Nagura K. et al. Suppression of hydroxyurea-induced centrosome amplification by NORE1A and down-regulation of NORE1A mRNA expression in non-small cell lung carcinoma. Lung Cancer. 2011;71:19-27
140. Weber RG, Bridger JM, Benner A. et al. Centrosome amplification as a possible mechanism for numerical chromosome aberrations in cerebral primitive neuroectodermal tumors with TP53 mutations. Cytogenet Cell Genet. 1998;83:266-269
141. Katsetos CD, Reddy G, Dráberová E. et al. Altered cellular distribution and subcellular sorting of gamma-tubulin in diffuse astrocytic gliomas and human glioblastoma cell lines. J Neuropathol Exp Neurol. 2006;65:465-477
142. Uccella S, Tibiletti MG, Bernasconi B. et al. Aneuploidy, centrosome alteration and securin overexpression as features of pituitary somatotroph and lactotroph adenomas. Anal Quant Cytol Histol. 2005;27:241-252
143. Caracciolo V, D'Agostino L, Dráberová E. et al. Differential expression and cellular distribution of gamma-tubulin and betaIII-tubulin in medulloblastomas and human medulloblastoma cell lines. J Cell Physiol. 2010;223:519-529
144. Loh JK, Lieu AS, Chou CH. et al. Differential expression of centrosomal proteins at different stages of human glioma. BMC Cancer. 2010;10:268
145. Saito T, Hama S, Izumi H. et al. Centrosome amplification induced by survivin suppression enhances both chromosome instability and radiosensitivity in glioma cells. Br J Cancer. 2008;98:345-355
146. Li L, Dutra A, Pak E. et al. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol. 2009;11:9-21
147. Hida K, Klagsbrun M. A new perspective on tumor endothelial cells: unexpected chromosome and centrosome abnormalities. Cancer Res. 2005;65:2507-2510
148. Roussel-Gervais A, Bilodeau S, Vallette S. et al. Cooperation between cyclin E and p27(Kip1) in pituitary tumorigenesis. Mol Endocrinol. 2010;24:1835-1845
149. Fukushi D, Watanabe N, Kasai F. et al. Centrosome amplification is correlated with ploidy divergence, but not with MYCN amplification, in neuroblastoma tumors. Cancer Genet Cytogenet. 2009;188:32-41
150. Slack AD, Chen Z, Ludwig AD. et al. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007;67:2448-2455
151. Sugihara E, Kanai M, Matsui A. et al. Enhanced expression of MYCN leads to centrosome hyperamplification after DNA damage in neuroblastoma cells. Oncogene. 2004;23:1005-1009
152. Sugihara E, Kanai M, Saito S. et al. Suppression of centrosome amplification after DNA damage depends on p27 accumulation. Cancer Res. 2006;66:4020-4029
153. Carroll PE, Okuda M, Horn HF. et al. Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene. 1999;18:1935-1944
154. Gustafson LM, Gleich LL, Fukasawa K. et al. Centrosome hyperamplification in head and neck squamous cell carcinoma: a potential phenotypic marker of tumor aggressiveness. Laryngoscope. 2000;110:1798-1801
155. Gisselsson D, Jonson T, Yu C. et al. Centrosomal abnormalities, multipolar mitoses, and chromosomal instability in head and neck tumours with dysfunctional telomeres. Br J Cancer. 2002;87:202-207
156. Reiter R, Gais P, Jütting U. et al. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:5136-5141
157. Reiter R, Gais P, Steuer-Vogt MK. et al. Centrosome abnormalities in head and neck squamous cell carcinoma (HNSCC). Acta Otolaryngol. 2009;129:205-213
158. Thirthagiri E, Robinson CM, Huntley S. et al. Spindle assembly checkpoint and centrosome abnormalities in oral cancer. Cancer Lett. 2007;258:276-285
159. Syed MI, Syed S, Minty F. et al. Gamma tubulin: a promising indicator of recurrence in squamous cell carcinoma of the larynx. Otolaryngol Head Neck Surg. 2009;140:498-504
160. Kaneko H, Ishikawa S, Sumida T. et al. Ultrastructural studies of a thymic carcinoid tumor. Acta Pathol Jpn. 1980;30:651-658
