Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The behavior of individual...
Sequential crosstalk among...
Spatial crosstalk among the...
Regulation mechanisms by PTMs
Deciphering the 'p53 code'
Future perspectives
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(5):672-684. doi:10.7150/ijbs.4283 This issue Cite
Review
Surf the Post-translational Modification Network of p53 Regulation
Bo Gu, Wei-Guo Zhu ![]()
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Biochemistry and Molecular Biology, Peking University Health Science Center, Beijing 100191, China.
Received 2012-2-26; Accepted 2012-5-7; Published 2012-5-10
Abstract
Among the human genome, p53 is one of the first tumor suppressor genes to be discovered. It has a wide range of functions covering cell cycle control, apoptosis, genome integrity maintenance, metabolism, fertility, cellular reprogramming and autophagy. Although different possible underlying mechanisms for p53 regulation have been proposed for decades, none of them is conclusive. While much literature focuses on the importance of individual post-translational modifications, further explorations indicate a new layer of p53 coordination through the interplay of the modifications, which builds up a complex 'network'. This review focuses on the necessity, characteristics and mechanisms of the crosstalk among post-translational modifications and its effects on the precise and selective behavior of p53.
Keywords: p53, post-translational modification, crosstalk, protein-protein interaction, semiotic system.
Introduction
Since the discovery of p53 in 1979 [1-3], numerous studies have been conducted related to its functions and regulatory mechanisms. Previous research has confirmed that p53 is able to coordinate a regulatory network that supervises and responds to a variety of stress signals. These signals include: DNA damage, aberrant oncogenic activation, telomere erosion, ribosomal stress, loss of cell-cell or cell-matrix adhesion, and hypoxia [4]. Regulating a vast pool of external stimuli, p53 exerts irreplaceable anti-neoplastic functions at homeostasis and thus is considered to be 'the guardian of the genome' [5]. Mutations of p53 or disruptions of p53 coordination, to a lesser extent, can disturb the normal physiological balance, and lead to cancer if genome disarrangement reaches a critical value.
Basic elements of the p53 coordination are its cellular localization, oligomerization [6] and concentration, which are tightly and exquisitely interrelated. Originally, p53 was thought to perform its functions in its tetrameric form in the nucleus by acting as a transcription factor or as a binding partner [7-10]. At homeostasis, the transcriptional activity of p53 is downregulated in three ways: 1. Ubiquitin-mediated proteasomal degradation of p53 in both cytoplasm and nucleus mainly through mouse double minute protein 2 (Mdm2) [11]; 2. Decrease in nuclear p53 levels through nuclear export by either the exposure of its nuclear export sequence (NES) [12, 13] or the NES of Mdm2 [14]; and 3. Transcriptional repression of chromatin-associated p53 by Mdm2-Mdmx-p53 complex formation [15-17].
Under stress, degradation and nuclear export of p53 are suppressed, and nuclear import of p53 is concomitantly enhanced, resulting in its nuclear accumulation. Recently proposed is another process involved in the activation of chromatin-bound p53 termed as 'anti-repression' [18]. Transcriptional levels of p53 downstream target genes can be generally increased by p53 nuclear accumulation and the release of chromatin-bound p53 from repression state. On the other hand, selective functions of p53 can be fulfilled through enhancement of p53's transactivation of specific target genes [19]. Although p53 primarily acts as a transcription factor, a transcription-independent role of cytosolic p53 to trigger apoptosis and inhibit autophagy has also been discovered [20-22]. Researchers during the past decades have discovered that, in either the homeostatic maintenance or stress-induced activation of p53, covalent modifications play pivotal roles (summarized in Figure 1).
Overview of p53 posttranslational modifications. The major domains of p53 and their distributions are depicted and only the modifications directly responsible for the listed effects are plotted. The modification sites within p53 are primarily updated from W Gu [164, 165].
