Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(1):108-117. doi:10.7150/ijbs.8.108 This issue Cite
Research Paper
Identification of Ghrelin Receptor Blocker, D-[Lys3] GHRP-6 as a CXCR4 Receptor Antagonist
Kalpesh Patel‡, Vishwa Deep Dixit†, Jun Ho Lee, Jie Wan Kim, Eric M. Schaffer*, Dzung Nguyen#, Dennis D. Taub ![]()
Laboratory of Molecular Biology and Immunology, National Institute on Aging, Intramural Program, NIH, 251 Bayview Boulevard, Baltimore, MD. 21224, USA.
‡ Present Address: The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, MD 21231.
† Present Address: Laboratory of Neuroendocrine-Immunology, Pennington Biomedical Research Center, LSU System, Baton Rouge, LA 70808.
* Present Address: BioSensors Branch, RDCB-DRB-S, Edgewood Chemical Biological Center, APGEA, MD 21010.
# Present Address: BioLegend, 1180 Roselle St., San Diego, CA 92121.
Received 2011-10-27; Accepted 2011-11-10; Published 2011-11-26
Abstract
[D-Lys3]-Growth Hormone Releasing Peptide-6 (DLS) is widely utilized in vivo and in vitro as a selective ghrelin receptor (GHS-R) antagonist. Unexpectedly, we identified that DLS also has the ability to block CXCL12 binding and activity through CXCR4 on T cells and peripheral blood mononuclear cells (PBMCs). Moreover, as CXCR4 has been shown to act as a major co-receptor for HIV-1 entry into CD4 positive host cells, we have also found that DLS partially blocks CXCR4-mediated HIV-1 entry and propagation in activated human PBMCs. These data demonstrate that DLS is not the specific and selective antagonist as thought for GHS-R1a and appears to have additional effects on the CXCR4 chemokine receptor. Our findings also suggest that structural analogues that mimic DLS binding properties may also have properties of blocking HIV infectivity, CXCR4 dependent cancer cell migration and attenuating chemokine-mediated immune cell trafficking in inflammatory disorders.
Keywords: CXCL12, CXCR4, HIV-1, Ghrelin, GHRP-6, GHS-R1a, Inflammation, Cancer
Introduction
Chemokines are small peptides that are known to exert potent regulatory effects on migration and activation of various immune and non hematopoietic cells via ligation to their seven transmembrane G-protein coupled receptors (GPCRs) [1, 2]. The CXC chemokine, CXCL12, also known as SDF-1α, is highly expressed in bone marrow stromal cells and potently stimulates the migration of T cells and monocytes via interactions with its cell surface receptor, CXCR4 receptor [3]. CXCR4 is widely expressed on hematopoietic stem cells, cells of the central nervous system, monocytes and T and B lymphocytes [4]. Interestingly, the chemokine receptors CXCR4 and CCR5 have attracted substantial interest because they form portals of cellular entry for the human immunodeficiency viruses (HIV-1 and HIV-2) and related simian or feline retroviruses[5]. While all the HIV-1 strains require CD4 to enter and infect cells, several laboratory and clinical HIV-1 variants utilize the chemokine receptors CXCR4 (for T-tropic/X4 strain or syncytium-inducing viruses), CCR5 (for M-tropic/R5 strain or non-syncytium-inducing viruses) and/or both receptors (for dual tropic R5X4 strains) for binding and entry. Several HIV-1 chemokine co-receptor antagonists have been recently discovered and are being utilized in human clinical trials [5]. In addition to pathogenesis of HIV, CXCR4 and CCR5 have been implicated in motility, invasion and metastasis of a wide variety of cancer cell types [2, 6]. Given the involvement of CXCR4 and CCR5 in HIV, cancers, stem cell mobilization and inflammation, these receptors have emerged as potential targets for therapeutic manipulation and intervention [5].
