Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Transcriptional regulation...
EZH2 and human breast cancer
Dynamic signaling regulated by...
A BRCA1-EZH2 connection
Genome-wide approaches to...
Moving forward using mouse...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(1):59-65. doi:10.7150/ijbs.8.59 This issue Cite
Review
EZH2 Methyltransferase and H3K27 Methylation in Breast Cancer
Kyung Hyun Yoo ![]() , Lothar Hennighausen
, Lothar Hennighausen
Laboratory of Genetics and Physiology, National Institutes of Health, Bethesda, MD, 20892, USA
Received 2011-8-16; Accepted 2011-9-12; Published 2011-11-18
Abstract
Histone modifications are thought to control the regulation of genetic programs in normal physiology and cancer. Methylation (mono-, di-, and tri-methylation) on histone H3 lysine (K) 27 induces transcriptional repression, and thereby participates in controlling gene expression patterns. Enhancer of zeste (EZH) 2, a methyltransferase and component of the polycomb repressive complex 2 (PRC2), plays an essential role in the epigenetic maintenance of the H3K27me3 repressive chromatin mark. Abnormal EZH2 expression has been associated with various cancers including breast cancer. Here, we discuss the contribution of EZH2 and the PRC2 complex in controlling the H3K27 methylation status and subsequent consequences on genomic instability and the cell cycle in breast cancer cells. We also discuss distinct molecular mechanisms used by EZH2 to suppress BRCA1 functions.
Keywords: EZH2, H3K27 methylation, Breast cancer, BRCA1
Transcriptional regulation modulated by the H3K27 methylation status
Post-translational modifications of the N-terminal tails of core histones, including methylation, acetylation, ubiquitylation and phosphorylation, influence chromatin configuration, which can modulate accessibility of transcription factors and transcriptional activity of nearby genes [1]. Four lysine residues in the N-terminal tail of histone H3, K4, K9, K27 and K36, are primary targets of specific histone methyltransferases and demethylases. Methylated H3K27 is abundant, and three different statues (mono-, di- or tri-methylation) exist. For example, based on quantitative mass spectrometry 50% of H3K27 in embryonic stem (ES) cells is dimethylated, 15% trimethylated and 15% monomethylated [2].
H3K27 di- and tri-methylation are characteristic of PcG (Polycomb group) target genes and are associated with transcriptional repression [3]. PRC1 (Polycomb repressive complex 1) and PCR2 are associated with chromatin condensation. PRC1 catalyzes the monoubiquitylation of histone H2A and PRC2 contributes to the methylation of H3K27 [4]. PRC2 is composed of several proteins, including EED, SUZ12, RbAP46/48 and either EZH1 or EZH2. EZH1 and 2 are characterized by a SET domain, which functions as a histone-lysine N-methyltransferases [5-8]. PRC2 complexes containing EZH1 or EZH2 affect repression of gene expression. While PRC2 complexes containing EZH2 are primarily responsible for H3K27 methylation, PRC2 complexes containing EZH1 can also catalyze H3K27 methylation [9], or compact chromatin through mechanisms other than histone methylation. However, these mechanisms remain to be fully explored (Figure 1A) [10].
Transcriptional regulation through H3K27 methylation. (A) Transcriptional repression regulated by the polycomb repressive complex 2 (PRC2) including EZH1 or EZH2. PRC2 complexes, which include EED, SUZ12, RbAP46/48 and EZH2, catalyze H3K27 di- and tri-methylation. This in turn leads to a more condensed chromatin state and transcriptional repression. PRC2 complexes containing EZH1 can also catalyze H3K27 methylation, or compact chromatin through other mechanism. G9a has H3K27 methyltransferase activity and also affects gene silencing. (B) Transcriptional activation regulated by UTX and JMJD3. UTX and JMJD3 are demethylases that form complexes with MLL, RbBP5 and WDR5. The removal of methyl groups from H3K27me3 leads to transcriptionally active chromatin.