161. Wu Q, Sahasrabudhe RM, Luo LZ. et al. Deficiency in myosin light-chain phosphorylation causes cytokinesis failure and multipolarity in cancer cells. Oncogene. 2010;29:4183-4193
162. Seifert HW. Electron microscopic investigation on cutaneous leiomyosarcoma. Arch Dermatol Res. 1978;263:159-169
163. Al-Romaih K, Bayani J, Vorobyova J. et al. Chromosomal instability in osteosarcoma and its association with centrosome abnormalities. Cancer Genet Cytogenet. 2003;144:91-99
164. Gisselsson D, Pålsson E, Yu C. et al. Mitotic instability associated with late genomic changes in bone and soft tissue tumours. Cancer Lett. 2004;206:69-76
165. Perucca-Lostanlen D, Rostagno P, Grosgeorge J, Marcié S, Gaudray P, Turc-Carel C. Distinct MDM2 and P14ARF expression and centrosome amplification in well-differentiated liposarcomas. Genes Chromosomes Cancer. 2004;39:99-109
166. Gong Y, Sun Y, McNutt MA. et al. Localization of TEIF in the centrosome and its functional association with centrosome amplification in DNA damage, telomere dysfunction and human cancers. Oncogene. 2009;28:1549-1560
167. Moskovszky L, Dezsö K, Athanasou N. et al. Centrosome abnormalities in giant cell tumour of bone: possible association with chromosomal instability. Mod Pathol. 2010;23:359-366
168. Koopal S, Furuhjelm JH, Järviluoma A. et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007;3:1348-1360
169. Verschuren EW, Klefstrom J, Evan GI. et al. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2:229-241
170. Verschuren EW, Hodgson JG, Gray JW. et al. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res. 2004;64:581-589
171. Pan H, Zhou F, Gao SJ. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 2004;64:4064-4068
172. Si H, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J Virol. 2006;80:697-709
173. Cuomo ME, Knebel A, Morrice N. et al. p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nat Cell Biol. 2008;10:723-730
174. Krämer A, Neben K, Ho AD. Centrosome aberrations in hematological malignancies. Cell Biol Int. 2005;29:375-383
175. Krämer A, Schweizer S, Neben K. et al. Centrosome aberrations as a possible mechanism for chromosomal instability in non-Hodgkin's lymphoma. Leukemia. 2003;17:2207-2213
176. Hensel M, Zoz M, Giesecke C. et al. High rate of centrosome aberrations and correlation with proliferative activity in patients with untreated B-cell chronic lymphocytic leukemia. Int J Cancer. 2007;121:978-983
177. Martín-Subero JI, Knippschild U, Harder L. et al. Segmental chromosomal aberrations and centrosome amplifications: pathogenetic mechanisms in Hodgkin and Reed-Sternberg cells of classical Hodgkin's lymphoma? Leukemia. 2003;17:2214-2219
178. Sánchez-Aguilera A, Montalbán C, de la Cueva P. et al. Tumor microenvironment and mitotic checkpoint are key factors in the outcome of classic Hodgkin lymphoma. Blood. 2006;108:662-668
179. Neben K, Ott G, Schweizer S. et al. Expression of centrosome-associated gene products is linked to tetraploidization in mantle cell lymphoma. Int J Cancer. 2007;120:1669-1677
180. Ottaggio L, Zunino A, Maric I. et al. The presence of high-risk chromosome aberrations in chronic lymphocytic leukaemia does not correlate with centrosome aberrations. Hematol Oncol. 2008;26:39-42
181. Ventura RA, Martin-Subero JI, Knippschild U. et al. Centrosome abnormalities in ALK-positive anaplastic large-cell lymphoma. Leukemia. 2004;18:1910-1911
182. Duensing S, Lee BH, Dal Cin P. et al. Excessive centrosome abnormalities without ongoing numerical chromosome instability in a Burkitt's lymphoma. Mol Cancer. 2003;2:30
183. Giehl M, Fabarius A, Frank O. et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19:1192-1197
184. Patel H, Gordon MY. Abnormal centrosome-centriole cycle in chronic myeloid leukaemia? Br J Haematol. 2009;146:408-417