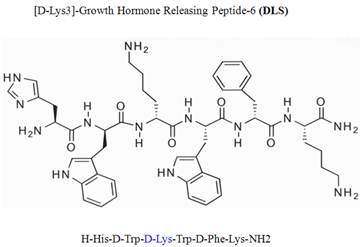
Growth hormone secretagogue receptor (GHS-R) belongs to the seven transmembrane GPCR family and serves as an endogenous ligand for the predominantly stomach-derived hormone ghrelin [7, 8]. Growth hormone releasing peptide-6 (GHRP-6) is one of the earliest synthetic peptidyl GHS-R agonist utilized to study the functions of GHS-R prior to the discovery of the endogenous ligand ghrelin [9]. Modification of GHRP-6 (H-His-D-Trp-Ala-Trp-D-Phe-Lys-NH2) from alanine to D-lysine resulted in a GHS-R antagonist, D-[Lys3]GHRP-6 (H-His-D-Trp-D-Lys-Trp-D-Phe-Lys-NH2). Currently, the D-[Lys3]GHRP-6 (DLS) is utilized in vitro and in vivo studies as a selective GHS-R antagonist [8, 9] (Figure 1).
Chemical structure of D-Lys3-GHRP-6.
However, no studies have yet addressed the specificity and efficacy of this compound on ghrelin-specific effects using human T lymphocytes, T cell lines, PBMCs or other immune cell subsets. However, while utilizing DLS as a control in some chemokine binding and functional experiments, we found that DLS partially inhibited CXCR4 functions and signaling. Given the potent effects of ghrelin on human T cell and monocyte responses [10-13], we have subsequently evaluated the specificity of DLS and its potential interactions with GHS-R1a as well as other immunologically relevant GPCRs of chemokine family. Here, we present evidence that DLS serves as a partial antagonist for the CXCR4 receptor and has an impact on receptor signaling, function and its ability to serve as an HIV-1 co-receptor.
Materials and Methods
Cell culture and Cell lines
GHOST-CXCR4 [14], H9 and Molt-4 cell lines, the CXCR4 receptor antagonist bicyclam JM-2987 (hydrobromide salt of AMD-3100) [15] and HIV1-IIIB [16] were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (GHOST-CXCR4 from Dr. Vineet N. KewalRamani and Dr. Dan R. Littman, H9 from Dr. Robert Gallo, Molt-4 from Dr. Ronald Desrosiers, HIV-IIIB from Dr. Robert Gallo). Leukapheresis packs were prepared from healthy male volunteers under an IRB-approved apheresis protocol between the age of 18 and 45 and the packs were subsequently processed for the isolation of PBMCs and T cells. PBMCs were obtained by Ficoll-Hypaque density centrifugation and T cells were obtained using R&D T-cell Enrichment columns (R&D Systems, Minneapolis, MN).
Intracellular calcium mobilization
Measurement of intracellular calcium release in response to CXCL12 was performed as described previously and as described in the figure legends [17]. T cells were incubated in PBS containing 5 mM Fura-2 acetoxymethyl ester (Molecular Probes) for 30 minutes at room temperature. The cells were subsequently washed and then resuspended at 1 x 106/ml in PBS. A total of 1.5 ml of the cell suspension was placed in a continuously stirring cuvette at room temperature in an LS50B spectrophotometer (Perkin-Elmer, Wellesley, Massachusetts, USA). Labeled T cells were treated with CXCL12 (100ng/ml) with or without DLS (Phoenix pharmaceuticals, Burlingame, California, USA) at various concentrations. Fluorescence was monitored at λex1 = 340 nm, λex2 = 380 nm, and λem = 510 nm. The data are presented as the relative ratio of fluorescence excited at 340 and 380 nm.
Fluorokine ligand binding assays
Fluorokine binding assay was performed as described previously and in the Figure legends [18]. Briefly, biotinylated CXCL12 (Fluorokine; R&D Systems) ligand binding was performed according to the R&D Systems kit protocol, with slight modifications. GHOST-CXCR4 cells were resuspended in PBS at 4 x 106cells/ml. 25μl of cells were treated with 1, 4 or 16 μg of DLS at 37°C for 30 min, then mixed with 20 µl of 2.5 µg/ml biotinylated CXCL12 and incubated at 4°C for 1 h. 20μl fluorescein-conjugated avidin (10 µg/ml) was added to the cells and incubated for an additional 30 min at 4°C. After incubation, cells were washed with 1x RDF-1 buffer (R&D Systems) and then fixed with 2% paraformaldehyde in PBS before being analyzed on a FACScan (BD Biosciences) flow cytometer.
