Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(5):640-649. doi:10.7150/ijbs.4096 This issue Cite
Research Paper
Characterization of the Complete Mitochondrial Genome Sequence of Spirometra erinaceieuropaei (Cestoda: Diphyllobothriidae) from China
Guo-Hua Liu1,2, Chun Li2,3, Jia-Yuan Li2,4, Dong-Hui Zhou2, Rong-Chuan Xiong5, Rui-Qing Lin4, Feng-Cai Zou6,2 ![]() , Xing-Quan Zhu2,1,6
, Xing-Quan Zhu2,1,6 ![]()
1. College of Veterinary Medicine, Hunan Agricultural University, Changsha, Hunan Province 410128, China;
2. State Key Laboratory of Veterinary Etiological Biology, Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, Gansu Province 730046, China;
3. Sichuan Center for Animal Disease Control and Prevention, Chengdu, Sichuan Province 610041, China;
4. College of Veterinary Medicine, South China Agricultural University, Guangzhou, Guangdong Province 510642, China;
5. Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China;
6. College of Animal Science and Technology, Yunnan Agricultural University, Kunming, Yunnan Province 650201, China.
Received 2012-1-15; Accepted 2012-4-20; Published 2012-4-27
Abstract
Sparganosis, caused by the plerocercoid larvae of members of the genus Spirometra, can cause significant public health problem and considerable economic losses. In the present study, the complete mitochondrial DNA (mtDNA) sequence of Spirometra erinaceieuropaei from China was determined, characterized and compared with that of S. erinaceieuropaei from Japan. The gene arrangement in the mt genome sequences of S. erinaceieuropaei from China and Japan is identical. The identity of the mt genomes was 99.1% between S. erinaceieuropaei from China and Japan, and the complete mtDNA sequence of S. erinaceieuropaei from China is slightly shorter (2 bp) than that from Japan. Phylogenetic analysis of S. erinaceieuropaei with other representative cestodes using two different computational algorithms [Bayesian inference (BI) and maximum likelihood (ML)] based on concatenated amino acid sequences of 12 protein-coding genes, revealed that S. erinaceieuropaei is closely related to Diphyllobothrium spp., supporting classification based on morphological features. The present study determined the complete mtDNA sequences of S. erinaceieuropaei from China that provides novel genetic markers for studying the population genetics and molecular epidemiology of S. erinaceieuropaei in humans and animals.
Keywords: Spirometra erinaceieuropaei, sparganosis, mitochondrial genome, mitochondrial DNA, phylogenetic analyses.
Introduction
Cestodes (Cestoda, Platyhelminthes) are a highly diversified group of parasites, and the majority of cestodes can cause serious diseases in humans and animals globally, such as dipylidiasis, hydatidosis, cysticercosis, diphyllobothriasis and sparganosis [1]. Human sparganosis is a neglected parasitic disease caused by the plerocercoid larvae (spargana) of members of the genus Spirometra [2]. Spirometra erinaceieuropaei and S. mansonoides are responsible for sparganosis in Asia and Americas, respectively [3].
Mitochondrial DNA (mtDNA) sequences have provided genetic markers for addressing questions in systematics and population genetics of Spirometra cestodes. A recent study showed that mtDNA cox1 sequences are useful genetic markers for the identification of species within Spirometra [4]. Employing mtDNA cox1 gene sequences, a previous study demonstrated that S. erinaceieuropaei and S. proliferum are distinct species [5]. Although the single gene or single region of mtDNA have been considered as useful molecular markers, the complete mt genome sequences have significantly improved the power and resolution of phylogenetic analysis compared with single gene or single region of mtDNA [6-8]. Additionally, the complete mt genome sequences allow for more accurate resolution of taxonomic relationships even at deep levels than single gene or single region of mtDNA [9].
Metazoan mitochondrial (mt) genomes contain 36-37 genes, including 12-13 protein-coding genes (PCGs), two ribosomal RNAs (rRNA) genes and 22 transfer RNAs (tRNA) genes necessary for translation of the proteins encoded by mt DNA (mtDNA) [10]. mtDNA sequences have been widely used as genetic markers not only for studies of population or ecological genetics, but also for phylogenetic and evolutionary analyses due to its maternal inheritance, rapid evolutionary rate, lack of recombination, and relatively conserved genome structures [11-15]. Although Cestoda is a large animal class, to date the complete mt genomes of only 19 species have been sequenced and deposited in GenBank. In particular, in the order Pseudophyllidea, the complete mt genomes of only three species have been reported, namely Diphyllobothrium latum and D. nihonkaiense [4], and S. erinaceieuropaei from Japan (GenBank accession No. NC_011037). The mt genome of S. erinaceieuropaei from a snake in Japan has been sequenced, but S. erinaceieuropaei isolated from different hosts and different geographic origins may have some differences because a previous study indicated that mt genomes for N. americanus from two endemic regions showed substantial sequence variation [16]. Moreover, the mt genome of S. erinaceieuropaei from Japan has not been characterized yet.
