Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(9):1311-1322. doi:10.7150/ijbs.7.1311 This issue Cite
Review
Regulating the Adaptive Immune Response to Blood-Stage Malaria: Role of Dendritic Cells and CD4+Foxp3+ Regulatory T Cells
Mary M. Stevenson ![]() , Rebecca Ing, Floriana Berretta, Jenny Miu
, Rebecca Ing, Floriana Berretta, Jenny Miu
Centre for the Study of Host Resistance and Centre for Host-Parasite Interactions, Research Institute of the McGill University Health Centre and Department of Medicine, McGill University Montreal, Quebec.
Received 2011-9-1; Accepted 2011-10-1; Published 2011-10-25
Abstract
Although a clearer understanding of the underlying mechanisms involved in protection and immunopathology during blood-stage malaria has emerged, the mechanisms involved in regulating the adaptive immune response especially those required to maintain a balance between beneficial and deleterious responses remain unclear. Recent evidence suggests the importance of CD11c+ dendritic cells (DC) and CD4+Foxp3+ regulatory T cells in regulating immune responses during infection and autoimmune disease, but information concerning the contribution of these cells to regulating immunity to malaria is limited. Here, we review recent findings from our laboratory and others in experimental models of malaria in mice and in Plasmodium-infected humans on the roles of DC and natural regulatory T cells in regulating adaptive immunity to blood-stage malaria.
Keywords: malaria, Plasmodium, immune responses, dendritic cells, regulatory T cells
Introduction
As a result of a massive scale-up in malaria control programs by the WHO as part of the Millennium Development Goals, the number of malaria deaths decreased worldwide by about 20% between 2000 and 2009 (1). Although this represents some progress in reducing the disease burden, malaria nevertheless remains a major global health threat and continues to cause high morbidity and mortality, especially in sub-Saharan Africa where almost 600 million people are at risk. Malaria persists due to the lack of an efficacious vaccine, the spread of drug-resistant parasites, and the emergence of insecticide-resistant Anopheles mosquito vectors as well as changing agricultural practices, global warming, and human migration (2,3). Refugees exposed to malaria and other infectious diseases in their country of origin or in camps who immigrate to developed countries may be important reservoirs and further hasten the spread of malaria to other regions (4-6). Of the Plasmodium parasites (P. falciparum, P. vivax, P. malariae, P. ovale curtisi, P. ovale wallikeri, and P. knowlesi) that cause human malaria, P. falciparum is the most virulent and deadly parasite (7, 8). Most deaths due to P. falciparum infection occur in sub-Saharan Africa, with 92% of the annual malaria deaths occurring among infants and young children <5 years of age (1,9-11). In this region, these individuals and pregnant women, especially primigravidae, are the groups with the highest risk for the development of severe and fatal complications of P. falciparum infection especially cerebral malaria (CM) and severe malarial anemia (SMA), (2,9-11). Unprotected travelers and other non-immune individuals are also extremely vulnerable to these complications.
Immunity to infection with blood-stage Plasmodium parasites is critically dependent on the type 1 cytokine IFN-γ and requires coordinate and timely innate and adaptive immune responses involving dendritic cells (DC), NK cells, CD4+ T helper cells, and B cells (12,13). Importantly, a balance between pro-inflammatory and anti-inflammatory responses is essential to limit the development of life-threatening immune-mediated pathology such as CM and SMA (14). Although a clearer understanding of the mechanisms involved in protective immunity and immunopathology is emerging, our understanding of the regulatory mechanisms required to maintain the balance between beneficial and deleterious responses during blood-stage malaria infection remains limited. Here, we review recent findings from our laboratory and others in experimental models of malaria in mice and in Plasmodium-infected humans on the activation and expansion of CD11c+ DC and Foxp3+ regulatory T cells and their possible roles in regulating adaptive immunity to blood-stage malaria.
Life cycle of the Plasmodium parasite and pathogenesis
The life cycle of the malaria parasite is complex with a sexual phase in the Anopheles mosquito vector and an asexual phase in the host (12). Sporozoites are transmitted to the host by the bite of an infected female mosquito and travel via the blood to the liver where they invade hepatocytes and initiate the exo-erythrocytic or liver stage, which is clinically silent and without any pathologic sequelae. Parasite multiplication in the liver results in merozoites that infect red blood cells (RBC), where they undergo mitosis and form erythrocytic schizonts containing many merozoites that are released and infect new RBC. As yet unknown factors trigger some merozoites to differentiate into male and female gametocytes that are taken-up during a blood meal by the mosquito for fertilization and sporozoite production (15). The major manifestations of malaria in infected individuals include fever, malaise, splenomegaly, and anemia and are due to the cyclical multiplication of merozoites in RBC (12,14). During P. falciparum infection, the spectrum of severe pathology is broad and includes metabolic acidosis, CM, and SMA, and is typically accompanied by hypoxia, hypoglycemia, and lactic acidosis due in part to the increased metabolic demands of the parasite (12,14).
Mouse malaria models
Human malaria is due to infection with 6 Plasmodium parasites. P. falciparum, the most virulent and deadly human parasite, is highly prevalent in sub-Saharan Africa (1). P. vivax is the most common species causing human infection and predominates outside Africa in South America and Asia while infections with P. malariae and P. ovale subspecies are usually mild. Recently, human infections with P. knowlesi, a parasite naturally occurring among monkeys, have been identified in malaria endemic areas (8). Numerous challenges such as the lack of access to appropriate tissue samples and the inability to manipulate the immune response to elucidate underlying mechanisms impede the study of malaria in humans (16). As an alternative, experimental models in non-human primates and rodent models have been used. However, the lack of availability and high cost as well as ethical issues limit the use of non-human primates to study malaria. Infection of inbred mice with various rodent Plasmodium species (P. chabaudi, P. berghei, P. vinckei and P. yoelii) that differ in virulence and pathology provide the most convenient experimental models (Table 1) (12,17). It is important to note that while no single rodent model replicates all of the features of human malaria in terms of pathophysiology or immune responses, these models have been essential in defining the basis of interactions between the host and the Plasmodium parasite. Studies in mouse malaria models have revealed important and novel information leading to a clearer understanding of the immune response in protection and pathogenesis, to identify previously unrecognized genes that regulate susceptibility to malaria, and to develop malaria vaccines and novel chemotherapeutic agents (12,17-21).