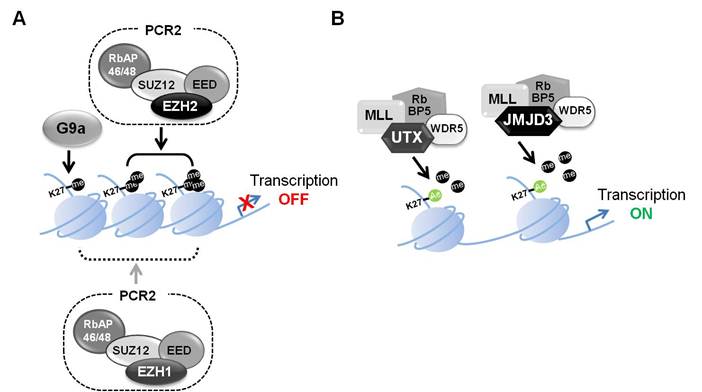
Experiments with cell-specific Ezh2 knockout mice have demonstrated that EZH2 influences proliferation and differentiation of stem cells and progenitor cells in three distinct cell types, adipocytes, keratinocytes, and neurons [11-13]. Experimental evidence from several systems suggests that H3K27 methylation is mainly achieved by EZH2 and to a lesser extent by EZH1. The critical importance of EZH2 is reflected by the fact that Ezh2-null mice die in utero, while Ezh1-null mice have no overt defects, possibly due to compensatory functions of EZH2. However, EZH1 can also compensate to some extent for EZH2 as loss of both EZH1 and 2 evoke more severe consequences than loss of EZH2 alone [14]. In addition, contribution to H3K27 monomethylation by G9a, a well-known H3K9 methyltransferase, was observed in vitro and in vivo [15, 16]. Although no changes in H3K27 di- and tri-methylation were observed between wild type and G9a-/- ES cells, the extent of monomethylation was significantly decreased in G9a -/- ES cells. It is feasible that G9a activity partially compensates for the loss of EZH2. It is possible that G9a introduces a monomethyl group on H3K27 in the absence of EZH2 and that this is sufficient the binding by Embryonic Ectoderm Development (EED), a component of the PRC2 complex, which contains the methyl-lysine histone-binding domain WD40.
JMJD3 (KDM6B, [jumonji-domain containing protein 3]) and UTX (KDM6A [Ubiquitously transcribed tetratricopeptide repeat gene on the X chromosome]) are demethylases acting on H3K27 (Figure 1B). They remove the gene-inactivating H3K27 di-methyl and tri-methyl marks and thereby presumably contribute to the maintenance of gene expression. Abberant of UTX and JMJD3 levels have been associated with cancer and defective differentiation programs [17].
EZH2 and human breast cancer
Excessive EZH2 concentrations have been reported as a marker of aggressive breast cancer [18, 19] and associated with invasion and cancer progression. Chinnaiyan and colleagues tested EZH2 protein levels in 280 breast cancer patients using high-density tissue microarray [20]. EZH2 levels were elevated in patients with invasive breast carcinoma relative to normal or atypical hyperplasia. Notably, increased expression was already observed in ductal carcinoma in situ (DCIS), a precursor of invasive carcinoma [21].
Dynamic signaling regulated by EZH2
Elevated EZH2 concentration has been associated with increased tumor cell proliferation in various cancers including breast, prostate and melanoma [22]. The two-layered regulatory loop includes transcriptional regulation of the EZH2 gene and ensuing consequences of associated H3K27me3 (Figure 2). Three well-defined transcription factors have been invoked in the regulation of EZH2. E2F, a target of the retinoblastoma protein (pRB), plays a critical role in regulating cell cycle progression through activating genes that control entry into the S phase and genes associated with DNA replication [23]. Upon phosphorylation of pRB, activated E2F binds to promoter regions of EZH2 and EED, and controls their expression. However, Helin and colleagues confirmed that the interrupt of EZH2 and EED expression was not enough to increase expression of negative regulators of cell cycle including p14ARF, but positive regulators of cell proliferation, cyclinD1 (CCND1), cyclinE1 (CCNE1), cyclinA2 (CCNA2) and cyclinB1 (CCNB1), significantly decreased in EZH2 lacking cells. They concluded that the PRC complex, including EZH2, is required for the activation or maintenance of the activated state of certain genes in proliferating cells through H3K27 methylation [24].