Internalization assay
Molt-4 cells were incubated with various concentrations of DLS and AMD for 30 min at 37ºC. Then, CXCL12 was added at a final concentration of 10nM and incubated for 90 min at 37ºC. Cells were washed with cold PBS and then FITC conjugated anti-human CXCR4 (12G5) antibody (BD Biosciences) added and incubated on ice for 30min. Again cells were washed with PBS and then fixed with 2% paraformaldehyde in PBS before being analyzed on a FACScan. Data is showed as a graph representing mean fluorescent intensity (MFI) of each treatment.
Western blot analysis
Activated T cells were treated with 20µM DLS or 2µM AMD for 30min at 37°C. Then CXCL12 was added at final concentration of 10nM and cells were washed with PBS and harvested and lysed in RIPA buffer supplemented with protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) after 1, 3 and 10minutes of addition of CXCL12. Protein concentrations of cell lysates were determined by Bradford assay. Protein lysates (30μg) were diluted with sample buffer and separated on 4-20% Tris HCl SDS-polyacrylamide gels (Biorad, Hercules, CA) and electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuell). The blots were then probed with mouse anti-phospho-ERK antibody (upper panel), stripped and again probed with rabbit anti-total-ERK antibody (lower panel) (Cell Signaling, Beverly, MA). Immune complexes were visualized by incubation with either an anti-rabbit or an anti-mouse HRP-conjugated secondary antibody (Amersham, Piscataway, NJ). The immunoreactive band was visualized by enhanced chemiluminescence (Perkin-Elmer).
Chemotaxis Assay
Fluorescence-based Transwell (Corning CoStar, Acton, MA) chemotaxis assays were performed to assess cell migration. Primary human T cells were labeled with 5 μg/ml Calcein AM (Molecular Probes) in complete RPMI culture media (cRPMI) for 30 min at 37 °C with or without various concentrations of DLS and AMD. The cells were then resuspended in cRPMI to a concentration of 1 × 107 /ml and 100 ul of Calcein AM labeled cells (106 cells) were added into the Transwell filter (5 μM pore size). cRPMI (0.6 ml) was added to the bottom wells of the 24-well plate with 10nM of CXCL12 with or without various concentrations of DLS and AMD. After 5 h incubation at 37°C, chambers were removed and the plate was read on a Fluoroskan Ascent FL fluorescence plate reader (Thermo Labsystems, Franklin, MA) at λex = 485 nm, and λem = 590 nm. Results are expressed as migration index calculated by subtracting the fluorescence intensity of the “No CXCL12” negative control and comparing the values to the fluorescence intensity (relative number) of cells migrated into the bottom chamber in the “CXCL12 alone” positive control, which is normalized to a value of 1. Fluorescence values were within the linear range of a standard dilution curve.
HIV infection of T cells
HIV1-IIIB was propagated in H9 cells and p24 was measured in sup by ELISA (SAIC, Frederick, USA). PBMCs were activated by 5μg/ml phytohemagglutinin (PHA) and 10units/ml IL-2 for three days. On day three, cells were washed and resuspended at one million per ml in complete RPMI culture (cRPMI) medium. Activated PBMCs were treated with various concentrations of DLS and AMD at 37°C for 30 minutes. Then, HIV1-IIIB was added to a final concentration of p24 of 10ng/ml. Cells were incubated at 37°C for three hours, washed to remove virus and resuspended at 500,000 cells/ml. Antagonist was added to the appropriate cell samples and cells were plated as triplicates in 24 well plates. Supernatants were collected at day 3, 6 and 9 and p24 levels in supernatants were measured by ELISA. Data graphs show % of p24 level over “virus only” controls at day 6 and 9.
Statistical Analysis
Statistical analysis performed using student t test for differences in the means of each parameter examined. For several of the studies shown, only representative data is shown as receptor expression on the T cell surface between donors or on T cell lines or the degree of calcium mobilization or cell migration varied in degree from experiment to experiment; however, ultimately the findings were reproducible and at least three experiments were performed for all studies as well as a number of additional experiments noted as “data not shown”.