The objectives of the present study were: (i) to characterise the mt genome of S. erinaceieuropaei from China, (ii) to compare this mt genome with that of S. erinaceieuropaei from snake in Japan, (iii) to test the hypothesis that S. erinaceieuropaei from different hosts and different geographic origins have significant nucleotide sequence differences in their mt genomes, and (iv) to re-construct the phylogenetic relationships of the class Cestoda using mtDNA sequence data.
Materials and Methods
Parasites and DNA extraction
An adult cestode representing S. erinaceieuropaei was obtained from the small intestine of an infected farmed dog at a slaughterhouse in Zhanjiang, Guangdong province, China. The cestode was washed in physiological saline, identified preliminarily to species based on host preference, morphological characters and predilection sites [17], fixed in 70% (v/v) ethanol and stored at -20°C until use.
Total genomic DNA was isolated form this tapeworm using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (Wizard® SV Genomic DNA Purification System, Promega). The identity of this specimen was confirmed as S. erinaceieuropaei by polymerase chain reaction (PCR) amplification and subsequent sequencing of the ITS-1 and ITS-2 rDNA using primers reported previously [18].
Amplification and sequencing of partial cox3, nad4, cox1 and rrnS
Initially, fragments of cox3, nad4 and rrnS genes were amplified individually from S. erinaceieuropaei (China isolate) by PCR with primers (Table 1) designed based on mtDNA sequences well-conserved in S. erinaceieuropaei (Japan isolate, GenBank accession No. NC_011037) (Table 2) by using Program Primer Primer 5.0 (PREMIER Biosoft International). Primers JB3 and JB4.5 [19] were utilized to amplify the partial cox1 gene of S. erinaceieuropaei (China isolate) (Table 1). PCR reactions (25 mL) were performed in 10 mM Tris-HCl (pH 8.4), 50 mM KCl, 4 mM MgCl2, 200 mM each of dNTP, 50 pmol of each primer and 2 U Taq polymerase (Takara) in a thermocycler (Biometra) under the following conditions: after an initial denaturation at 94 °C for 5 min, then 36 cycles at 94 °C for 30 s (denaturation), 55 °C (for cox1) or 50 °C (for cox3) or 54 °C (for nad4 and rrnS) for 30 s (annealing), 72 °C for 30 s (extension), followed by 72 °C for 5 min (final extension). One microliter (5-10 ng) of genomic DNA was added to each PCR reaction. Each amplicon (5 µL) was examined by (1%) agarose gel electrophoresis.
Sequences of primers used to amplify PCR fragments from Spirometra erinaceieuropaei.
| Name of primer | Sequence (5' to 3') |
|---|---|
| Short-PCR | |
| cox3-F | TGCATTTTGGTTATTTATTCTTAG |
| cox3-R | ACGATAGGCCCCGGCTGAAG |
| nad4-F | GGTTCCGTTATTTCCATTTCA |
| nad4-R | TACTACCCTCAAAAGACTCACCACG |
| JB3 | TTTTTTGGGCATCCTGAGGTTTAT |
| JB4.5 | TAAAGAAAGAACATAATGAAAATG |
| rrnS-F | ATTGCGTAGTGAGGGGGATTA |
| rrnS-R | CTGGGGCTACCTTGTTACGACTTA |
| Long-PCR | |
| cox3-F | TGCATTTTGGTTATTTATTCTTAG |
| nad4-R | TACTACCCTCAAAAGACTCACCACG |
| SEND41F | CTGCGAGGTATTTGTGCTGTCTTCTTCA |
| SECO11R | CACAGGCTCACGCAACGAAACACGACTA |
| JB3 | TTTTTTGGGCATCCTGAGGTTTAT |
| rrnS-R | CTGGGGCTACCTTGTTACGACTTA |
| SERNS1F | CCTGTAATGGTTTATGTTTTAGGACTTG |
| SECO31R | CAAAATGTCAATACCAAGTAACTAAAG |
Mitochondrial genome organization of Spirometra erinaceieuropaei from China and Japan.