Rodent malaria parasites and their pathogenesis
| P. chabaudi AS | Malarial anemia |
| P. berghei ANKA | Cerebral malaria |
| P. vinckei | Hyperparasitemia |
| P. yoelii 17XL | Hyperparasitemia |
Because P. chabaudi AS is considered to share many biological and immunological features in common with P. falciparum, infection of mice with this parasite is widely used as a tool to study immune responses to blood-stage malaria (12,17,19). Infection of resistant C57BL/6 (B6) mice with 104-106 P. chabaudi AS infected RBC (iRBC) results in a course of parasitemia characterized by parasite replication during the first week post infection (p.i.) culminating in a moderate peak parasitemia of 30-35% between days 8-10 p.i., with control of parasitemia and clearance by about 4 weeks p.i. (12,17,19). In a seminal study, Grun and Weidanz demonstrated in mice infected with P. chabaudi that: 1) the control of peak parasitemia is dependent on a cell-mediated immune response that we now know requires CD4+ T cells, and 2) the requirement for B cells in the clearance and elimination of the parasite (12). Later studies from our laboratory showed that control of acute P. chabaudi AS infection is critically dependent on IL-12-dependent IFN-γ production by NK cells and CD4+ Th1 cells indicating the importance of both innate and adaptive immune responses (12). The importance of cognate interactions between CD4+ Th cells and B cells for malaria-specific antibody production for efficient parasite elimination later during infection was demonstrated by Langhorne and her colleagues (12,17). Identification of the importance of NK cells, CD4+ T cells, and B cells in mediating control and resolution of P. chabaudi AS infection has provided a framework to delineate the accessory cells and cytokines involved in activating innate immunity and triggering adaptive immunity to malaria. Studies in mice infected with avirulent and virulent strains of P. yoelii as well as in mice infected with P. berghei ANKA have also provided important information about the role of the immune response in protection and immunopathogenesis (14,17,21).
Dendritic cells and blood-stage malaria
DC are a heterogeneous and highly specialized population of antigen presenting cells (APC) that provide a central link between innate and adaptive immune responses and play an important role at the host-pathogen interface including in response to Plasmodium parasites (22-25). DC reside in almost all tissues where they play important roles in surveillance and sensing foreign antigens. Pattern recognition receptors, such as Toll-like receptors (TLRs), Nod-like receptors, and C-type lectins, expressed by these cells are important in distinguishing a diverse but overlapping repertoire of conserved microbial molecules (26). DC precursors are generated from pluripotent stem cells in the bone marrow under the influence of lineage specific growth factors and are released into the blood for transit to lymphoid organs, including secondary lymphoid tissues such as the spleen as well as non-lymphoid tissues such as the skin, where they reside as immature cells (22). Following encounter with antigen, immature DC undergo maturation in response to stimulation by pathogens or cytokines and other soluble mediators produced by various cell types. DC maturation results in a decreased ability to capture and process antigen and increased surface expression of MHC-II and co-stimulatory (CD40, CD80, CD86) and adhesion molecules (22-27). DC maturation is critical in immunity to pathogenic microorganisms because of the potent ability of these cells to polarize the differentiation of naïve CD4+ T cells to Th cell subsets including Th1, Th2, Th17, follicular Th cells, and induced regulatory T cells (28,29). DC also function as accessory cells and are important in activating NK cells for cytotoxicity and cytokine secretion in response to viruses and tumors (30). As a result of interactions with T cells, NK cells and other effector cells, the ability of DC to secrete cytokines is enhanced. DC, thus, are not only important in antigen presentation and activating CD4+ T cells but also function as accessory cells to regulate and amplify innate and adaptive immune responses.
The relationship between DC maturation and malaria has been investigated in studies using peripheral blood monocytes from humans and murine DC (25,31). Initially, it was shown that DC function is compromised during malaria based on the in vitro observation that binding of P. falciparum iRBC to human monocyte-derived DC inhibits maturation and reduces the capacity of the DC to function as an APC (32). Analysis of DC recovered from African children with severe P. falciparum malaria revealed that the frequency of myeloid-derived CD11c+BDCA3+ DC is significantly increased during acute infection (33). A recent study comparing DC subpopulations in individuals infected with P. falciparum or P. vivax and residing in areas with similar endemicity found a significant increase in plasmacytoid CD123+ DC and a decreased ratio of myeloid to plasmacytoid DC during infection, regardless of the infecting species (34). In this study, the maturation of peripheral blood DC was observed to be impaired ex vivo as shown by low CD86 expression in a proportion of the P. vivax-infected patients, but there was no evidence that DC maturation was affected in P. falciparum-infected individuals. These data suggest that DC subpopulations are differentially affected in humans during blood-stage malaria and that the variability observed in the effects of malaria infection on DC function may be due to the Plasmodium species, the severity of the infection, and the patient population studied.
Similar observations of diverse effects of malaria infection on DC have been made in studies using various combinations of parasites and inbred mouse strains. Distinct subpopulations of DC including myeloid-derived DC (CD11c+CD4+, CD11c+CD8α+, CD11c+CD8α-), plasmacytoid DC (CD11c+B220+), and a regulatory subset of CD11clowCD45RBhigh DC have been observed to expand in the spleens of mice infected with various Plasmodium parasites (27,31,35-38). The development of transgenic mice expressing a T cell receptor (TCR) specific for a peptide of the P. chabaudi merozoite surface protein-1 (MSP1) has provided a novel tool to elucidate the role of DC and other cells in the immune response to malaria (39). Using transgenic MSP1-specific TCR mice infected with P. chabaudi AS, Sponaas et al provided direct evidence that CD11c+ DC in the spleen activate Plasmodium-specific CD4+ T cells (38). This study also provided evidence that splenic DC subpopulations have distinct antigen presenting functions during malaria. Although both CD8+ and CD8- DC present malaria peptides and induce IFN-γ production by CD4+ T cells, only CD8- DC stimulate antigen-specific proliferation of CD4+ T cells and secretion of IL-4 and IL-10.