The biology of EZH2 in breast cancer. EZH2 gene expression is regulated by the HIF, E2F, and RAF/ERK/Elk pathways. Increased levels of EZH2 have been linked to H3K27 trimethylation (H3K27me3) on promoter regions of several genes (RAD51, RUNX3, CDKN1C (p57 KIP2), FOXC1, and CDH1 (E-cadherin)) whose expression is decreased. Reduced levels of RAD51 lead to the activation of Raf1/ERK and β-catenin signaling. Transcription of the CDKN1A gene is under RUNX3 control and lower concentrations of RUNX3 result in lower p21WAF/Cip1 levels, which fail to fully block the cell cycle. The CDKN1C gene encodes the cyclin-dependent kinase inhibitor p57KIP2, a strong inhibitor of cyclinE1 and CDK2 complexes. Expression of the CDKN1C gene is regulated through H3K27me3 of its promoter. Reduction of p57KIP2 leads to accelerated G1-S transition, which has been suggested to support breast cancer progression. Accumulation of H3K27me3 in the promoters of the FOXC1 gene, a fork head family transcription factor used in developmental programs, and the CDH1 gene, that encodes E-cadherin, has been observed in breast cancer cell lines. A decrease of FOXC1 and E-cadherin would be in agreement with enhanced cell invasion and metastasis.
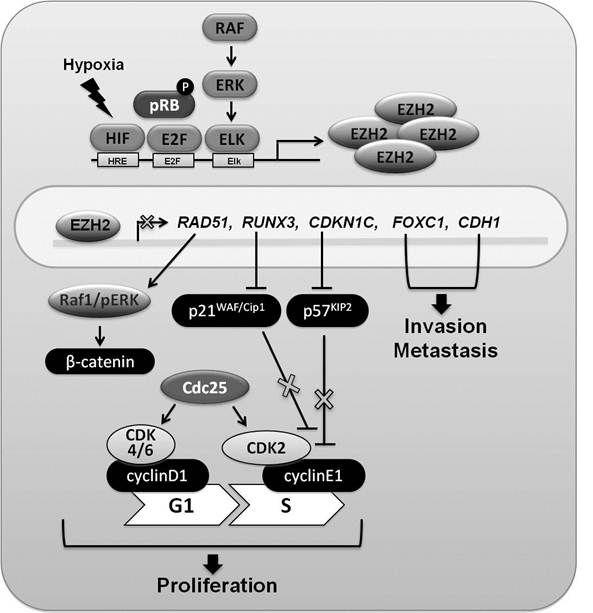
EZH2 expression is also controlled by hypoxia through HIF response elements (HRE) in the EZH2 gene promoter [25]. In hypoxic microenvironment, EZH2 expression is increased and thereby promotes proliferation of breast tumor initiating cells. Lastly, MEK/ERK/Elk pathway can also leads to EZH2 overexpression in ERBB2-overexpressing breast cancer cell lines [26].