Results
DLS inhibits CXCL12 binding to CXCR4
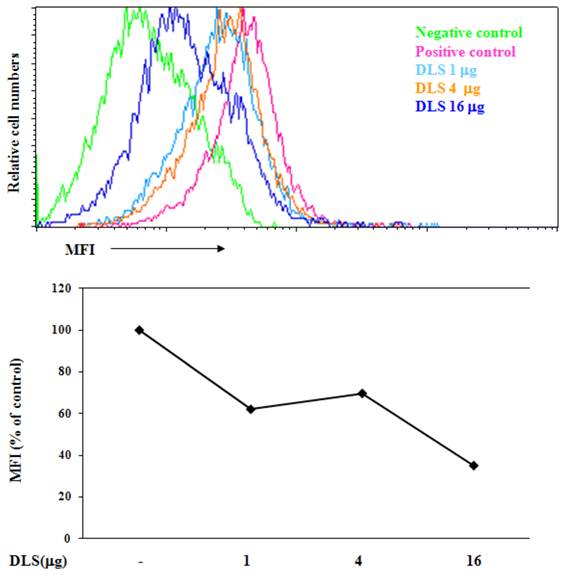
To determine if DLS blocks CXCL12 binding to CXCR4, we utilized a whole cell ligand binding assay with a biotinylated CXCL12, followed by binding of an avidin-fluorescein isothiocyanate (FITC) conjugate, and subsequent examination by using flow cytometric analysis. Upon DLS treatment, the percent maximal binding of CXCL12 in the GHOST-CXCR4 cells was reduced by 20 to 60% depending on the concentration of DLS being examined (Figure 2). These data suggest that DLS may directly block ligand binding to the CXCR4 receptor, although, microgram quantities of DLS was required for the observed inhibition while significantly less AMD3100 (between 500 ng-1 µg), a CXCR4 specific antagonist, was required to mediate similar inhibitory effects (data not shown).
DLS blocks chemokine ligand binding to receptor-positive cells. 25µl of 4 million cells/ml T cells were incubated with or without different amount of DLS (1/4/16μg) for 30 minutes at 37°C. 20 μl of biotinylated-CXCL12 was added and cells were incubated for 1hr at 4°C. After incubation, 20 µl of avidin-fluorescein was added to the tubes and incubated for 30 minutes at 4°C. The cells were subsequently washed with 1xRDF1 buffer, fixed with 2%PFA and analyzed on the FACScan. Histogram shows mean fluorescence intensity (MFI) for each treatment. Data shown is representative of at least four separate experiments.
D-Lys3-GHRP-6 inhibits CXCL12 induced intracellular calcium release from human T cells
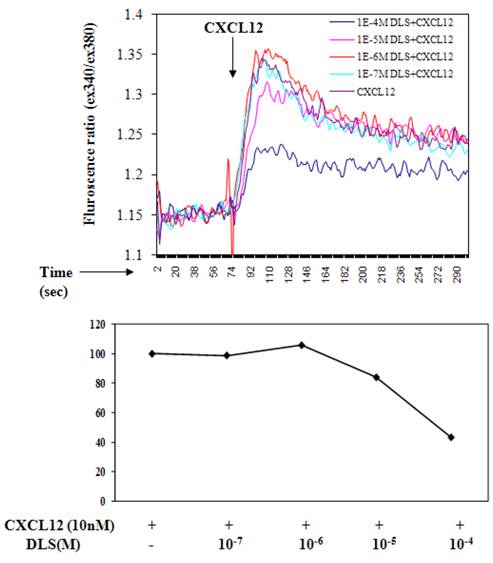
Ligation of seven transmembrane GPCRs typically results in calcium mobilization from the intracellular stores by generation of inositol triphosphate [1, 3]. CXCL12 binding to CXCR4 is known to elicit a potent release of calcium from intracellular sources. The direct effects of DLS on CXCL12- induced calcium mobilization were evaluated on primary human T cells labeled with Fura-2AM. CXCL12 at a final concentration of 10nM caused a significant increase in intracellular calcium and this CXCL12-induced calcium flux was markedly inhibited in a dose dependent fashion by DLS (Figure 3).
D-Lys3-GHRP-6 inhibits CXCL12-induced intracellular calcium release from human T cells. T cells were loaded with Fura-2-AM dye, washed, and treated with different concentrations of DLS for 20 minutes. 1.5 ml cells were analyzed for Ca++ release after addition of CXCL12 (final concentration of 10nM) in a luminescence spectrometer. Each graph shows the 340nm/380nm excitation ratio at 509nm absorption. The data reveals that DLS partially inhibits CXCL12-induced Ca++ release (range of 20-60% inhibition). Data shown is representative of three separate experiments.