Recently, Lundie et al reported another strategy to study the immune response to malaria that involves the generation of genetically-modified strains of P. berghei ANKA expressing MHC-I- and MHC-II-restricted T cell epitopes specific for cognate antigens, such as OVA, linked to reporter GFP (40). Using the genetically modified parasites to infect OT-I or OT-II transgenic mice, these investigators observed that both CD8α-CD4+ and CD8α+ but not CD8α-CD4- DC present P. berghei-expressed antigens to CD4+ T cells during malaria infection, while the CD8α+ subpopulation induces pathogenic CD8+ T cells via cross-presentation (40). In a separate study, blood-stage P. berghei ANKA infection was found to cause suppression of MHC-I-restricted immunity to non-malarial antigens as a possible consequence of systemic DC activation (41). This observation suggests that DC are not directly suppressed by malaria parasites, but that they follow their normal maturation pathway after activation by iRBC or parasite products. Subsequent findings by Lundie et al showed that DC isolated from P. berghei-infected mice efficiently present P. berghei-expressed antigens to CD4+ and CD8+ T cells on days 2 and 3 p.i., but their ability to present parasite antigens as well as MHC-I- and MHC-II-restricted, non-malarial antigens is drastically impaired by day 4 p.i. (42). These findings are consistent with earlier studies by Perry et al in P. yoelii-infected mice which showed that DC become refractory to TLR-ligand-stimulated IL-12 and TNF-α production with increased IL-10 secretion as the infection progresses (43). DC from infected mice were found to retain the capacity to stimulate OVA-specific proliferation of naïve OT-II CD4+ T cells throughout P. yoelii infection, but the responding T cell phenotype changed later during infection from CD4+IFN-γ+ to CD4+IL-10+ T cells. However, functional impairment of DC at early times p.i. during lethal infections due to P. yoelii YM, P. vinckei or P. berghei ANKA has been observed based on the inability of DC from infected mice to secrete high levels of IL-12 and prime naïve CD4+ T cells (31). It was reported that the decreased function of DC in mice infected with P. vinckei or P. berghei is mediated in part by TNF-α (31). In contrast, Lundie and colleagues failed to demonstrate a role for TNF-α in impaired DC function during P. berghei ANKA infection (42). As will be described below, spleen DC recovered from P. chabaudi AS-infected B6 mice during the first week p.i. are fully functional APC (31,44). Altogether, studies in mouse models essentially confirm the findings in human studies and provide further explanation that the conflicting data obtained in human studies may be due to several factors including the parasite species and strain, the time after infection, and the size of the inoculum.
Role of DC as accessory cells
Studies performed using human peripheral blood mononuclear cells and in Plasmodium-infected mice indicate that NK cells are an early and crucial source of IFN-γ during blood-stage malaria (12). Findings in experimental models of infection, especially viral infections, tumors, and inflammatory diseases, provide important evidence that optimal IFN-γ production by NK cells requires contact-dependent and -independent interactions with myeloid-derived accessory cells such as monocytes, macrophages, and DC (30). As will be described below, we investigated if an accessory cell is necessary to promote IFN-γ secretion by NK cells during P. chabaudi AS infection and questioned if CD11c+ DC are involved.
Previous studies in B6 mice showed that during early P. chabaudi AS infection DC, macrophages, and NK cells rapidly accumulate and expand in the spleen where they undergo maturation and functional activation in response to iRBC captured in the spleen by phagocytosis (reviewed in 44). During early infection, CD11c+ DC migrate from the marginal zone to the CD4+ T cell-rich periarteriolar lymphoid sheaths while macrophages expand and remain in the red pulp (45). Importantly, within the first week p.i., spleen DC from P. chabaudi AS-infected B6 mice display up-regulation of MHC-II and co-stimulatory molecule expression, secrete pro-inflammatory cytokines including high levels of IL-12, and stimulate naïve CD4+ T cell proliferation and IFN-γ production (25,27,31,45). In contrast, splenic macrophages from P. chabaudi AS-infected mice suppress T cell proliferation and IL-2 secretion (46,47). Together, these findings provide compelling evidence that CD11c+ DC play an important role in innate immunity during blood-stage malaria and trigger an adaptive type 1 immune response necessary for control and rapid resolution of infection.
To determine if DC have a role in activating NK cells during blood-stage malaria, we compared the maturation and function of CD11c+ DC purified by magnetic beads from the spleens of infected wild-type (WT) B6 mice and IL-12p40-/- and IL-15-/- mice that lack cytokines involved in NK cell proliferation, survival and activation (44). In previous studies, we demonstrated that the course of P. chabaudi AS infection is more severe in IL-12p40-/- mice compared to WT and IL-15-/- mice (12, reviewed in 44). We observed that the uptake of iRBC by CD11c+ DC early after infection was similar in WT, IL-12p40-/-, and IL-15-/- mice. DC from infected IL-15-/- and WT mice expressed similar levels of CD40, CD80, and CD86 while DC from infected IL-12p40 -/- mice expressed significantly lower levels of co-stimulatory molecules. Analysis of cytokine production in response to in vitro stimulation with iRBC revealed that DC recovered from IL-12p40-/- mice on day 4 p.i. produced significantly lower levels of pro-inflammatory cytokines compared to WT DC but secreted significantly more IL-2 while the levels of IL-10 secreted by WT, IL-12p40-/-, and IL-15-/- DC were similar. These data indicate that CD11c+ DC from infected IL-12p40-/- mice are as efficient as DC from infected WT and IL-15-/- mice in phagocytizing iRBC but that DC maturation is impaired in infected IL-12p40-/- mice.
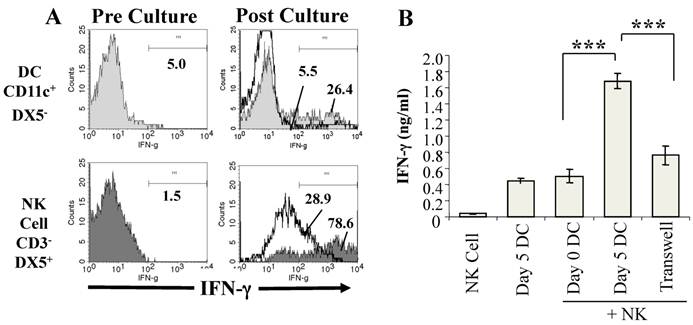
To investigate if DC from P. chabaudi AS-infected mice promote IFN-γ production by NK cells, we established co-cultures of purified CD11c+ DC from infected WT mice with CD3-DX5+ NK cells purified from the spleens of naïve WT mice and analyzed NK cell expression of intracellular IFN-γ expression before and after co-culture with DC by flow cytometry (44). IFN-γ levels in the supernatants of the co-cultures were also determined. As shown in Fig. 1A, few CD3-DX5+ NK cells from naïve WT mice expressed intracellular IFN-γ prior to co-culture with DC from infected WT mice. After co-culture with DC from infected mice, there was a dramatic increase in the frequency of DX5+IFN-γ+ NK cells. Although the frequency of CD11c+IFN-γ+ DC also increased in the co-cultures, the major source of IFN-γ was observed to be NK cells, a finding corroborated when DC from infected WT mice were co-cultured with NK cells from naïve IFN-γ-/- mice (44). Determination of the levels of IFN-γ in the supernatants confirmed that WT DC from infected mice stimulated significantly higher IFN-γ secretion than WT DC from naïve mice (Fig. 1B).