A set of genes associated with cell proliferation and invasion is regulated by EZH2. Among them RAD51, RUNX3, and CDKN1C (p57KIP2) regulate cell proliferation, and FOXC1 and CDH1 (E-cadherin) have been linked to metastasis. The runt-related (RUNX) family is associated with normal development and neoplasia and RUNX3 is an established tumor suppressor gene in gastric cancer [46]. Inactivation of RUNX3 expression through DNA hypermethylation has been reported in various cancers, including those of prostate, lung and pancreas. Notably, upon reducing EZH2 expression in the MCF-7 breast cancer cell line, RUNX3 expression was recovered. RUNX3 expression also increased upon treatment with the deacetylase inhibitor TSA [27]. It has been suggested that RUNX3 down-regulation is controlled by H3K27me3 through EZH2 and histone deacetylation through HDAC1. Even though DNA methylation affects repression of RUNX3, H3K27me3 by EZH2 has a critical role in down-regulation of RUNX3. RUNX3 reduction leads to the decrease of the cyclin-dependent kinase inhibitor p21WAF/Cip1 expression, which results in the induction of cell proliferation in breast cancer [28].
FOXC1, a member of the Forkhead box transcription factor family, plays an important role in differentiation. Increased EZH2 influences transcriptional repression of FOXC1 through the accumulation of H3K27me3 and diminished acetylation of H3/H4 in MDA-MB-231 cell lines. Elevated FOXC1 concentrations lead to reduced migration and invasion in vitro and in vivo [29]. The cyclin-dependent kinase (CDK) inhibitor, CDKN1C (p57KIP2) encodes the tumor suppressor p57KIP2 protein. Reduced CDKN1C expression in various breast cancer cell lines was associated with EZH2 overexpression and increased H3K27me3 [30]. Upon treatment with the histone methylation inhibitor 3-deazaneplanocin A (DZNep), recovery of CDKN1C expression was observed suggesting a direct link between H3K27me3 and transcriptional suppression. The transmembrane receptor E-cadherin maintains epithelial cellular adhesion and integrity and it has been proposed that its down-regulation leads to enhanced invasion in cancer. In EZH2-overexpressing breast epithelial cells, reduced CDH1 (E-cadherin) expression correlated with H3K27me3 levels in the promoter. However, elevated expression of EZH2 was not sufficient to increase enzymatic activity for H3K27me3 at the CDH1 promoter, which also required HDAC activity. Increased HDAC activity leads to the removal of acetyl groups from H3K27, a prerequisite for EZH2-controlled histone methylation. Increased EZH2 induces reduction of CDH1 expression, and it leads to metastasis and invasion in breast cancer as well as pancreatic cancer and ovarian cancer [31-33].
EZH2 also appears to regulate breast tumor initiating cells (BTICs) through suppressing RAD51 expression. PHO and GAGA motifs, which are targets for PRC2 complexes, are located in the RAD51 promoter and H3K27me3 coincides with these sites. Reduction of RAD51 affects chromosome breaks, deletion and translocation as well as increase of centrosome number and genome instability through activated RAF/pERK/ ß-catenin signaling [25].
A BRCA1-EZH2 connection
Mutations in BRCA1 are a cause of basal like breast carcinomas that are ER, PR and Her2-neu negative (triple negative) and increased EZH2 concentrations were found mainly in basal type breast carcinomas [34]. Germline Brca1 mutations predispose women to develop breast cancer, and patients have a poor prognosis, in part because BRCA1's role in DNA repair and genomic stability [35]. Even though the majority of basal breast cancers do not carry Brca1 germline mutations, reduced concentration of BRCA1 protein and mRNA levels were reported in these patients [36]. It has been speculated that the underlying cause is promoter methylation or transcriptional repression. However, the mechanisms causing decreased BRCA1 expression in basal like carcinomas remains to be established. Figure 3 proposes mechanisms linking EZH2 overexpression and BRCA1 in basal like breast cancer.