| Gene/region | Positions and nt size (bp) | Ini/Ter codon | Anticodons | ||
|---|---|---|---|---|---|
| SEC | SEJ | SEC | SEJ | ||
| tRNA-Tyr (Y) | 1-68 (68) | 1-68 (68) | GTA | ||
| Non-coding region (NC) | 69-272 (204) | 69-272 (204) | |||
| tRNA-LeuCUN (L1) | 273-340 (68) | 273-340 (68) | TAG | ||
| tRNA-SerUCN (S2) | 343-407 (65) | 343-407 (65) | TGA | ||
| tRNA-LeuUUR (L2) | 412-476 (65) | 412-476 (65) | TAA | ||
| tRNA-Arg (R) | 492-548 (57) | 492-548 (57) | ACG | ||
| nad5 | 552-2117 (1566) | 552-2120 (1569) | ATG/TAA | ATG/TAA | |
| Non-coding region (NR) | 2118-2291 (174) | 2121-2294 (174) | |||
| tRNA-Gly (G) | 2292-2368 (67) | 2295-2361 (67) | TCC | ||
| cox3 | 2362-3004 (643) | 2365-3007 (643) | GTG/T | GTG/T | |
| tRNA-His (H) | 3005-3073 (69) | 3008-3076 (69) | GTG | ||
| cytb | 3077-4186 (1110) | 3080-4189 (1110) | ATG/TAA | ATG/TAA | |
| nad4L | 4191-4451 (261) | 4194-4454 (261) | ATG/TAG | ATG/TAG | |
| nad4 | 4412-5665 (1254) | 4415-5668 (1254) | ATG/TAG | ATG/TAG | |
| tRNA-Gln (Q) | 5666-5729 (64) | 5669-5732 (64) | TTG | ||
| tRNA-Phe (F) | 5726-5789 (64) | 5729-5792 (64) | GAA | ||
| tRNA-Met (M) | 5786-5853 (68) | 5789-5856 (68) | CAT | ||
| atp6 | 5857-6372 (516) | 5860-6375 (516) | ATG/TAA | ATG/TAA | |
| nad2 | 6380-7252 (873) | 6383-7255 (873) | ATG/TAG | ATG/TAG | |
| tRNA-Val (V) | 7263-7327 (65) | 7266-7330 (65) | TAC | ||
| tRNA-Ala (A) | 7345-7405 (61) | 7348-7408 (61) | TGC | ||
| tRNA-Asp (D) | 7411-7474 (64) | 7414-7477 (64) | GTC | ||
| nad1 | 7475-8365 (891) | 7478-8368 (891) | ATG/TAA | ATG/TAA | |
| tRNA-Asn (N) | 8371-8436 (66) | 8374-8439 (66) | GTT | ||
| tRNA-Pro (P) | 8443-8508 (66) | 8446-8510 (65) | TGG | ||
| tRNA-Ile (I) | 8514-8577 (64) | 8516-8579 (64) | GAT | ||
| tRNA-Lys (K) | 8584-8646 (63) | 8586-8648 (63) | CTT | ||
| nad3 | 8650-8995 (346) | 8652-8997 (346) | ATG/T | ATG/T | |
| tRNA-SerAGN (S1) | 8996-9054 (59) | 8998-9056 (59) | GCT | ||
| tRNA-Trp (W) | 9057-9122 (66) | 9059-9124 (66) | TCA | ||
| cox1 | 9130-10695 (1566) | 9132-10697 (1566) | ATG/TAG | ATG/TAG | |
| tRNA-Thr (T) | 10686-10755 (70) | 10688-10757 (70) | TGT | ||
| rrnL | 10756-11728 (973) | 10758-11730 (973) | |||
| tRNA-Cys (C) | 11729-11793 (65) | 11731-11795 (65) | GCA | ||
| rrnS | 11794-12523 (730) | 11796-12525 (730) | |||
| cox2 | 12524-13093 (570) | 12526-13095 (570) | ATG/TAA | ATG/TAA | |
| tRNA-Glu (E) | 13099-13163 (65) | 13101-13165 (65) | TTC | ||
| nad6 | 13168-13635 (468) | 13170-13637 (468) | ATG/TAA | ATG/TAA | |
SEC: S. erinaceieuropaei (China isolate), SEJ: S. erinaceieuropaei (Japan isolate).
Ini/Ter codons: initiation and termination codons.
Long-PCR amplification and sequencing
The obtained partial cox1, cox3, nad4 and rrnS sequences were compared with the corresponding sequences of S. erinaceieuropaei mt genome (Japan isolate, NC_011037) using the program Clustal X 1.83 [20], and then primers (Table 1) were designed in the conserved regions to amplify the complete mt genome of S. erinaceieuropaei (China isolate). Long-PCR reactions (25 μl) were performed in 2 mM MgCl2, 0.2 mM each of dNTPs, 2.5 μl 10× rTaq buffer, 2.5 μM of each primer, 1.25 U rTaq polymerase (Takara), and 1 μl of DNA sample in a thermocycler (Biometra) under the following conditions: 92 °C for 2 min (initial denaturation), then 92 °C for 10 s (denaturation), 50 °C or 55 °C for 30 s (annealing), and 60 °C for 10 min (extension) for 10 cycles, followed by 92 °C for 10 s, 50 °C or 55 °C for 30 s, and 60 °C for 10 min for 20 cycles, with a cycle elongation of 10 s for each cycle and a final extension at 60 °C for 10 min. Samples not containing DNA (no-DNA controls) were included in each amplification run, and in neither case were amplicons detected in the no-DNA controls (data not shown). Each amplicon (5 µL) was examined by agarose (1%) gel electrophoresis, stained with ethidium bromide and photographed using a gel documentation system (UVItec). PCR products were sent to Sangon Company (Shanghai, China) for sequencing from both directions using a primer walking strategy.