DC from infected mice activate NK cells to produce IFN-γ. CD11c+ DC enriched by magnetic beads from the spleens of P. chabaudi AS infected B6 mice on day 5 p. i. and NK cells (CD3-DX5+) enriched by magnetic beads from the spleens of naïve B6 mice were cultured separately or co-cultured together for 24-36 hrs. (A) The expression of intracellular IFN-γ was analyzed by flow cytometry in gated CD11c+DX5- and CD3-DX5+ cells before co-culture (Pre-culture, left panel) and after culture separately (solid lines) or together in co-cultures (shaded areas) (Post-culture, right panel). (B) IFN-γ levels in the supernatants of NK cells or DC from day 5 infected mice cultured separately or of co-cultures of NK cells and DC from mice at day 0 or 5 p.i. were determined by ELISA. Transwell indicates that DC and NK cells were separated using a transwell insert. ***, p<0.001 indicates significant differences compared to DC from day 5 infected B6 mice. Originally published in part in Infection and Immunity (Ref. 44), Copyright® 2009, The American Society for Microbiology.
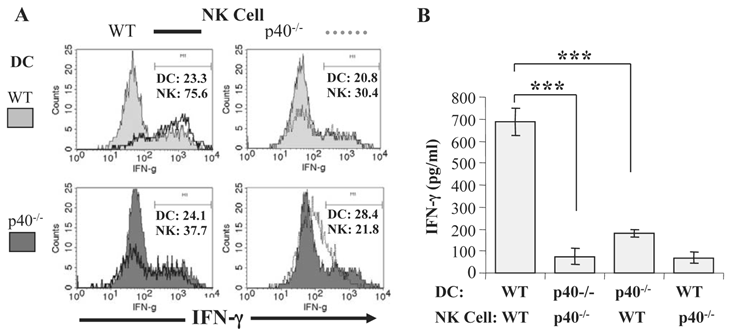
We also determined the relative contribution of IL-12 and IL-15 to DC-NK cell interactions during malaria by co-culturing purified CD11c+ DC from infected IL-12p40-/- or IL-15-/- mice with NK cells from naïve WT mice. DC from infected IL-15-/- mice stimulated a similar percentage of WT NK cells to express intracellular IFN-γ as WT DC, and IFN-γ levels were similar in supernatants of WT NK cells co-cultured with WT DC and IL-15-/- DC. In contrast, DC from infected IL-12p40-/- mice are unable to promote IFN-γ expression by NK cells from naïve WT mice (Fig. 2A and 2B). These data are consistent with the observations described above linking lethal malaria infections in mice with functional impairment of DC and indicate that soluble mediators, especially IL-12, are required to induce optimal IFN-γ secretion by NK cells during blood-stage malaria. We also observed that DC-NK cell interaction is dependent on cell-cell contact since significantly lower IFN-γ production was observed when NK cells and DC were separated by a transwell insert.
IL-12 secretion by DC is required for optimal NK cell IFN-γ production during blood-stage malaria. CD11c+ DC enriched by magnetic beads from the spleen of P. chabaudi AS-infected WT B6 and IL-12p40-/- mice were co-cultured with NK cells enriched by magnetic beads from the spleens of naïve WT or IL-12p40-/- mice. (A) The expression of intracellular IFN-γ was analyzed by flow cytometry in gated CD11c+DX5- and CD3-DX5+ cells in co-cultures of DC from infected WT or IL-12p40-/- mice and NK cells from naïve WT (left panel) or IL-12p40-/- (right panel) mice. (B) IFN-γ levels in the supernatants of co-cultures of DC from infected WT or IL-12p40-/- mice and NK cells from naïve WT or IL-12p40-/- mice were determined by ELISA. ***, p<0.001 indicates significant differences compared to co-cultures of DC from infected WT mice and NK cells from naïve WT mice. Originally published in part in Infection and Immunity (Ref. 44), Copyright® 2009, The American Society for Microbiology.
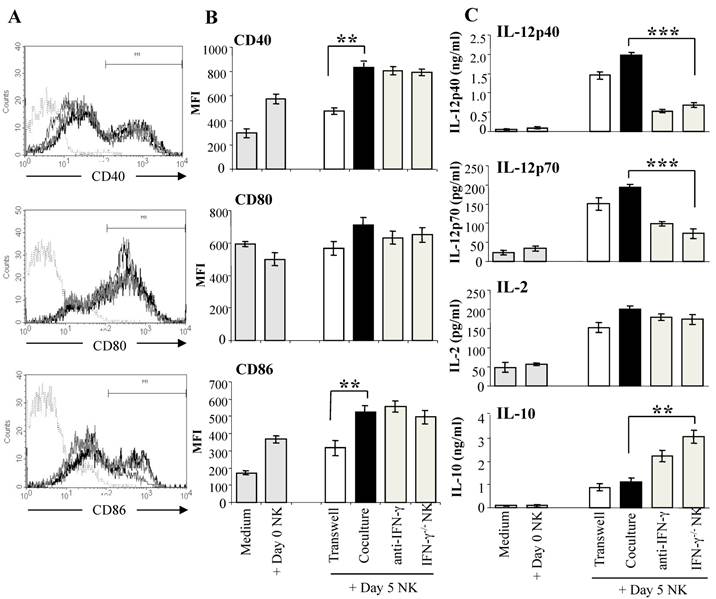
Reciprocal crosstalk between DC and NK cells has been demonstrated in a number of studies, with evidence of complex interactions between the two cell types (30,48). NK cells activated by tumors, bacterial products, or IL-2 have been shown to induce DC maturation and secretion of the Th1-promoting cytokine IL-12 (30,48). To determine if malaria-activated NK cells induce DC maturation, we co-cultured purified DX5+ NK cells from day 5 infected WT mice with CD11c+ DC from naïve WT mice which were exposed to iRBC for 1 hr prior to co-culture with NK cells to prevent lysis of immature DC by the activated NK cells (44). DC from naïve mice co-cultured with NK cells from infected WT mice displayed significantly increased CD40 and CD86 expression compared to control DC cultured alone or with NK cells from naïve WT mice (Fig. 3A). Up-regulation of co-stimulatory molecule expression on DC co-cultured with malaria-activated NK cells was found to be dependent on cell contact. Co-culture with malaria-activated NK cells but not NK cells from naïve mice also stimulated DC to secrete high levels of cytokines especially IL-12 (Fig. 3B). DC separated from NK cells by a transwell insert showed similar levels of cytokine production as control co-cultures indicating that soluble factors produced by NK cells and not cell-cell contact are responsible for enhanced IL-12 production by DC. The importance of NK cell-derived IFN-γ in inducing enhanced production of Th1-promoting IL-12 by DC was demonstrated by co-culture of WT DC with NK cells from P. chabaudi AS-infected IFN-γ-/- mice and by the addition of anti-IFN-γ monoclonal antibody to co-cultures of WT DC and WT NK cells. In the absence of NK cell-derived IFN-γ, DC produced significantly higher levels of the immunoregulatory cytokine IL-10. Importantly, DC stimulated by malaria-activated NK cells were found to be fully licensed APC and primed naïve CD4+ T cells to proliferate and produce the type 1 cytokine IFN-γ indicating Th1 polarization (44).