185. Giehl M, Fabarius A, Frank O. et al. Expression of the p210BCR-ABL oncoprotein drives centrosomal hypertrophy and clonal evolution in human U937 cells. Leukemia. 2007;21:1971-1976
186. Fabarius A, Giehl M, Frank O. et al. Induction of centrosome and chromosome aberrations by imatinib in vitro. Leukemia. 2005;19:1573-1578
187. Fabarius A, Giehl M, Frank O. et al. Centrosome aberrations after nilotinib and imatinib treatment in vitro are associated with mitotic spindle defects and genetic instability. Br J Haematol. 2007;138:369-373
188. Fabarius A, Giehl M, Rebacz B. et al. Centrosome aberrations and G1 phase arrest after in vitro and in vivo treatment with the SRC/ABL inhibitor dasatinib. Haematologica. 2008;93:1145-1154
189. Giehl M, Leitner A, Haferlach C. et al. Detection of centrosome aberrations in disease-unrelated cells from patients with tumor treated with tyrosine kinase inhibitors. Eur J Haematol. 2010;85:139-148
190. Neben K, Giesecke C, Schweizer S. et al. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood. 2003;101:289-291
191. Neben K, Tews B, Wrobel G. et al. Gene expression patterns in acute myeloid leukemia correlate with centrosome aberrations and numerical chromosome changes. Oncogene. 2004;23:2379-2384
192. Kearns WG, Yamaguchi H, Young NS. et al. Centrosome amplification and aneuploidy in bone marrow failure patients. Genes Chromosomes Cancer. 2004;40:329-333
193. Ching YP, Chan SF, Jeang KT. et al. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat Cell Biol. 2006;8:717-724
194. Nitta T, Kanai M, Sugihara E. et al. Centrosome amplification in adult T-cell leukemia and human T-cell leukemia virus type 1 Tax-induced human T cells. Cancer Sci. 2006;97:836-841
195. Peloponese JM Jr, Haller K, Miyazato A. et al. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc Natl Acad Sci U S A. 2005;102:18974-18979
196. Pumfery A, de la Fuente C, Kashanchi F. HTLV-1 Tax: centrosome amplification and cancer. Retrovirology. 2006;3:50
197. Maxwell CA, Keats JJ, Belch AR. et al. Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res. 2005;65:850-860
198. Dementyeva E, Nemec P, Kryukov F. et al. Centrosome amplification as a possible marker of mitotic disruptions and cellular carcinogenesis in multiple myeloma. Leuk Res. 2010;34:1007-1011
199. Chng WJ, Braggio E, Mulligan G. et al. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood. 2008;111:1603-1609
200. Chng WJ, Fonseca R. Centrosomes and myeloma; aneuploidy and proliferation. Environ Mol Mutagen. 2009;50:697-707
201. Carpinelli P, Moll J. Is there a future for Aurora kinase inhibitors for anticancer therapy? Curr Opin Drug Discov Devel. 2009;12:533-542
202. Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663-678
203. Lee SC, Chan JY, Pervaiz S. Spontaneous and 5-fluorouracil-induced centrosome amplification lowers the threshold to resveratrol-evoked apoptosis in colon cancer cells. Cancer Lett. 2010;288:36-41
204. Wang X, Liu L, Montagna C. et al. Haploinsufficiency of Parp1 accelerates Brca1-associated centrosome amplification, telomere shortening, genetic instability, apoptosis, and embryonic lethality. Cell Death Differ. 2007;14:924-931
205. Duensing A, Chin A, Wang L. et al. Analysis of centrosome overduplication in correlation to cell division errors in high-risk human papillomavirus (HPV)-associated anal neoplasms. Virology. 2008;372:157-164
Author contact
![]() Corresponding author: Dr Jason Yongsheng Chan (MBBS, Singapore), Ministry of Health Holdings, Singapore. Tel.: +65-8100-4804; E-mail: jasoncyscom.sg
Corresponding author: Dr Jason Yongsheng Chan (MBBS, Singapore), Ministry of Health Holdings, Singapore. Tel.: +65-8100-4804; E-mail: jasoncyscom.sg