Molecular interplay between EZH2 and BRCA1 in basal like breast cancer. In ER-negative breast cancer cells, elevated EZH2 concentrations block the phosphorylation of BRCA1 (Ser1423). Once the tyrosine phosphatase Cdc25C is phosphorylated, it dephosphorylates the cyclinB-bound Cdc2 (CDK1) and thereby triggers entry into mitosis. Accumulation of Cdc2 and cyclinB1 results in increased cell proliferation. In addition to its canonical function as a H3K27 methyltransferase, EZH2 most likely also has non-canonical functions. EZH2 can phosphorylate Akt-1 (Ser473), but not Akt-2 and -3, and interact with Akt-1. It leads to reduced nuclear retention of BRCA1. In breast cancer cell lines this leads to increased centrosome numbers and genome instability. The EZH2 inhibitor DZNep is more effective on BRCA1-deficient cells, and can thus serve as one arm in a therapeutic regimen.
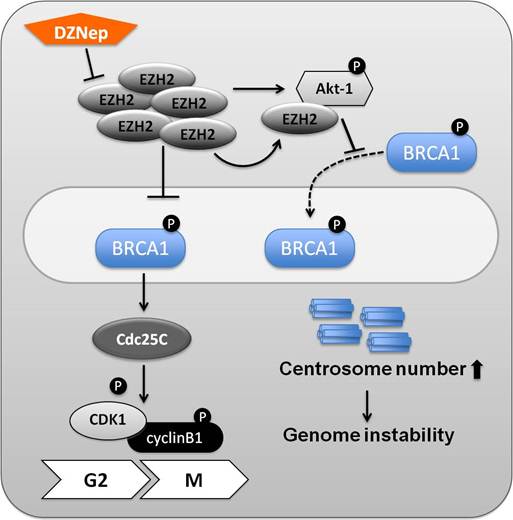
EZH2 is frequently overexpressed in ER-negative breast cancer cell lines, and a reduction of EZH2 results in suppressed cell proliferation. Biological effects of EZH2 knockdowns were observed in vivo and in vitro system using xenograft transplantations. Reduced tumor growth was confirmed in xenograft mouse models injected with EZH2 knockdown cells [37]. EZH2 overexpression inhibits BRCA1 phosphorylation (Ser1423) and thereby promotes an increase of Cdc25C, an essential player for G2/M checkpoint control. Once Cdc25C is activated, it induces the activity of the Cdc2/cyclinB1 complex. In summary, elevated EZH2 concentrations in ER-negative breast cancer cell lines confer increased cell proliferation in part possibly through the inhibition of BRCA1 phosphorylation [37].
BRCA1 is a nuclear protein involved in numerous activities, including DNA repair and estrogen receptor modulation. It has been proposed that Akt-1 (Ser473), but not Akt-2 and 3, are phosphorylated and activated by EZH2 in the EZH2 overexpressing breast cancer cell line CAL51, and nuclear location of BRCA1 protein is suppressed in these cells by a yet to be determined mechanism (Figure 3). This in turn results in aberrant mitoses with extra centrosomes, and genomic instability [38]. Elevated EZH2 concentrations were observed in mouse mammary epithelium from which the Brca1 gene had been deleted. As a result gene expression related to differentiation was suppressed and excessive cell proliferation was promoted. DZNep, a well known as EZH2 inhibitor [47], affects EZH2 concentrations. It was confirmed that BRCA1-deficient cells are more sensitive to DZNep, a potential treatment, when compared with BRCA1-proficient cells [39].
Genome-wide approaches to explore PRC
Genome-wide approaches have been applied to identify genomic localizations and target genes of PRC complexes in human and mouse ES cells. This system has been explored using a combination of chromatin immunoprecipitation (ChIP), ChIP on CHIP and microarray analyses [40-43]. Transcription factors related with developmental regulation, including homeobox protein (HOX), GATA binding protein (GATA), and runt related transcription factor (RUNX) are regulated by the PRC2 complex in human ES cells. [42]. In mouse ES cells, 512 genes regulated by PRC1 and 2 were identified. These include genes encoding the transcription factor families FOX, SOX, GATA and TBX [40]. Moreover, enriched H3K27me3 was found in proximal promoter regions of these genes, which were also derepressed in Suz12- and Eed-null cells [40, 42] supporting the notion that their suppression is dependent on the PRC complex. Moreover, the differentiation of ES cells resulted in the activation of these genes.