DLS inhibits CXCL12-induced T cell chemotaxis
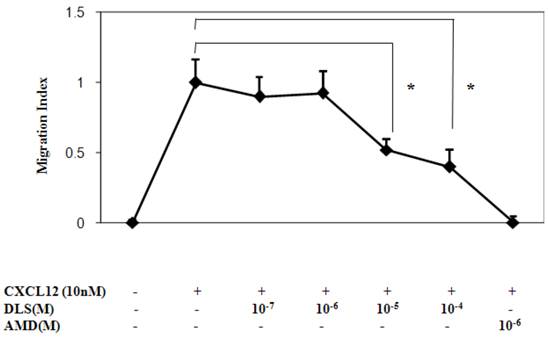
We next determined the functional consequences of inhibition of CXCL12 binding to CXCR4 by DLS in a Transwell chamber migration assay. Primary human T cells, upon optimal CXCL12 treatment (10nM), exhibited robust chemotaxis and pre-treatment with DLS led to marked dose dependent inhibition of CXCL12-induced T cell migration (Figure 4). The CXCR4 specific antagonist, AMD, resulted in almost 100% inhibition of CXCL12-induced migration, while DLS treatment resulted in a 20-60% reduction depending on the DLS concentration being tested. The combination of AMD and DLS failed to have any additive or synergistic effects on blocking CXCL12-mediated migration (data not shown).
DLS inhibits CXCL12-induced chemotaxis. Activated T cells were loaded with Calcein AM dye, treated with different concentration of DLS or the CXCR4 specific antagonist, AMD, for 30 minutes and were then examined for their ability to migrate in response to CXCL12 (10nM) in 24-well Transwell migration chambers. After 5 hours of incubation, the chambers were removed and the plates were read at 485/590 excitation/emission in a Cytofluor fluorimeter. The graph shows Migration Index or the ratio of migration of inhibitor plus CXCL12-induced cells over CXCL12-only induced cell migration. The results demonstrate that DLS inhibits CXCL12 induced activated T cell chemotaxis. Data shown are mean ±SEM and plotted as summary of three experiments, each done with three replicates. (* p<0.05)
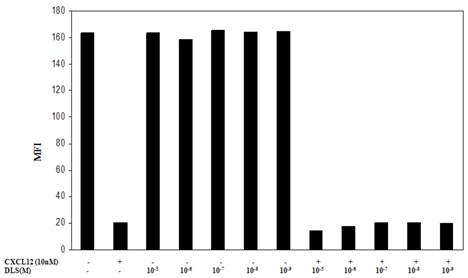
One possible mechanism for this inhibition of chemokine binding and signaling is the possible effects of DLS on CXCR4 internalization. To address this possibility, the Molt-4 T cell line was utilized, as it expresses high surface levels of CXCR4, and treated with different concentrations of DLS for 30 minutes after which the cells were treated with or without CXCL12 (10nM) for 90min to facilitate receptor internalization. The cells were then harvested, stained with anti-CXCR4-PE antibody, fixed and then run on a FACScan. The results (Figure 5) demonstrate that DLS by itself failed to induce CXCR4 internalization and does not interfere with CXCL12-induced CXCR4 internalization. AMD alone or in combination with DLS also failed to mediate CXCR4 internalization alone or interfere with CXCL12-induced CXCR4 internalization (data not shown).
DLS does not influence CXCL12-induced CXCR4 internalization. Molt-4 cells were treated with different concentrations of DLS for 30 minutes, then treated with or without CXCL12 (10nM final concentration) for 90min. The cells were then harvested in cold FACS buffer and stained with anti-CXCR4-PE (12G5) antibody for 45 minutes at 4°C. The stained cells were then washed and fixed with 2%PFA and samples acquired in FACScan. The graph shows average of two replicate's mean fluorescence intensity (MFI) for each cell treatment. The results demonstrate that DLS and AMD (data not shown) do not internalize CXCR4 by itself and do not interfere with CXCL12-induced CXCR4 internalization.