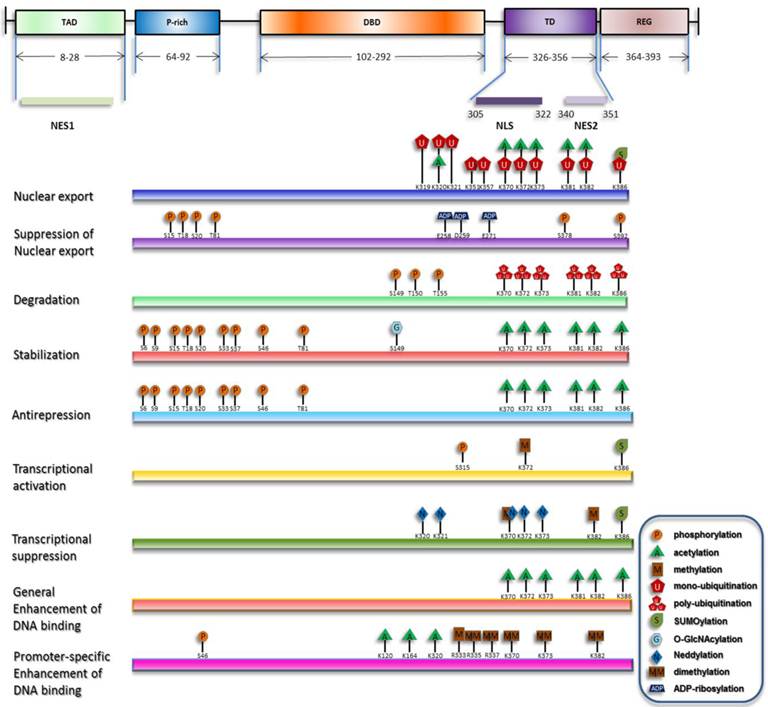
Although relatively unified findings related to the functions of post-translational modifications (PTMs) were obtained in vitro, the in vivo data are somehow contradictory, indicating a variable behavior of p53. This variability is characterized by cell type- and tissue-dependent [23, 24], genotype and stimuli-specific responses [25-27].
The stimuli-specific response has received intensive investigations, for it provides a potential model to study the discriminative behavior of p53 pathway. The prominent features of the stimuli-specific responses are distinct elevation manners and different gene expression profiles [28]. Because the concentration of p53 is tested indirectly by the antibody, Differences in the elevation manner of p53 can be partly explained by the occupation of p53 antibody binding epitopes by PTMs. Different gene expression profiles is confirmed to be a result of combinatorial expression of specific sets of p53 target gene. This can be accomplished either through the promoter selection by p53 or the dissimilar assembly manner of transcription complexes by chromatin-bound p53 [29]. Promoter selectivity is attributed to different binding affinities for different response elements (REs) [30], presumably due to PTM marks on p53 [31-33]. Likewise, different assembly manners of transcription complexes, such as chromatin remodelers, histone modifiers or RNA polymerase, can also be ascribed to p53 modifications [34, 35]. It is important to note that different types of stresses can result in different sets of modifications, which bolsters the relationship between covalent modifications and the variability of p53 response [36, 37].
Overall, PTM exerts both general and distinctive role in regulating p53 behavior. However, the contradictions between the results of the in vitro and in vivo experiments call for more in-depth studies and raise some open problems concerning the real regulatory network of PTMs.
The behavior of individual modifications
p53 harbors an array of amino acids subject to various kinds of PTMs, which are mainly concentrated in the tetramerization domain (TD) and C-terminal domain (CTD). The earliest-discovered behavior of individual modifications of p53 is the redundancy of many N-terminal and C-terminal modifications [18], which is characterized by either the flexible correspondence between the enzymes and modifications, or the subtle effects by the mutation of single site [38]. This can be explained by either the complementarity among the modifications, or an additive and synergistic performance of the modifications. Both mechanisms illustrate the significance of the crosstalk among the modifications.