ChIP-seq analyses with antibodies against EZH2, Ring1B, H3K4me3 and H3K27me3 have been performed to identify the conservation of chromatin states in orthologous genomic loci in human and mouse ES cells [44]. Bivalent promoters, which carry H3K4me3 and H3K27me3 marks, are highly conserved between mouse and human ES cells. In addition to PRC2, PRC1 binding was identified in the above genes. In general, H3K27me3 retention was most pronounced in regions that were occupied by both PRC1 and 2, while poor retention was detected on sequences occupied by PRC2 only. These findings suggest that PRC2 catalyzes and regulates H3K27 methylation but is not sufficient to maintain the methylation status of respective target genes.
Genome-wide approaches have been used to predict the importance of the PRC2 complex in the regulation of developmental transcription through H3K27 methylation. However, it is not clear what concentration of EZH2 is required to regulate H3K27 methylation, and whether excess EZH2 has other functions besides its methyltransferase activity.
Moving forward using mouse genetics
The suggestion that excess EZH2, and thereby altered histone modifications, leads to a changed epigenetic genomic landscape, and thereby contributes to cancer progression, is mainly based on correlative studies and cell culture investigations. Although biochemical studies are capable of linking elevated EZH2 concentrations with altered genetic programs in cancer, clear genetic evidence is scarce. It will be necessary to conduct decisive genetic experiments in mice to link EZH2 to the H3K27 methylation status and subsequent genetic programs in relevant breast cancer models, such as mice carrying mutant Brca1 [45]. It will be necessary to explore breast cancer initiation and progression in relevant mouse models in which H3K27 methylation is disrupted. By ablating the Ezh1 and Ezh2 genes in mammary epithelium the mammary genome will lack H3K27me3 marks and upon deletion of the Utx and Jmjd3 genes a persistent H3K27me3 should be expected. Such experiments, although labor intensive and time-consuming, shall provide definitive answers on the role of H3K27 methylation in development and the etiology of cancer.
Acknowledgements
This work was supported by the Intramural program of the National Institutes of Diabetes, Digestive and Kidney Diseases (NIDDK). Kyung Hyun Yoo is a recipient of a Korea NRF fellowship (NRF-2011-357-C00087).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Aagaard-Tillery KM, Suter MA, Harris A. et al. Epigenetics and reproduction and the developmental origins of health and disease. Anim Reprod. 2010;7:103-16
2. Peters AH, Kubicek S, Mechtler K. et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577-89
3. Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nature Rev Genet. 2007;8:9-22
4. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-9
5. Cao R, Wang L, Wang H. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039-43
6. Pasini D, Bracken AP, Jensen MR. et al. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061-71
7. Sewalt RGAB, van der Vlag J, Gunster MJ. et al. Characterization of interactions between the mammalian polycomb-group proteins Enx1/EZH2 and EED suggests the existence of different mammalian polycomb-group protein complexes. Mol Cell Biol. 1998;18:3586-95
8. Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57-67
9. Shen X, Liu Y, Hsu YJ. et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491-502
10. Margueron R, Li G, Sarma K. et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503-18
11. Wang L, Jin Q, Lee JE. et al. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc Natl Acad Sci U S A. 2010;107:7317-22
12. Ezhkova E, Pasolli HA, Parker JS. et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136:1122-35
13. Pereira JD, Sansom SN, Smith J. et al. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci U S A. 2010;107:15957-62
14. Ezhkova E, Lien WH, Stokes N. et al. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011;25:485-98
15. Tachibana M, Sugimoto K, Fukushima T. et al. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309-17