Sequence analyses
Sequences were assembled manually and aligned against the complete mt genome sequence of S. erinaceieuropaei (Japan isolate) using the computer program Clustal X 1.83 to identify gene boundaries. Sequence differences between the S. erinaceieuropaei China and Japan isolates were calculated by pairwise comparisons using the formula D = 1 - (M/L), where M is the number of alignment positions at which the two sequences have a base in common, and L is the total number of alignment positions over which the two sequences are compared [21]. Gene annotation, genome organization, translation initiation, translation termination codons and the boundaries between PCGs of the mt genome sequence of S. erinaceieuropaei (China isolate) were identified based on comparison with the mt genome sequence of S. erinaceieuropaei (Japan isolate) reported previously. The amino acid sequences deduced from open-reading frames were aligned with those of S. erinaceieuropaei (Japan isolate) by using Clustal X 1.83. Based on pairwise comparison, amino acid identity (%) was calculated for homologous genes. For analyzing ribosomal RNA genes, putative secondary structures of 22 tRNA genes were identified using tRNAscan-SE [22], or by recognizing potential secondary structures and anticodon sequences by eye by comparison with that of S. erinaceieuropaei (Japan isolate).
Sliding window analysis of nucleotide variation
To detect variable nucleotide sites, pairwise alignments of the complete genomes including tRNA and all intergenic spacers were performed using the program Clustal X 1.83. The complete alignment of nucleotides of the 4 Pseudophyllidea cestode mtDNAs was used to effect sliding window analyses using DnaSP v5 [23]. A sliding window of 300 bp and steps of 10 bp were used to estimate nucleotide diversity Pi (π) for the complete alignment. Nucleotide diversity for the complete alignment was plotted against mid-point positions of each window, and gene boundaries were indicated.
Phylogenetic analyses
Phylogenetic relationships among 19 cestode species were reconstructed using Ascaris suum (GenBank accession No. HQ704901), an ascaridoid nematode, as the outgroup, based on amino acid sequences of 12 PCGs. Each gene was translated into amino acid sequence using the invertebrate mitochondrial genetic code in MEGA 5.0 [24] , and aligned on the basis of its amino acid sequence, using default settings, and ambiguously aligned regions were excluded using the Gblocks online server [25] (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using the options for a less stringent selection. The final amino acid sequences of the 12 PCGs were then concatenated into single alignments for phylogenetic analyses.
Two inference methods, namely, Bayesian inference (BI) and maximum likelihood (ML) were used for phylogenetic re-constructions. BI analyses were conducted with four independent Markov chains run for 1,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generations in MrBayes 3.1.1 [26]. The first 2,500 trees were omitted as burn-in and the remaining trees were used to calculate Bayesian posterior probabilities (PP). ML analyses were performed using PhyML 3.0 [27], and the JTT+G+F (lnL = −50057.3646, Gamma distribution parameter α = 0.5034) model with its parameter for the concatenated dataset was determined for the ML analysis using MEGA 5.0 based on the Akaike information criterion (AIC). Bootstrap support for ML trees was calculated using 100 bootstrap replicates. Phylograms were drawn using the Tree View program version 1.65 [28].
Results and Discussion
Genome organization and structure
The complete mt genome of S. erinaceieuropaei (China isolate) was amplified in four overlapping long fragments between cox3 and nad4 (approximately 3.1 kb), between nad4 and cox1 (approximately 4.6 kb), between cox1 and rrnS (approximately 2.7 kb) and between rrnS and cox3 (approximately 3.5 kb). The complete mt genome sequence of S. erinaceieuropaei (China isolate) was 13,641 bp in size (Fig. 1), and it has been deposited in the GenBank under the accession number JQ267473. The S. erinaceieuropaei (China isolate) mt genome sequence (13,641 bp) was 2 bp shorter than that of S. erinaceieuropaei (Japan isolate). The slight discrepancy of nucleotide size is likely due to difference in their hosts, developmental stages or geographical locations. The S. erinaceieuropaei China specimen (adult) was collected from a dog, while the S. erinaceieuropaei Japan isolate (larva) was collected from a snake. The mt genome of S. erinaceieuropaei (China isolate) contains 12 PCGs (cox1-3, nad1-6, nad4L, atp6 and cytb), 22 transfer RNA genes, two ribosomal RNA genes and two non-coding regions (Table 2). All genes are transcribed in the same direction as other cestode species sequenced to date. These results indicate that the organization of mt genomes among sequenced cestodes is conserved, which may provide mtDNA evidence for the validity of the Cestoda as a unique animal class.
Arrangement of the mitochondrial genome of Spirometra erinaceieuropaei (China isolate). Gene scaling is only approximate. All genes are coded by the same DNA strand and are transcribed clockwise. All genes have standard nomenclature except for the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the two leucine- and serine-specifying tRNA (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGN and UCN, respectively). ''NR'' refers to a small noncoding region. ''NC'' refers to a large noncoding region.