There also exist switch-like behaviors of individual sites. The individual modifications involved in the transactivation by p53 reflect this behavior, of which the most scrutinized is acetylation. As an example, lysine 320 (K320) acetylation is necessary in antagonizing apoptotic activity of p53 [39]; in contrast, acetylation of K373 and K120 dominantly favors the activation of proapoptotic genes [31, 40]. Besides the acetylation, serine 46 (S46) phosphorylation is found to play critical roles in p53-mediated proapoptic gene induction but not in the induction of cell-cycle arrest [41-43]. Thus, it is possible that individual modifications with a predominant preference for specific physiological outcomes serve as 'binary switches' of different cell fates. Furthermore, with structural biological methods, threonine 18 (T18) has been found to exert 'on-off'-switch role controlling the binding of p53 with Mdm2 [44-46]. This suggests that development of test methods also influences the determination of the functions of modifications. However, switch-like behavior of the individual modifications is mostly identified by mutation assay. This method only proves the essentiality but not the sufficiency of these modifications in initiating specific effects. So it remains to be determined whether there is a simple correlation between an individual modification and a specific effect. In fact, another characteristic of the modifications is their multi-potency—that modification of one site exerts various effects in different contexts and under different stresses, even if the effects are seemingly conflicting. For example, our group found that p21waf1/cip1, a canonical cell cycle regulating gene, is activated by K373/K382 acetylation after a specific histone deacetylase inhibitor (HDACi)—depsipeptide—is administered [47]. This finding contradicts previous results that K373 acetylation has a preference for proapoptotic genes [40]. Of note, depsipeptide also induces T18 and S37 phosphorylation. This specific combination pattern of phosphorylation and acetylation is a likely cause of the contrasting results. Similarly, it is shown that K120, with a widely accepted transcription-dependent apoptotic activity, has a transcription-independent proapoptotic function [48]. While a transcription-dependent activity requires prior nuclear accumulation, nuclear export is the prerequisite for transcription-independent apoptotic activity. S315 phosphorylation both increases p53 transactivation potential through nuclear retention, and promotes Mdm2-dependent proteolysis of p53 [49-51]. This raises the question of how a single modification is able to choose between two contrasting fates.
Therefore, an individual modification of p53 is far from discriminating p53 isoforms in deciding biological effects. Instead, certain combinations of modifications can expand the functional scope of individual modifications and explain the results from the functional studies. In this respect, combined with other modifications, an individual modification can exert various functions, which interprets the multi-potency of the individual modifications.
Sequential crosstalk among modifications
While individual modifications show little significance in coordinating the vast pool of upstream stresses and the downstream repertoire of the target genes, crosstalk among different modifications may provide a way to guarantee the complexity of the p53 network. In normal cells during cell cycle progression, a modification cascade of p53 exists. Phosphorylation of S9, 15, 20 and 372 peaks during G1, whereas S37 and S392 phosphorylation peak during G2/M. S37 is the only site to be phosphorylated during S phase and acetylation is mostly abundant at G0 [52]. This demonstrates that at homeostasis, p53 modification is a dynamic and transient event which may be predicted under controlled conditions.
Under stress, p53 is modified more extensively. At the center of the p53 activation are acetylation and phosphorylation. Phosphorylation of the N terminus serves as the initial wave of response to stress, which shows strong inter-dependence between one another. For example, T18 is phosphorylated in vitro and in vivo subject to the prior phosphorylation of S15, which is a prerequisite of S20 phosphorylation [36, 53]. Thus, N-terminal phosphorylations can be classified into several clusters. For each cluster, one site is directly modified by the kinases, i.e. nucleating sites, whereas others are modified followed by the nucleating sites. Not only does sequential inter-site dependence exist among the N-terminal phosphorylation sites, C-terminal phosphorylation sites are also involved [54]. Thus, for phosphorylation, inter-site dependence and activation cascades set a new level for more comprehensive and precise coordination of different types of stress.
Phosphorylation is also influenced by other upstream modifications, such as the addition of O-linked β-N-acetylglucosamine (O-GlcNAcylation) and poly (ADP-ribosylation) [55-57]. poly (ADP-rybosylation) of different nuclear acceptors by poly (ADP-ribose) polymerase 1 (PARP-1) is believed to be a damage sensing modification, and thus may bridge the gap between the DNA damage sensing and p53 stabilization. Considering the scarcity of the sites subject to either O-GlcNAcylation or poly (ADP-rybosylation), they may merely exert subtle effects or perform switch-like roles in p53 activation.