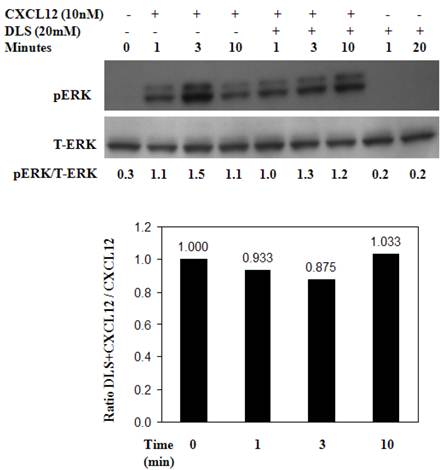
DLS inhibits CXCL12-mediated signaling in activated T cells
Activated human T cells were pretreated with either DLS (20μM) for 30 minutes at 37°C. CXCL12 was subsequently added and the cells were harvested and examined for phosphorylated ERK at the indicated times. These results (Figure 6) reveal that DLS very modestly blocks ERK activation in response to CXCL12 treatment in human T cells. Similar experiments were performed using GHOST-CXCR4 cell line with similar results (data not shown). Moreover, in separate control experiments, AMD3100 blocked ERK activation by 80% after CXCL12 treatment in activated human T cells at a 10-6M dose and combinations of AMD and DLS failed to mediate any additional inhibition (data not shown).
DLS inhibits CXCL12-mediated signaling in activated T cells. Approximately 10 million activated T cells per 0.5ml were utilized per treatment. DLS (20μM) was added to respective tubes and then incubated for 30 minutes at 37°C. CXCL12 (10nM) was subsequently added to the indicated tubes. The cells were then harvested in RIPA after washing with cold PBS at the indicated times. 30μg of protein per sample were run on a 4-20% Bis-Tris gel, transferred and probed with mouse anti-phospho-ERK antibody (upper panel) stripped and again probed with rabbit anti-total ERK antibody (lower panel). The ratio of phospho vs. total ERK is shown below the top panel and the bottom graph shows levels of pERK as compared to only CXCL12 control over various times following CXCL12 addition. Data shown here is of one experiment from total of three separate experiments.
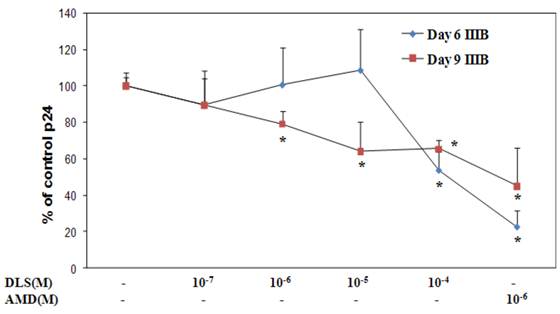
DLS decreases HIV-1 propagation in vitro in activated PBMCs cells
Given the ability of DLS to partially abrogate chemokine binding and signaling, we sought to determine if DLS could exhibit any inhibitory effects in HIV-1 infectivity in human PBMCs. Activated PBMCs were treated with DLS or the CXCR4 receptor antagonist, AMD3100, for 30 minutes at 37°C. HIV1-IIIB (CXCR4 tropic) was added to the cultures at 37°C for 3 hours. The non-bound virus was washed off the treated cells and the cells were cultured for 3, 6 or 9 days to examine HIV-1 viral output by the infected cells. Supernatants were collected from each well at day 3, 6 and 9 and p24 levels were measured using ELISA assays. The results reveal that DLS inhibits X4 tropic HIV-1 propagation in PBMCs (Figure 7).