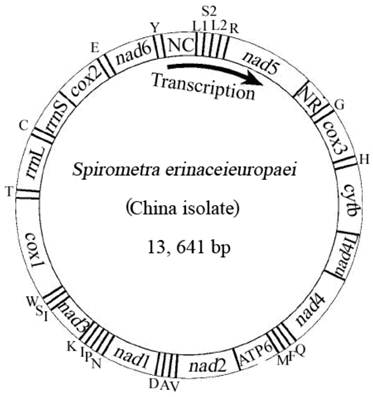
The mt genome sequence of S. erinaceieuropaei (China isolate) has high T content and low C content (T=45.9%, A=20.4%, G=22.6%, C=11.1%), and its A+T content was 66.3%, in accordance with mt genomes of other Pseudophyllidea species sequenced to date [4]. The identity of the nucleotide sequences between mt genomes of S. erinaceieuropaei (China isolate) and S. erinaceieuropaei (Japan isolate) is 99.1%. Interestingly, the mt genomes of S. erinaceieuropaei (China and Japan isolates) have 20 intergenic regions, excluding the non-coding regions (Table 2). Even more noteworthy, the sizes and locations of these intergenic regions of both mt genomes were identical (Table 2). Furthermore, previous studies have shown that the intergenic regions of mt genome of metazoans contain signals for transcription initiation and replication [29].
Protein-coding genes, transfer RNA genes, ribosomal RNA genes and non-coding regions
The boundaries between PCGs of mt genome of S. erinaceieuropaei (China isolate) were determined by aligning their sequences and by identifying translation initiation and termination codons with comparison to those of S. erinaceieuropaei (Japan isolate). The longest genes are cox1 and nad5 genes (Table 2).
A total of 3356 amino acids are encoded in the mt genome of S. erinaceieuropaei (China isolate). All PCGs use ATG or GTG as their initiation codon, which encodes methionine and valine that is consistent with those of S. erinaceieuropaei (Japan isolate). Although ATG is the most common initiation codon, unusual initiation codons such as GTG and GTT were also reported for some cestodes [30, 31]. All genes have complete termination codon, except for cox3 and nad3 genes which uses incomplete termination codon (T) as termination codon. 6 genes (cox2, nad1, nad6, nad5, cytb and atp6) use TAA and 4 genes (cox1, nad2, nad4L and nad4) use TAG as termination codon. Incomplete termination codons (T or TA) as termination codon are also present in other cestodes [4, 32].
The sizes of 22 tRNA genes identified in the mt genome of S. erinaceieuropaei (China isolate) ranged from 57 to 69 nucleotides (nt) with differences in stem and loop sizes of dihydrouridine (D) and TCC arms. Their putative secondary structures (not shown) are similar to those of other Pseudophyllidea cestodes [4], indicating their similar functions.
The rrnL and rrnS genes of S. erinaceieuropaei (China isolate) were identified by comparison with that of S. erinaceieuropaei (Japan isolate). The rrnL is located between tRNA-Thr and tRNA-Cys, and rrnS is located between tRNA-Cys and cox2. The sizes of the rrnL and rrnS genes are 973 bp and 730 bp both for S. erinaceieuropaei (China isolate) and S. erinaceieuropaei (Japan isolate), respectively (Table 2). Sequence identity in the rrnL and rrnS genes are 98.6% and 99.3% between S. erinaceieuropaei (China isolate) and S. erinaceieuropaei (Japan isolate), respectively.
mtDNA sequences of most cestodes contain two non-coding regions with significant size difference. The large non-coding region (NC) in the mt genome of the S. erinaceieuropaei (China isolate) is located between the tRNA-Tyr and tRNA-LeuCUN, its size is 204 bp, and its A+T content is 71.6%, whereas the small non-coding region (NR) in the mt genome of the S. erinaceieuropaei (China isolate) is located between the nad5 and tRNA-Gly, its length is 174 bp, and its A+T content is 63.8%, which are identical to those of S. erinaceieuropaei (Japan isolate).
Moreover, the non-coding regions in S. erinaceieuropaei (both China and Japan isolates) contain continuous A or T base, similar to those of some Taenia species [13]. The non-coding regions are known to be related to the origin of replication in both vertebrates and invertebrates [10].