Specific phosphorylation patterns induce the acetylation of the C terminus and initiate a phosphorylation-acetylation cascade [58]. Consistently, with the use of specific DNA damage agents, we and other groups testified this hypothesis [59-61]. This suggests that this cascade is generally implicated in all circumstances. It is now confirmed that Mdm2, CREB binding protein (CBP)/p300 (specific enzymes for the acetylation of p53) and p53 form a ternary complex in unstressed cell, and phosphorylation of S15, T18 and S20 increase p53's affinity for CBP/p300 [62]. Phosphorylation of the C terminus also differentially influences the acetylation status. For example, phosphorylation of S378 and T377 reduces the acetylation of K373, K382 and K320, and phosphorylation of S366 and T387 enhances the C-terminal acetylation [54, 58]. Since S378 is constitutively phosphorylated in unstressed cells, T377 and S378 phosphorylation may suppress p53 activation through inhibition of acetylation. Moreover, C-terminal phosphorylation also mediates ubiquitination by Mdm2 [51, 63]. Therefore, p53 degradation may be accomplished through a series of interlocking processes, which is initiated by C-terminal phosphorylation [51, 63], relayed by inhibition of C-terminal acetylation and ended up with the ubiquitination (Figure 2). In addition to acetylation, other modifications including mono-ubiquitination, poly-ubiquitination [64, 65] and ubuiquitin-like modifications [66], are also included in various cascades.
Summary of the sequential interplay of modifications of p53. This figure shows the modification cascades which can be classified into short-range and long-range influences, which is reminiscent of the model raised by X-J Yang [127]. In addition, effects on the downstream modification sites can be either negative (indicated by arrows with a '-' in a circle) or positive (indicated by arrows with a '+' in a circle). According to the sequential order of the modifications, they are crudely classified into two cassettes. Modifications in the initializing cassette are responsible for the sensing and distinguishing of the stresses and can transmit the signals to the functional cassette. Modification combinations in the functional cassette can induce specific biological outcome.
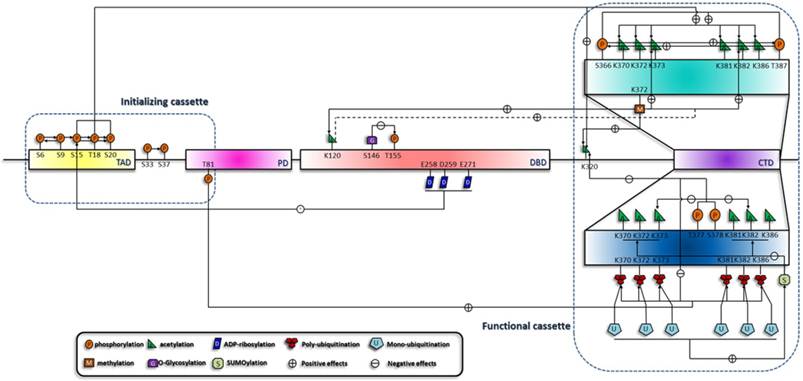
Acetylation is the hub of p53 transactivation and is contained within a network of various upstream and downstream modifications. Loss of Set7/9 in mouse embryonic fibroblasts cells (MEFs) prevents acetylation at K117, K317, K370 and K379 (human homologous sites are K120, K320, K373 and k382), suggesting a general effect of K372 methylation on the acetylation status of p53 [67]. Consequently, acetylated p53 can recruit coactivators to the binding promoters and mediate the acetylation of histone H4 [68, 69]. In addition, SUMOylation also influences the acetylation of the C terminus and illustrates a unidirectional cascade. K386 SUMOylaiton of at least one subunit of p53 tetramer inhibits the consecutive acetylation by p300/CBP, whereas prior acetylation by p300 remains permissive for the SUMOylation machinery [70]. This demonstrates distinct behaviors of a pathway cascading in opposite directions and a coordination mechanism of a level higher than the primary structure of p53.
Therefore, the modification of p53 is a dynamic process that rapidly relays the sequential signals to the final target during the course of p53 activation. (Summarized in Figure 2).