Malaria-activated NK cells stimulate DC to mature and secrete cytokines. NK cells enriched by magnetic beads from the spleens of day 5 P. chabaudi AS-infected B6 mice were co-cultured with CD11c+ DC enriched by magnetic beads from the spleens of naïve B6 mice. DC were exposed to iRBC prior to co-culture to limit the cytotoxic effects of NK cells. DC were analyzed for co-stimulatory molecule expression and cytokine production. To determine the role of NK secreted IFN-γ, DC were co-cultured with NK cells from IFN-γ-/- mice or with WT NK cells in the presence of anti-IFN-γ monoclonal antibody or isotype control antibody (data not shown). DC were also co-cultured with NK cells in wells with a transwell insert to prevent cell-cell contact. Panel A shows the strategy to determine the mean fluorescence intensity (MFI) of CD40, CD80 and CD86. Histogram in dotted line represents isotype control, grey line, DC co-cultured with day 0 NK, black line, DC co-cultured with day 5 NK. Panel B shows the CD40, CD80 and CD86 expression (MFI) after culture alone or co-culture with NK cells from naïve mice (Day 0 NK) as controls or co-culture with WT or IFN-γ-/- NK cells from day 5 infected mice (Day 5 NK). The asterisks indicate a significant difference between DC separated from day 5 NK cells by a transwell insert (Transwell) or DCs co-cultured with day 5 NK cells (Coculture); **, p <0.01. (C) Cytokine levels in supernatants of DC cultured alone or with day 0 or day 5 NK cells were determined by ELISA. The data are representative of three experiments, each containing three or four replicates per group. The asterisks indicate significant differences between WT NK cells (coculture) and IFN-γ-/- cells; **, p <0.01 and ***, p <0.001. The error bars indicate SEM. Originally published in part in Infection and Immunity (Ref. 44), Copyright® 2009, The American Society for Microbiology.
Our observations in the mouse model of P. chabaudi AS infection confirmed in vitro studies by Newman et al in human peripheral blood mononuclear cells stimulated with P. falciparum iRBC showing that IFN-γ production by NK cells only occurs following multiple contact-dependent and cytokine-mediated signals derived from myeloid-derived DC and monocytes (49). Importantly, reciprocal activation of accessory cell maturation by malaria-activated NK cells was observed in the studies with PBMC and in our studies in mice. These findings indicate that interactions between accessory cells and NK cells during malaria are bi-directional similar to virus and tumor models (30,44,48,49). To address if NK cells are required to induce DC maturation and function in vivo, we treated P. chabaudi AS-infected WT mice with anti-asialo-GM1 antibody and examined DC maturation and Th cell polarization. As we observed previously, infected B6 mice treated with anti-asialo-GM1 antibody mice display a course of infection characterized by a significantly higher peak parasitemia and significantly higher recrudescent parasitemia during the chronic phase of infection (12,44). It should be noted that a similar course of P. chabaudi AS infection is also evident in IL-12p40-/- mice which are deficient in NK cell secretion of IFN-γ during early infection (50). During infection, CD11c+ DC purified from the spleens of NK cell-depleted WT mice expressed lower levels of CD40 and CD86 and produced significantly less IL-12 and more IL-2 in response to iRBC in vitro than DC from isotype control-treated WT mice. Impairment of DC maturation and IL-12 production in infected NK cell-depleted WT mice resulted in inefficient priming of naïve CD4+ T cells for malaria antigen-specific proliferation and IFN-γ secretion compared to DC from isotype control-treated WT mice. Together, these data provide in vivo evidence that DC-NK cell crosstalk is required for DC maturation and function and optimal IFN-γ production by NK cells and CD4+ T cells during P. chabaudi AS infection.
Natural CD4+Foxp3+ regulatory T cells and blood-stage malaria
Recent studies have focused on the role of CD4+ regulatory T cells in regulating adaptive immune responses during Plasmodium infection. In particular, natural CD4+Foxp3+ regulatory T cells have been shown to be important in determining the outcome of infection with various intracellular pathogens, including protozoan parasites (51,52). CD4+Foxp3+ T cells have been shown to inhibit effector T cell responses during infection, resulting in inadequate immune control of the pathogen and persistence of low-level infection. It has also been observed that the potent ability of natural regulatory T cells to modulate effector T cell responses is important in limiting the development of tissue damage during infection with protozoan parasites (51,52).
Studies in individuals with malaria and in mice infected with various rodent Plasmodium species have revealed an important expansion of CD4+CD25+Foxp3+ T cells (53-55). Experimental P. falciparum infection in human volunteers showed that the magnitude of expansion of this T cell population correlates with high parasitemia levels and low pro-inflammatory responses, findings that suggest a possible link between natural regulatory T cells and the clinical outcome of malaria (53). Experiments to determine the impact of CD4+CD25+Foxp3+ T cells in Plasmodium-infected mice by depleting CD25+ T cells have led to inconsistent findings (53-55). Although there are several possible explanations for the inconsistencies, including the concern that antibody depletion of CD25+ T cells may result in the loss of naïve and effector T cells, the results of these studies are consistent with the notion that natural regulatory T cells suppress pro-inflammatory responses involved in protective immunity as well as immunopathology during malaria (53-55).
We investigated the role of CD4+Foxp3+ regulatory T cells during P. chabaudi AS infection using several approaches as alternatives to in vivo depletion of CD25+ T cells (56). First, we compared the course and outcome of P. chabaudi AS infection and malaria-specific immune responses in WT mice and transgenic mice generated by Khattri et al (57) that overexpress the Foxp3 transcription factor. Compared to infected WT mice, infected Foxp3 transgenic mice develop fulminant peak parasitemia levels and 100% of these mice succumb to the infection in association with deficient malaria-specific immune responses. Adoptive transfer of regulatory T cell populations purified from Foxp3 transgenic or WT mice to recipient WT mice confirmed that high numbers of these cells compromised immune control of malaria.