Levels of variability in mtDNAs among Pseudophyllidea cestodes
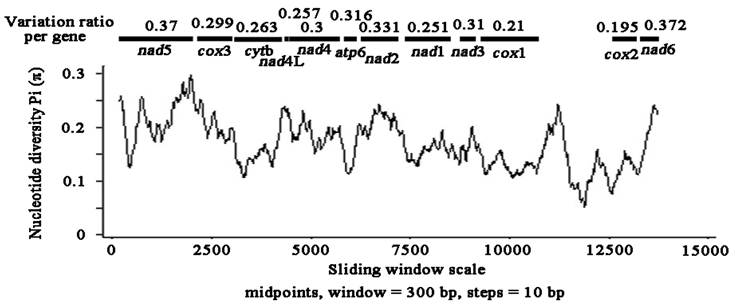
Sliding window analysis of the complete nucleotide alignment of 4 available Pseudophyllidea cestode mtDNAs provided an indication of nucleotide diversity Pi (π) within and between mt genes (Fig. 2). In the curve, the nucleotide variation within and between mt genes among the aligned Pseudophyllidea cestode mt genomes was intuitively displayed for any given window of 300 bp and steps of 10 bp, with the π ranging from 0.055 to 0.297. Coupled with computation of the number of variable positions per unit length of gene, the sliding window indicated that the PCGs with high sequence variability included nad6 (0.372), nad5 (0.37), nad2 (0.331), atp6 (0.316), nad3 (0.31) and nad4 (0.3), while the genes with low sequence variability included cox3 (0.299), cytb (0.263), nad4L (0.257), nad1 (0.251), cox1 (0.21) and cox2 (0.195). In this study, the cox2 and cox1 genes are the most conserved PCGs, and nad6 and nad5 are the least conserved PCGs. Given the lowest and the highest sequence variability, cox2 (0.195) and nad6 (0.372) can be considered as the best markers in future studies of phylogenetics, population genetics and diagnostics of the Pseudophyllidea cestodes. Obviously, these genes are also more variable than the cox1 gene in the present study (0.21 for cox1), not consistent with that of previous studies [33,34]. Currently, mt genes such as cox1 and cytb have been used as targets for PCR-based approaches for the identification and diagnosis of cestodes [35,36]. These results suggest that cox1 gene may not be the most informative or most suitable for all cestode taxa although it has commonly been optioned as molecular markers. Our resultes indicated that sliding window analyses could define genetic markers for population genetics and systematics studies of cestodes.
Sliding window analysis of the alignment of complete mtDNAs of Pseudophyllidea cestodes. The black line shows the value of nucleotide diversity Pi (π) in a sliding window analysis of window size 300 bp with step size 10, and the value is inserted at its mid-point. Gene boundaries are indicated with a variation ratio per gene.
Nucleotide variation in mtDNA sequences between S. erinaceieuropaei from China and Japan
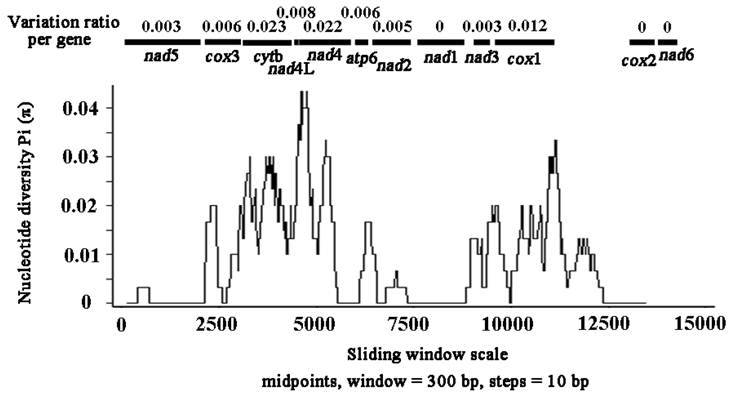
Sliding window analysis of the nucleotide alignment of S. erinaceieuropaei mtDNA from China and Japan provided an indication of nucleotide diversity (π) within or between mt genes (Fig. 3). The plot readily shows the high degree of nucleotide variation within or between genes amongst the aligned S. erinaceieuropaei genomes for any given window of 300 bp (π) ranges from 0 to 0.043). Sliding window analysis indicates that genes with high sequence variability included cytb (0.023), nad4 (0.022) and cox1 (0.012). Genes with no sequence variability include nad1, cox2 and nad6. Based on these results, it seemed that nad1 and cox2 are the most conserved protein-coding genes, and cytb and nad4 show high sequence variability between S. erinaceieuropaei from China and Japan. These results indicated that cytb and nad4 sequences provide alternative new genetic markers for population genetic studies of S. erinaceieuropaei from different hosts and different geographic origins.
Sliding window analysis of the alignment of complete mtDNAs of Spirometra erinaceieuropaei from China and Japan. The black line shows the value of nucleotide diversity Pi (π) in a sliding window analysis of window size 300 bp with step size 10, and the value is inserted at its mid-point. Gene boundaries are indicated with a variation ratio per gene.