Spatial crosstalk among the modifications
In addition to sequential crosstalk, there is also spatial crosstalk among the PTMs, which is characterized by combinations of multisite modifications to trigger p53 response cooperatively (i.e. combinatorial behaviors). Series of combinatorial behaviors of different modifications on other proteins have already been characterized, including Forkhead Box protein O (FoxO) family [71-74], tubulin and the C-terminal domain of RNA polymerase II [75, 76]. Analogous with the interplay of histone modifications and the none-histone protein modifications [77, 78], p53 modifications also demonstrate spatial crosstalk. The simplest behavior is the competition of the same site by different kinds of modifications. Mostly influenced are lysines located in the CTD of p53, especially the 6 lysines acetylated by p300/CBP and K320 acetylated by P300/CBP-associated factor (PCAF). All the acetylation sites are ubiquitination targets [79], with some of them competing with methylation, SUMOylation and neddylation, suggesting mutual exclusivity of these modifications. Moreover, functions of the modifications of the same sites vary according to the number of moieties added. This is exemplified by methylation of K370, K373 and K382 [80-82] and the competitive mono- and polyubiquitination of several lysines in the C terminus [83]. Besides the competition for the same amino acid, crosstalk among adjacent sites in the primary amino acid sequence or higher order structure of protein also exists. The spatial crosstalk has both antagonistic and synergistic effects. Antagonism is exemplified by the interplay of modifications on the CTD, such that methylation of K370 and K372 and phosphorylation of S315 and S392 occur in a mutually exclusive way [84, 85]. Synergy is characterized by the sites functioning or modified simultaneously. As shown in Figure 3, the key problem resolved by this hypothesis is the multisite phosphorylation of the N terminus. The multiplicity may indicate the need for a critical amount of phosphorylation sites to reach an activation threshold, forming a multisite switch, which is reminiscent of the regulation of Ste5 by cyclin-cyclin dependent kinase complex (Cln/CDK) [86]. In addition, simultaneous phosphorylation and acetylation of several sites is essential for the interaction of p53 with Pin1 and TAF1 respectively [87, 88]. Apart from addition of covalent moieties, deletion is also indispensable. For example, simultaneous phosphorylation of S378, S366 and T387 and dephosphorylation of S376 results in p53 tetramerization and transcriptional activation [89-91].
Generalization of the synergistic manner of different modifications and their binding partners. The functions of the p53 modifications are mutually dependent. A given combination of modifications can exert their specific functions simultaneously, and a specific binding partner, usually protein, mediates the function of modifications. Dashed lines and questions marks highlight that these modifications are likely to function synergistically.
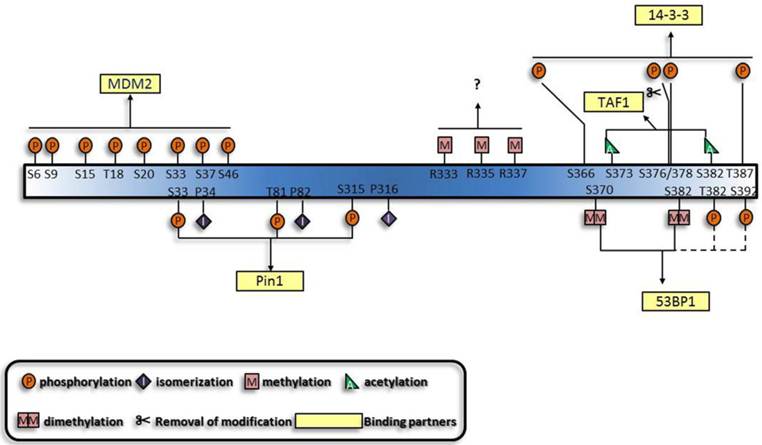
Based on the established cooperative manners of modifications, lots of other modifications are predicted to perform synergistically. Multisite-monoubiquitination that was previously thought to function redundantly, now seems to strengthen the binding affinity between proteins [92-94]. Likewise, methylation of three arginines in the C terminus may also perform simultaneously [95]. Furthermore, the dual modification pattern in the interaction between 14-3-3 and histone through phospho-acetylation [96] is indicative of the interaction between 14-3-3 and p53. Apart from the established role of K370 and K382 di-methylation [80, 81] in the interaction between p53 and p53 binding protein 1 (53BP1), phosphorylation shows great potential to be involved [97-100]. Furthermore, motifs which can associate with several modifications and proteins with multiple modification-recognizing motifs have also been characterized [101]. On the whole, protein-protein interactions appear to underlie the majority of cooperative regulations by modifications.
Both sequential and spatial crosstalk represents combinatorial performance of covalent modifications. Due to this combinatoriality, the selectivity and variability of p53 functions are yielded.
Regulation mechanisms by PTMs
PTM cascade of p53 is always accompanied by a binding partner cascade, indicating a role of the modifications to mediate the interaction between p53 and its partners. This has raised tantalizing questions of how the interactions are regulated. In fact, there are various underlying mechanisms for the interaction which should be discussed.
Conformational changes
Conformation is the most important feature of protein structure, which dramatically influences the function of the protein. Since PTMs can elicit significant effect on protein function through conformational changes [102-104], different combinations of PTMs may yield distinct protein conformations, resulting in the ensuing specific interaction [76].