It is well established that an intact spleen is required for the development of protective immune responses to malaria infection in mice (58). To determine if regulatory T cells localize to this tissue during P. chabaudi AS infection, we adoptively transferred CD4+CD25+ or CD4+CD25- T cells purified from the spleens of UBI-GFP/B6 reporter mice and analyzed the frequency of CD4+GFP+ T cells in various tissues in recipient WT mice infected with P. chabaudi AS one day later. This experiment demonstrated that GFP+CD4+ T cells preferentially accumulate in the spleen as opposed to the blood or draining lymph nodes (56). We also observed that significantly higher numbers of GFP+CD4+CD25+ compared to GFP+CD4+CD25- T cells accumulate in the spleens of infected recipient mice. Immunohistochemical staining of spleen sections from infected WT mice confirmed that Foxp3+ regulatory T cells accumulate in the spleen during malaria and demonstrated that this T cell population localizes almost exclusively within the T cell areas of the white pulp.
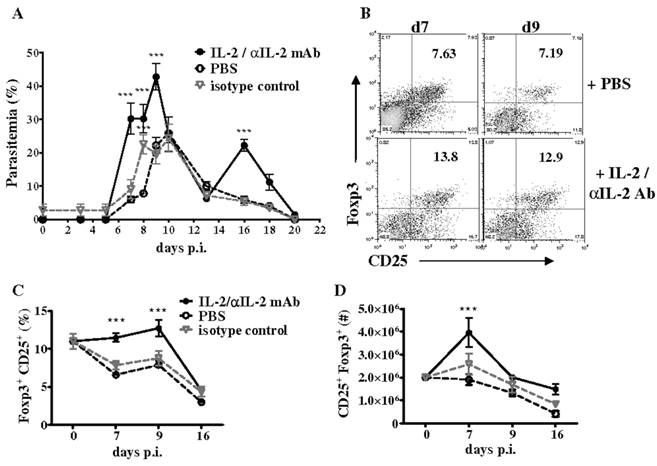
Additionally, we investigated if CD4+CD25+Foxp3+ T cells expand in the spleen of WT mice during P. chabaudi AS infection by analyzing the frequency of regulatory T cells at various times p.i. by flow cytometry. Consistent with previous observations, we observed a significant but transient increase in the numbers of CD4+CD25+Foxp3+ T cells in the spleen of infected WT mice, which was followed by a significant and sustained decrease due to reduced proliferation and apoptosis of CD4+Foxp3+ T cells (56). The T cell growth factor IL-2, a cytokine produced by activated CD4+ T cells, is required for the expansion, survival, and function of natural regulatory T cells and especially to maintain Foxp3 expression by these cells (59). Decreases in IL-2 secretion by effector CD4+ T cells has been shown to be important for contraction of Foxp3+ regulatory T cells in mice infected with Toxoplasma gondii (52). During P. chabaudi AS infection in WT mice, we observed that the kinetics of IL-2 secretion by effector CD4+Foxp3- T cells coincided with changes in CD4+Foxp3+ cells and the differentiation of CD4+T-bet+IFN-γ+ cells required for immune control of blood-stage malaria. As shown in Fig. 4B, the administration of an immune complex consisting of recombinant murine IL-2 and anti-IL-2 monoclonal antibody (clones JES6-1) to infected WT mice increased the severity of P. chabaudi AS infection and promoted the expansion of Foxp3+ regulatory T cells (56,60).
Treatment with IL-2/anti-IL-2 complex increases the severity of P. chabaudi AS infection by promoting the expansion of CD4+Foxp3+ regulatory T cells. Infected B6 mice were treated daily by intraperitoneal injection for 3 days (days 0 to 2 p.i.) with 200 μl IL-2/JES6-1 monoclonal antibody (1:10) complex, isotype control antibody or PBS. (A) Course of parasitemia in treated and control mice. Spleen cells recovered from naïve B6 mice and the three groups of infected mice were analyzed for expression of CD25 and intracellulear Foxp3 in gated CD4+ cells by FACS. Representative dot plots of the (B) frequency of CD4+CD25+Foxp3+ cells in PBS-treated and IL-2/antiIL-2 complex-treated mice on days 7 and 9 p.i., and the (C) frequency and (D) number of Foxp3+CD25+ in gated CD4+ cells in PBS control and complex-treated mice during infection are shown. Data are expressed as mean ± SEM of 5 mice per group from one of two replicate experiments. ***, p <0.001, complex-treated compared to control mice. Originally published in The Journal of Immunology (Ref. 56), Copyright®2011, The American Association of Immunologists, Inc.
Together, our findings demonstrate that a tight balance between CD4+Foxp3+ regulatory T cells and effector CD4+ Th1 cells is necessary to effectively control and eliminate P. chabaudi AS infection and that this balance is dependent in part on IL-2. Although our findings provide convincing evidence that shifting the balance between natural regulatory T cells and effector CD4+ T cells in favor of CD4+Foxp3+ T cells severely compromises immune control of blood-stage malaria, it has not been established if this balance is perturbed in individuals infected with P. falciparum in endemic areas (61). Nor has the contribution of IL-2 to expansion of CD4+Foxp3+ T cells in humans infected with malaria been investigated.
Conclusions
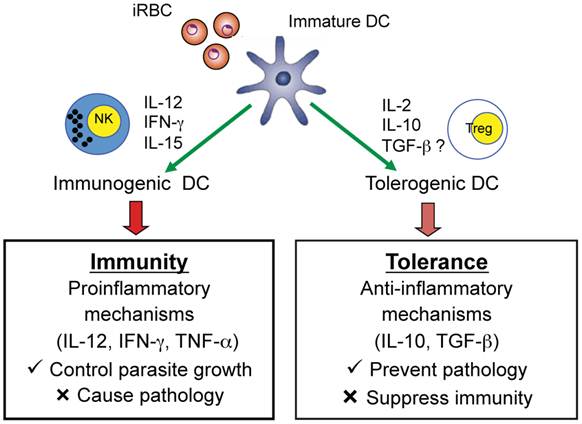
Studies in malaria-infected humans and in mouse models provide strong evidence that Plasmodium parasites modulate DC maturation and function and induce the expansion of CD4+Foxp3+ regulatory T cells. In turn, these regulatory T cells may modulate effector CD4+ T cell responses to malaria infection suppressing protective immune responses but preventing immune-mediated pathology. Accumulating data indicate that immune control of parasite growth is a likely down-stream effect of induction of functionally mature DC that produce IL-12 and induce IFN-γ production by NK cells and CD4+ Th1 cells (Fig. 5). Recent studies also indicate that myeloid-derived CD11c+ DC induce CD4+ T cells to secrete IFN-γ and lead to the development of experimental CM in mice infected with P. berghei ANKA suggesting that malaria-activated DC are also involved in mediating pathology during malaria (62). More extensive studies are required to provide a clearer understanding of the role of DC that are deficient in producing the Th1-promoting cytokine IL-12 and result in a lethal infection. The possibility that such DC have a tolerogenic phenotype and secrete cytokines such as IL-2, IL-10, and TGF-β which may induce regulatory T cells requires further investigation. Indeed, it has been observed that populations of tolerogenic human and murine DC induce or promote CD4+Foxp3+ regulatory T cells via TGF-β- and IL-2-dependent mechanisms (63-66). Increased knowledge concerning DC and CD4+Foxp3+ regulatory T cells may provide strategies for devising novel immunotherapeutic approaches for manipulating the immune response to malaria and tilting the balance between protective and immunopathogenic responses in favor of the host and prevention of severe disease.