DLS decreases HIV1-IIIB propagation in vitro in activated PBMCs. Activated PBMCs were washed and treated with DLS or AMD3100 for 30 min at 37°C. 10ng (p24) of HIV1-IIIB (CXCR4 tropic) was added per 106 cells/ml, and then the cells were incubated at 37°C for 3hrs. Cells were washed to remove virus and resuspended to a concentration of 500,000 cells/ml. Antagonists were added to appropriate tubes and cells were plated in triplicate in 24-well plates and incubated at 37°C. Supernatants were collected from each well at day 3 (data not shown), 6 and 9 and p24 levels were measured using ELISA. The graphs show % of p24 level over virus only controls at day 6 & 9. The results reveal that DLS inhibits X4 tropic HIV1-IIIB propagation in PBMCs. Data shown are mean ±SEM and plotted as summary of three experiments, each performed using different donors with three replicates in all cultures.(* p<0.05)
Discussion
Migration of immune cells to sites of inflammation is a multistep process mediated largely by interactions of various chemokines to their G protein linked seven transmembrane receptors [1, 3]. CXCR4 is a chemokine receptor critical for cellular migration and is used in association with CD4 by human immunodeficiency virus (HIV) to enter its target cells. These co-receptors are important determinants of viral tropism, pathogenesis and virulence and are widely believed to be important drug targets to prevent HIV infections [5]. Currently, AMD3100 a selective CXCR4 inhibitor has been successfully utilized to block CXCR4 mediated HIV viral entry [19] and also blocks glioma cell invasion [6] and metastasis of breast [20] and pancreatic carcinoma [21] and decreases allergy and collagen induced arthritis [22]. In addition CXCR4 antagonists have recently emerged as a potential candidate drugs to increase hematopoietic stem cell (HSC) mobilization from bone marrow [23]. Typically, a high concentration of CXCL12, the natural CXCR4 ligand in bone marrow, is critical in retention of HSCs in bone marrow. The AMD3100's Phase II and planned Phase III clinical program is designed to evaluate the ability of AMD3100 to increase stem cells available for transplant in cancer patients.
The synthetic peptidyl compound, D-[Lys3]GHRP-6 (H-His-D-Trp-D-Lys-Trp-D-Phe-Lys-NH2), is currently believed to be a selective antagonist of GHS-R. However, no studies exist on the potential interaction of this compound with other clinically relevant GPCRs of chemokine family. We have discovered that DLS can also modestly inhibit the binding and activity of CXCR4 receptor in human T cells. While DLS may be blocking or hindering CXCL12 binding to the ligand binding site of the CXCR4 surface receptor, it is also feasible that DLS may also act indirectly by activating intracellular signaling and/or phosphorylation events that alters CXCR4 conformation and thus reducing its affinity for CXCL12. We have previously demonstrated that the conformation of CXCR4 can influence ligand binding in T cells as well as HIV-1 infectivity [18]. Given the modest inhibition of calcium mobilization and ERK activation, DLS appears only to be a partial receptor antagonist to CXCR4 and may be blocking ligand binding by non-traditional mechanisms. Nonetheless, these data do support that DLS may not be the exclusive GHS-R1a antagonist that has been reported in the literature.
DLS has also been utilized experimentally in rodent models without any adverse side effects and repeated administration have been found to reduce body weight in obese mice and improve their glycemic control and insulin resistance, all presumably through it's blocking of acylated ghrelin binding to the GHS-R [24]. Furthermore, DLS reduced the size of abdominal fat pads without affecting the muscle mass in these mice. There is accumulating clinical evidence linking currently used HIV inhibitors to the pathogenesis of insulin resistance, dyslipidemia, lipodystrophy and atherosclerosis in AIDS patients [25]. Thus, DLS along with its potential HIV inhibitory properties may attenuate, alone or in combination with other therapeutics, the metabolic effects associated with HAART therapy in AIDS patients. Moreover, DLS does not affect food intake in the fed state when circulating ghrelin levels are low, thus permitting its potential use post-prandially. Our findings of DLS as a modest CXCR4 inhibitor thus suggests that further development of the DLS peptide may lead to effective classes of anti-cancer and anti-HIV agents. However, in its current form and potency, our data with DLS does not suggest a clinical role for this compound.
Therapy for HIV-1 infected subjects includes reverse transcriptase and protease inhibitors. These inhibitors have achieved sustained suppression of viral replication in HIV-1-infected individuals, although the HIV-1 virus may develop resistance to one or more agents. These inhibitors are costly and require long-term use. Despite the efficacy of these agents, there is still dire need to discover new anti-HIV agents to inhibit viral infectivity. CXCR4 and CCR5 are the major co-receptors for HIV-1 entry into the CD4 positive cells. Several of the SNPs and specific deletions in CXCR4 and CCR5 genes have been shown to result in a resistance to or slower progression of HIV-1 infection without any substantial immune (functional) defect in humans. Thus, it would appear that chemokine receptors, more specifically CXCR4 and CCR5, are attractive targets to block HIV-1 binding, fusion and infectivity. AMD3100 has been found to be a potent CXCR4 [15] blocking agent. However, these antagonists have had little success in clinical trials. Recent work by Won-Tak Choi and colleagues [26] have revealed that D-amino acid (aa) modified CXCL12 demonstrates some specific gp120 blocking activity compared to an unmodified peptide. As we are using DLS in these studies (a six amino acid peptide that is comprised of three D-aa), we hypothesize that the D-aa in DLS may be contributing to the decreased propagation of HIV-1 in vitro through its interaction with chemokine receptors. We are currently testing new sets of peptides with additional D-aa as well as D-aa-free peptides to assess the potential role of the D forms in our observed activities.