Phylogenetic analyses
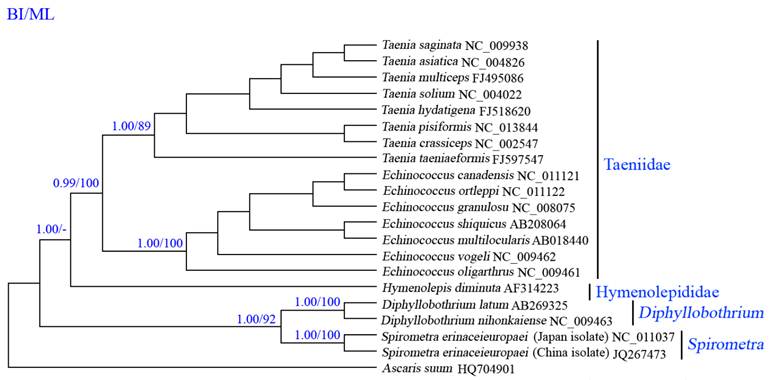
Phylogenetic analyses of cestodes using two methods (BI and ML) yielded identical tree topologies based on concatenated amino acid sequences of 12 protein genes, all revealed distinct groups with high statistical support, and demonstrated that S. erinaceieuropaei is closely related to Diphyllobothrium spp. (Fig. 4), consistent with that of a previous study using cox1 sequences [37], supporting classification based on morphological features [38]. In this tree, Cyclophyllidea and Pseudophyllidea form monophyletic groups. Within the Cyclophyllidea clade, Hymenolepididae and Taeniidae form monophyletic groups. Within the Cyclophyllidea clade, Echinococcus and Taenia are sister genera.
Inferred phylogenetic relationships among cestodes based on amino acid sequences of 12 mitochondrial protein-coding genes using Bayesian inference (Bayes) and maximum likelihood (ML) analysis, using Ascaruis suum (GenBank accession number HQ704901) as the outgroup. The numbers along branches indicate posterior probability (PP) and bootstrap probability (BP) values resulting from different analyses in the order of Bayes/ML. Values lower than 70 are given as "-".
In conclusion, the present study determined and characterized the complete mtDNA sequence of S. erinaceieuropaei from China, which is slightly shorter (2 bp) than that of S. erinaceieuropaei from Japan, and both mt genomes differed by 0.9% only, refuted our hypothesis. These mtDNA sequences should provide novel genetic data for addressing further questions in systematics and population genetics of these and other related cestodes of socio-economic significance.
Acknowledgements
This work was supported, in part, by the Program for Outstanding Scientists in Agricultural Research, the Open Funds of the State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Grant Nos. SKLVEB2011KFKT011, SKLVEB2010KFKT009, SKLVEB2009KFKT008, SKLVEB2011KFKT010 and SKLVEB2011KFKT004) and the Yunnan Provincial Program for Introducing High-level Scientists (Grant No. 2009CI125).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Spakulová M, Orosová M, Mackiewicz JS. Cytogenetics and chromosomes of tapeworms (Platyhelminthes, Cestoda). Adv Parasitol. 2011;74:177-230
2. Li MW, Song HQ, Li C, Lin HY, Xie WT, Lin RQ, Zhu XQ. Sparganosis in mainland China. Int J Infect Dis. 2011;15:e154-156
3. Anantaphruti MT, Nawa Y, Vanvanitchai Y. Human sparganosis in Thailand: an overview. Acta Trop. 2011;118:171-176
4. Nakao M, Abmed D, Yamasaki H, Ito A. Mitochondrial genomes of the human broad tapeworms Diphyllobothrium latum and Diphyllobothrium nihonkaiense (Cestoda: Diphyllobothriidae). Parasitol Res. 2007;101:233-236
5. Miyadera H, Kokaze A, Kuramochi T, Kita K, Machinami R, Noya O, Alarcón de Noya B, Okamoto M, Kojima S. Phylogenetic identification of Sparganum proliferum as a pseudophyllidean cestode by the sequence analyses on mitochondrial COI and nuclear sdhB genes. Parasitol Int. 2001;50:93-104
6. Santamaria M, Lanave C, Vicario S, Saccone C. Variability of the mitochondrial genome in mammals at the inter-species/intra-species boundary. Biol Chem. 2007;388:943-946
7. Zardoya R, Meyer A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol Biol Evol. 1996;13:933-942
8. Douglas KC, Halbert ND, Kolenda C, Childers C, Hunter DL, Derr JN. Complete mitochondrial DNA sequence analysis of Bison bison and bison-cattle hybrids: function and phylogeny. Mitochondrion. 2011;11:166-175
9. Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301-120
10. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
11. Li MW, Lin RQ, Song HQ, Wu XY, Zhu XQ. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics. 2008;9:224
12. Lin RQ, Qiu LL, Liu GH, Wu XY, Weng YB, Xie WQ, Hou J, Pan H, Yuan ZG, Zou FC, Hu M, Zhu XQ. Characterization of the complete mitochondrial genomes of five Eimeria species from domestic chickens. Gene. 2011;480:28-33
13. Liu GH, Lin RQ, Li MW, Liu W, Liu Y, Yuan ZG, Song HQ, Zhao GH, Zhang KX, Zhu XQ. The complete mitochondrial genomes of three cestode species of Taenia infecting animals and humans. Mol Biol Rep. 2011;38:2249-2256
14. Li H, Gao J, Liu H, Liu H, Liang A, Zhou X, Cai W. The architecture and complete sequence of mitochondrial genome of an assassin bug Agriosphodrus dohrni (Hemiptera: Reduviidae). Int J Biol Sci. 2011;7:792-804
15. Li H, Liu H, Cao L, Shi A, Yang H, Cai W. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int J Biol Sci. 2012;8:93-107