Role of DC in inducing adaptive immune responses that mediate protection as well as pathology or regulatory T cells that prevent pathology but suppress protective immunity. An iRBC is shown interacting with an immature DC and the possible consequences on the adaptive immune response to malaria.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR) (MOP-81169) and the Fonds Québecois de la Recherche sur la Nature et les Technologies (FQRNT) (PR12007) to M.M.S. R.I was supported by Studentships from CIHR, the Research Institute of the MUHC, and the Department of Medicine, McGill University. J.M. was supported by Fellowships from the Research Institute of the MUHC and the Fonds de la Recherche en Santé de Québec. F.B. gratefully acknowledges Travel Awards from the Centre for Host-Interactions funded by the FQRNT.
We gratefully acknowledge the excellent technical assistance of MiFong Tam and her assistance in preparing this manuscript.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. World Malaria Report 2010. Who. http://www.who.int./malaria/world_malaria_report_2010/en/index.html
2. Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680-685
3. Good MF. Vaccine-induced immunity to malaria parasites and the need for novel strategies. Trends Parasitol. 2005;21:29-34
4. Matisz CE, Naidu P, Shokoples SE. et al. Post-arrival screening for malaria in asymptomatic refugees using real time PCR. Am J Trop Med Hyg. 2011;84:161-165
5. Ndao M, Bandyayera E, Kokoskin E. et al. Comparison of blood smear, antigen detection, and nested-PCR methods for screening refugees from regions where malaria is endemic after a malaria outbreak in Quebec, Canada. J Clin Microbiol. 2004;42:2694-2700
6. Ndao M, Bandyayera E, Kokoskin E. et al. Malaria “epidemic” in Quebec: diagnosis and response to imported malaria. Can Med Assoc J. 2005;172:46-60
7. Oquike MC, Betson M, Burke M. et al. Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in Africa communities. Int J Parasitol. 2011;41:677-683
8. Baird KJ. Malaria zooneses. Travel Med Infect Dis. 2009;7:269-277
9. Snow RW, Trape J-F, Marsh K. The past, present and future of childhood mortality in Africa. Trends Parasitol. 2001;17:593-597
10. Steketee RW, Nahlen BL, Priase ME, Menendez C. The burden of malaria in pregnancy in malaria-endemic areas. Am J Trop Med Hyg. 2001;64:28-35
11. Roca-Feltrer A, Carneiro I, Smith L. et al. The age patterns of severe malaria syndromes in sub-Saharan Africa across a range of transmission intensities and seasonality setting. Malaria J. 2010;9:282
12. Stevenson MM, Riley EM. Innate immunity to malaria. Nature Rev Immunol. 2004;4:169-180
13. McCall MB, Sauerwein RW. Interferon-γ: central mediator of protective immune responses against the pre-erythrocytic and blood stage of malaria. J Leuk Biol. 2010;88:1131-1143
14. Artavanis-Tsakonas K, Tongren JE, Riley EM. The war between the malaria parasite and the immune system: immunity, immunoregulation and immunopathology. Clin Exp Immunol. 2003;133:145-152
15. Nakazawa S, Culleton R, Yoshimasa M. In vivo and in vitro gametocyte production of Plasmodium falciparum isolates from Northern Thailand. Int J Parasitol. 2011;41:317-323
16. Langhorne J, Buffet P, Galinski M, Good M, Harty J, Leroy D, Mota MM, Pasini E, Renia L, Riley E, Stins M, Duffy P. The relevance of non-human and rodent malaria models for humans. Malaria J. 2011;10:23
17. Langhorne J, Quin SJ, Sanni LA. Mouse models of blood-stage malaria infections: immune responses and cytokines in protection and pathology. Chem Immunol. 2002;80:204-228
18. Fortin A, Stevenson M, Gros P. Susceptibility to malaria as a complex trait: huge pressure from a tiny creature. Hum Mol Genet. 2002;11:2469-2478
19. Chang KH, Stevenson MM. Malarial anaemia: mechanisms and implications of insufficient erythropoiesis during blood-stage malaria. Intl J Parasitol. 2004;24:1501-1516
20. Wykes MN, Good MF. What have we learnt from mouse models for the study of malaria? Eur J Immunol. 2009;39:2004-2007
21. De Souza JB, Hafalla JC, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitol. 2010;137:755-772
22. Banchereau J, Briere F, Davoust J. et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767-811
23. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255-258
24. Rescigno M, Borrow P. The host-pathogen interaction: new themes from dendritic cell biology. Cell. 2001;106:267-270
25. Urban BC, Ing R, Stevenson MM. Early interactions between blood-stage Plasmodium parasites and the immune system. Curr Topics Microbiol Immunol. 2005;297:25-70
26. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291-295
27. Ing R, Segura M, Thawani N, Tam M, Stevenson MM. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J Immunol. 2006;176:441-450
28. Zhu J, Paul WE. CD4 T cells: fates, function, and faults. Blood. 2008;112:1557-1569
29. Coquerell C, Moser M. DC subsets in positive and negative regulation of immunity. Immunol Rev. 2010;234:317-334
30. Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nature Immunol Rev. 2007;7:279291
31. Wykes MN, Good MF. What really happens to dendritic cells during malaria? Nature Revs Microbiol. 2008;6:864-870
32. Urban BC, Ferguson DJ, Pain A. et al. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400:73-77
33. Urban BC, Cordery D, Shafi MJ, Bull PC, Newbold CI, Williams TN, Marsh K. The frequency of BDCA3-positive dendritic cells is increased in the peripheral circulation of Kenyan children with severe malaria. Infect Immun. 2006;74:6700-6706
34. Gonçalves RM, Salmazi KC, Santos BAN, Bastos MS, Rocha SC, Boscardin SB, Silber AM, Kallás EG, Ferreira MU, Scopel KKG. CD4+CD25+Foxp3+ regulatory T cells, dendritic cells, and circulating cytokines in uncomplicated malaria: do different parasite species elicit similar host responses? Infect Immun. 2010;78:4763-4772