16. Wu H, Chen X, Xiong J. et al. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011;21:365-7
17. Hübner MR, Spector DL. Role of H3K27 Demethylases Jmjd3 and UTX in Transcriptional Regulation. Cold Spring Harb Symp Quant Biol. 2011;75:43-9
18. Kunju LP, Cookingham C, Toy KA. et al. EZH2 and ALDH-1 mark breast epithelium at risk for breast cancer development. Mod Pathol. 2011;24:786-93
19. Gong Y, Huo L, Liu P. et al. Polycomb group protein EZH2 is frequently expressed in inflammatory breast cancer and is predictive of worse clinical outcome. Cancer. 2011 [Epub ahead of print]
20. Kleer CG, Cao Q, Varambally S. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606-11
21. Ding L, Erdmann C, Chinnaiyan AM. et al. Identification of EZH2 as a molecular marker for a precancerous state in morphologically normal breast tissues. Cancer Res. 2006;66:4095-9
22. Bachmann IM, Halvorsen OJ, Collett K. et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268-73
23. Ren B, Cam H, Takahashi Y. et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245-56
24. Bracken AP, Pasini D, Capra M. et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323-35
25. Chang CJ, Yang JY, Xia W. et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell. 2011;19:86-100
26. Fujii S, Tokita K, Wada N. et al. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene. 2011 [Epub ahead of print]
27. Fujii S, Ito K, Ito Y. et al. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J Biol Chem. 2008;283:17324-32
28. Chi XZ, Yang JO, Lee KY. et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21WAF1/Cip1 expression in cooperation with transforming growth factor ß-activated SMAD. Mol Cell Biol. 2005;25:8097-107
29. Du J, Li L, Ou Z. et al. FOXC1, a target of polycomb, inhibits metastasis of breast cancer cells. Breast Cancer Res Treat. 2011 [Epub ahead of print]
30. Yang X, Karuturi RK, Sun F. et al. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One. 2009;4:e5011
31. Cao Q, Yu J, Dhanasekaran SM. et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27:7274-84
32. Toll AD, Dasgupta A, Potoczek M. et al. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum Pathol. 2010;41:1205-9
33. Rao ZY, Cai MY, Yang GF. et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis. 2010;31:1576-83
34. Puppe J, Drost R, Liu X. et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11:R63
35. Phillips KA. Immunophenotypic and pathologic differences between BRCA1 and BRCA2 hereditary breast cancers. J Clin Oncol. 2000;18:107S-12S
36. Pejovic T, Koul A, Olsen D. et al. No BRCA1 germline mutation in a family with uterine papillary serous carcinoma: a case report. Eur J Gynaecol Oncol. 2001;22:336-8
37. Gonzalez ME, Li X, Toy K. et al. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene. 2009;28:843-53
38. Gonzalez ME, DuPrie ML, Krueger H. et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360-70
39. Puppe J, Drost R, Liu X. et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11:R63
40. Boyer LA, Plath K, Zeitlinger J. et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349-53
41. Bracken AP, Dietrich N, Pasini D. et al. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123-36
42. Lee TI, Jenner RG, Boyer LA. et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301-13
43. Squazzo SL, O'Geen H, Komashko VM. et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16:890-900
44. Ku M, Koche RP, Rheinbay E. et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242
45. Shukla V, Coumoul X, Cao L. et al. Absence of the full-length breast cancer-associated gene-1 leads to increased expression of insulin-like growth factor signaling axis members. Cancer Res. 2006;66:7151-7
46. Ito K, Lim AC, Salto-Tellez M. et al. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell. 2008;14:226-37
47. Fiskus W, Wang Y, Sreekumar A. et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733-43
Author contact
![]() Corresponding author: Kyung Hyun Yoo, Laboratory of Genetics and Physiology, NIH/NIDDK, Bldg. 8, Rm. 107, Bethesda, MD 20892, USA. ryuknih.gov
Corresponding author: Kyung Hyun Yoo, Laboratory of Genetics and Physiology, NIH/NIDDK, Bldg. 8, Rm. 107, Bethesda, MD 20892, USA. ryuknih.gov