Acknowledgements
We would like to thank Mr. Gary Collins for his assistance in the completion of this work. This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol. 2004;4:360-370
2. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540-550
3. Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58-71
4. Cascieri MA, Springer MS. The chemokine/chemokine-receptor family: potential and progress for therapeutic intervention. Curr Opin Chem Biol. 2000;4:420-427
5. Castagna A, Biswas P, Beretta A, Lazzarin A. The appealing story of HIV entry inhibitors: from discovery of biological mechanisms to drug development. Drugs. 2005;65:879-904
6. Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100:13513-13518
7. Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J. et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974-977
8. Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495-522
9. Smith RG. Development of growth hormone secretagogues. Endocr Rev. 2005;26:346-360
10. Taub DD. Novel Connections between the Neuroendocrine and Immune Systems: The Ghrelin Immunoregulatory Network. Vit Horm. 2008;77:325-346
11. Redelman D, Welniak L, Taub DD, Murphy WJ. Neuroendocrine hormones such as growth hormone (GH) and prolactin (PRL) are integral members of the immunological cytokine network. Cell Immunol. 2008;252(1-2):111-121
12. Patel K, Taub DD. Role of neuropeptides, hormones, and growth factors in regulating thymopoiesis in middle to old age. F1000 Biology Reports. 2009;1:42
13. Taub DD, Murphy WJ, Longo DL. Rejuvenation of the Aging Thymus: GH- and Ghrelin-Signaling Pathways. Curr Opin Pharm. 2010;10(4):408-424.13
14. Morner A, Bjorndal A, Albert J, Kewalramani VN, Littman DR, Inoue R, Thorstensson R, Fenyo EM, Bjorling E. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J Virol. 1999;73:2343-2349
15. Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, Bridger G, Henson GW. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44:1667-1673
16. Popovic M, Read-Connole E, Gallo RC. T4 positive human neoplastic cell lines susceptible to and permissive for HTLV-III. Lancet. 1984;2:1472-1473
17. Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW Jr, Taub DD. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57-66
18. Nguyen DH, Taub D. CXCR4 function requires membrane cholesterol: implications for HIV infection. J Immunol. 2002;168:4121-4126
19. De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581-587
20. Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604-8612
21. Marchesi F, Monti P, Leone BE, Zerbi A, Vecchi A, Piemonti L, Mantovani A, Allavena P. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004;64:8420-8427
22. Lenoir M, Djerdjouri B, Perianin A. Stroma cell-derived factor 1alpha mediates desensitization of human neutrophil respiratory burst in synovial fluid from rheumatoid arthritic patients. J Immunol. 2004;172:7136-7143
23. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB. et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307-1318
24. Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, Kasuga M. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut. 2003;52:947-952
25. Kino T, Chrousos GP. AIDS-related insulin resistance and lipodystrophy syndrome. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:111-117
26. Choi WT, Tian S, Dong CZ, Kumar S, Liu D, Madani N, An J, Sodroski JG, Huang Z. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J Virol. 2005;79:15398-15404
Author contact
![]() Corresponding author: Dennis D. Taub, PhD, Clinical Immunology Section, Laboratory of Molecular Biology and Immunology, National Institute on Aging-Intramural Research Program, NIH, 251 Bayview Boulevard, Baltimore, MD, 21224. Phone: (410) 558-8428; Fax: (410) 558-8284; E mail: Taubdnia.nih.gov
Corresponding author: Dennis D. Taub, PhD, Clinical Immunology Section, Laboratory of Molecular Biology and Immunology, National Institute on Aging-Intramural Research Program, NIH, 251 Bayview Boulevard, Baltimore, MD, 21224. Phone: (410) 558-8428; Fax: (410) 558-8284; E mail: Taubdnia.nih.gov