35. Bettiol E, Carapau D, Galan-Rodriguez C, Ocaña-Morgner C, Rodriguez A. Dual effect of Plasmodium-infected erythrocytes on dendritic cell maturation. Malaria J. 2010;9:64
36. Voisine C, Mastellic B, Sponaas AM, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol. 2010;40:711-719
37. Wong KA, Rodriguez A. Plasmodium infection and endotoxic shock induce the expansion of regulatory dendritic cells. J Immunol. 2008;180:716-726
38. Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O'Garra A, Langhorne J. Malaria infection changes the ability of splenic dendritic cell populations to stimulated antigen-specific T cells. J Exp Med. 2006;203:1427-1433
39. Stephans R, Albano FR, Quin S, Pascal BJ, Harrison V, Stockinger B, Kioussis D, Weltzien HU, Langhorne J. Malaria-specific transgenic CD4+ T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood. 2005;106:1676-1684
40. Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR. et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8α+ dendritic cells. Proc Natl Acad Sci USA. 2008;105:14509-14514
41. Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, de Koning-Ward TF, Betz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross presentation and antiviral immunity. Nat Immunol. 2006;7:165-172
42. Lundie RJ, Young LJ, Davey GM, Villadangos JA, Carbone FR, Heath WR, Crabb BS. Blood-stage Plasmodium berghei infection leads to short-lived parasite-associated antigen presentation by dendritic cells. Eur J Immunol. 2010;40:1674-1681
43. Perry JA, Olver CS, Burnett RC, Avery AC. The acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol. 2005;174:5921-5925
44. Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun. 2009;77:770-782
45. Leisewitz AL, Rockett KA, Gumede B, Jones M, Urban B, Kwiatkowski DP. Response of the splenic dendritic cell population to malaria infection. Infect Immun. 2004;72:4233-4239
46. Ahvazi BC, Jacobs P, Stevenson M.M. Role of macrophage-derived nitric oxide in suppression of lymphocyte proliferation during blood-stage malaria. J. Leuk Biol. 1995;58:23-31
47. Scorza T, Magez S, Brys L, De Baeseller P. Hemozoin is a key factor in induction of malaria-associated immunosuppression. Parasite Immunol. 1999;21:545-554
48. Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005;5:112-124
49. Newman KC, Korbel DS, Hafalla JC, Riley EM. Cross-talk with myeloid accessory cells regulates human natural killer cell interferon-gamma responses to malaria. PLoS Pathog. 2006;2:e118
50. Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J Immunol. 2002;168:1348-1355
51. Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions. Annu Rev Immunol. 2009;27:551-589
52. Oldenhove G, Bouladoux EA, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Nataraian S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772-786
53. Finney OC, Riley EM, Walther M. Regulatory T cells in malaria: friend or foe? Trends Immunol. 2010;31:63-70
54. Scholzen A, Minigo G, Pleblanski M. Heroes or villains? T regulatory cells in malaria infection. Trends Parasitol. 2010;26:16-25
55. Hansen DS, Schofield L. Natural regulatory T cells in malaria: host or parasite allies? PLoS Pathog. 2010;6:31000771
56. Berretta F, St-Pierre J, Piccirillo CA, Stevenson MM. IL-2 contributes to maintaining a balance between CD4+Foxp3+ regulatory T cells and effector CD4+ T cells required for immune control of blood-stage malaria infection. J Immunol. 2011;186:4862-4871
57. Khattri R, Cox T, Ysayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337-342
58. Engwerda CR, Beattie L, Amante FH. The importance of the spleen in malaria. Trends Parasitol. 2005;21:75-80
59. Brandenburg S, Takahashi T, de la Rosa M, Janke M, G Karsten, Muzzulini T, Orinska Z, Bulfone-Paus S, Scheffold A. IL-2 induces in vivo suppression by CD4+CD25+Foxp3+ regulatory T cells. Eur J Immunol. 2008;38:1643-1653
60. Boyman O, Kovar M, Rubinstein MP, Surth D, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924-1927
61. Walther M, Jeffries D, Finney OC, Njie M, Ebonyi A, Deininger S, Lawrence E, Ngwa-Amambua A, Javasooriva S, Cheeseman IH, Gomez-Escobar N, Okebe J, Conway DJ, Riley EM. Distinct roles for FOXP3+ and FOXP3- CD4+ T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog. 2009;5:e1000364
62. deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KPA, Hill GR, Engwerda CR. Conventional dendritic cells are critical APC required for the induction of experimental cerebral malaria. J Immunol. 2007;178:6033-6037
63. Tateosian NL, Reiteri RM, Amiano NO, Costa MJ, Villalonga X, Guerrieri D, Maffia PC. Neutrophil elastase treated dendritic cells promote the generation of CD4+FOXP3+ regulatory T cells in vitro. Cell Immunol. 2011;269:128-134
64. Yamazaki S, Ivoda T, Tarbell K, Olson K, Velinzon K, Inaba K, Steinman RM. Direct expansion of functional CD25+CD4+ regulatory T cells by antigen-presenting dendritic cells. J Exp Med. 2003;198:235-247
65. Oldenhove G, de Heusch M, Urbain-Vansanten G, Urbain J, Maliszewski C, Leo O, Moser M. CD4+CD25+ regulatory T cells control T helper cell type 1 responses to foreign antigens induced by mature dendritic cells in vivo. J Exp Med. 2003;198:259-266
66. Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM. Dendritic cells are specialized accessory cells along with TGF-β for the differentiation of Foxp3+CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood. 2007;110:4293-4302
Author contact
![]() Corresponding author: Mary M. Stevenson, Ph.D. Research Institute of the McGill University Health Centre Rm. L11-409, 1650 Cedar Avenue, Montreal, Quebec. H3G 1A4 Canada Tel: 514-934-1934 (x44507) Fax: 514-934-8332 mary.stevensonca
Corresponding author: Mary M. Stevenson, Ph.D. Research Institute of the McGill University Health Centre Rm. L11-409, 1650 Cedar Avenue, Montreal, Quebec. H3G 1A4 Canada Tel: 514-934-1934 (x44507) Fax: 514-934-8332 mary.stevensonca