16. Hu M, Chilton NB, Abs El-Osta YG, Gasser RB. Comparative analysis of mitochondrial genome data for Necator americanus from two endemic regions reveals substantial genetic variation. Int J Parasitol. 2003;33:955-963
17. Schmidt GD. Handbook of tapeworm identification. Boca Raton, FL: CRC Press. 1986
18. Dai RS, Liu GH, Song HQ, Lin RQ, Yuan ZG, Li MW, Huang SY, Liu W, Zhu XQ. Sequence variability in two mitochondrial DNA regions and internal transcribed spacer among three cestodes infecting animals and humans from China. J Helminthol. 2012 in press
19. Gasser RB, Zhu XQ, McManus DP. NADH dehydrogenase subunit 1 and cytochrome c oxidase subunit I sequences compared for members of the genus Taenia (Cestoda). Int J Parasitol. 1999;29:1965-1970
20. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876-4882
21. Chilton NB, Gasser RB, Beveridge I. Differences in a ribosomal DNA sequence of morphologically indistinguishable species within the Hypodontus macropi complex (Nematoda: Strongyloidea). Int J Parasitol. 1995;25:647-651
22. Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
23. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451-1452
24. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731-2739
25. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564-577
26. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572-1574
27. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
28. Page RD. TREEVIEW: an application to display phylogenetic trees on personal. Comp Appl Biosci. 1996;12:357-358
29. Goddard JM, Wolstenholme DR. Origin and direction of replication in mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res. 1980;8:741-757
30. Nakao M, Yokoyama N, Sako Y Fukunaga M, Ito A. The complete mitochondrial DNA sequence of the cestode Echinococcus multilocularis (Cyclophyllidea: Taeniidae). Mitochondrion. 2002;1:497-509
31. von Nickisch-Rosenegk M, Brown WM, Boore JL. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that platyhelminths are eutrochozoans. Mol Biol Evol. 2001;18:721-730
32. Nakao M, Sako Y, Ito A. The mitochondrial genome of the tapeworm Taenia solium: a finding of the abbreviated stop codon U. J Parasitol. 2003;89:633-635
33. Jia WZ, Yan HB, Guo AJ, Zhu XQ, Wang YC, Shi WG, Chen HT, Zhan F, Zhang SH, Fu BQ, Littlewood DT, Cai XP. Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics. 2010;11:447
34. Nkouawa A, Sako Y, Nakao M, Nakaya K, Ito A. Loop-mediated isothermal amplification method for differentiation and rapid detection of Taenia species. J Clin Microbiol. 2009;47:168-174
35. González LM, Montero E, Morakote N, Puente S, Díaz De Tuesta JL, Serra T, López-Velez R, McManus DP, Harrison LJ, Parkhouse RM, Gárate T. Differential diagnosis of Taenia saginata and Taenia saginata asiatica taeniasis through PCR. Diagn Microbiol Infect Dis. 2004;49:183-188
36. Yamasaki H, Allan JC, Sato MO, Nakao M, Sako Y, Nakaya K, Qiu D, Mamuti W, Craig PS, Ito A. DNA differential diagnosis of taeniasis and cysticercosis by multiplex PCR. J Clin Microbiol. 2004;42:548-553
37. Okamoto M, Iseto C, Shibahara T, Sato MO, Wandra T, Craig PS, Ito A. Intraspecific variation of Spirometra erinaceieuropaei and phylogenetic relationship between Spirometra and Diphyllobothrium inferred from mitochondrial CO1 gene sequences. Parasitol Int. 2007;56:235-238
38. Bray RA, Jones A, Andersen KI. Order Pseudophyllidea Carus, 1863. In: (ed.) Khalil LF, Jones A, Bray RA. Key to the Cestode Parasites of Vertebrates. Wallingford, UK: CBA International. 1994:205-247
Author contact
![]() Corresponding author: Prof. Xing-Quan Zhu, State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, CAAS, Lanzhou, Gansu Province 730046, China. Phone: 86-931-8342837; Fax: 86-931-8340977; Email: xingquanzhu1com or Assoc Prof. Feng-Cai Zou, College of Animal Science and Technology, Yunnan Agricultural University, Kunming, Yunnan Province 650201, China. Phone: 86-871-5222291; Fax: 86-871-5222291; Email: zfc1207163.com.
Corresponding author: Prof. Xing-Quan Zhu, State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, CAAS, Lanzhou, Gansu Province 730046, China. Phone: 86-931-8342837; Fax: 86-931-8340977; Email: xingquanzhu1com or Assoc Prof. Feng-Cai Zou, College of Animal Science and Technology, Yunnan Agricultural University, Kunming, Yunnan Province 650201, China. Phone: 86-871-5222291; Fax: 86-871-5222291; Email: zfc1207163.com.