As for p53, modification confirmed to influence its conformation is the phosphorylation-dependent isomerization by Pin1 [105], which is supported by the fact that p53 needs to form a complex with Pin1 to exert its functions [87, 105, 106]. This indicates that other modifications of p53 may also show great potential in converting PTMs into conformational changes.
p53 CTD can either positively or negatively regulate p53's transactivation ability, and its acetylation can potentiate p53 sequence-specific binding in vitro [19, 107, 108]. The underlying mechanism for sequence-specific binding was suggested to be allosteric activation [19, 58, 109], which either exposes the DNA binding domain (DBD) of p53, or influences the interaction between other proteins. DBD of p53 mainly mediates its direct binding with its consensus sequence and is heavily influenced by conformation. This is consolidated by the structural study revealing that p53 sequence-specific binding involves a conformational switch in its DBD [110]. Unlike DBD of FoxO family whose binding affinity is substantially influenced by its phosphorylation and acetylation [111], DBD of p53 shows poor access for covalent modifications. However, since the mechanism for K164 and K120 acetylation of p53 remains unclear, a conformational change similar to FoxO may cause this effect. Likewise, ubiquitination, especially mono-conjugated, shows great potential to regulate p53 conformation, possibly owing to its function as a chaperone to promote folding of nascent proteins [112].
Conformation is closely related to the energy of the molecule and serves as a major regulator of backbone structure. By virtue of the conformational change, the otherwise buried docking sites or catalytic sites can be exposed and thus induce the interaction between proteins, DNA and chromatin. Since these modifications can induce significant structural changes, they may serve as simple 'on/off' switches to regulate the qualitative responses of p53. However, as have been suggested, conformational changes cannot be incorporated into specific kind of protein during evolution, which limits the generality of this mechanism [101].
Combinations of docking motifs
Histone tails are heavily modified and can be read by effector proteins through direct binding. It was hypothesized that this effect is mediated by the covalent modifications embedded in specific motifs [113]. In support of this hypothesis, protein modules specific for recognizing modifications on histone tails are identified. These include bromodomain [114, 115], chromodomain [116, 117] and the more recent plant homeodomain (PHD) [118-121], Tudor and MBT [122] domains. Furthermore, it is confirmed that a protein can contain more than two modification recognition domains [123-125], and some protein complexes can embrace subunits with distinct modification recognition motifs. This provides a novel way to recognize cooperatively the modification signals [126].
The functions of non-histone protein modifications can be extrapolated from the histone modifications. Consistently, protein modules that specifically recognize modifications on non-histone proteins are characterized [127]. Together with the flanking sequences, modifications can mediate specific binding of p53 with the modification recognizing modules (summarized in Table 1). Therefore, docking motifs appear to be a precise commander enabling dynamic and specific binding of p53 with other partners.
Modifications as docking motifs and their binding proteins.
| Modification | Sites | Binding protein | domain | Reference |
|---|---|---|---|---|
| phosphorylation | S46, S33, T81, S127, T150, S315 | Pin1 | WW | Ref 105 |
| dephosphorylation | S376 | 14-3-3 | Ref 95 | |
| acetylation | K382 | P300/CBP | bromo | Ref 159 |
| di-acetylation | K373, K382 | TAF1 | Tadem bromo | Ref 88 |
| SUMOylation | K386 | SIM | Ref 160, 161 | |
| di-methylation | K382, K370 | 53BP1 | Tudor | Ref 80, 81 |
| Ubiquitination | UBD | Ref 83, 162 | ||
| monomethylation | K382 | L3MBTL1 | MBT | Ref 163 |
| K372 | Tip60 | Chromo | Ref 67 |
Bulk electrostatics
Intrinsically disordered regions of proteins which are quite frequent in nature perform important functions in cells [128]. These regions always serve as the linkers between different domains of the proteins. There are three unstructured regions in p53: the linker between N-terminal transactivation domain (TAD) and DBD, the linker between DBD and TD, and CTD [129, 130].
Many kinds of modifications, including phosphorylation, acetylation and ADP-ribosylation, can change the overall charge of the amino acid residues, and in turn contribute to the electrostatic force-mediated interactions [131]. The first to be mentioned is the multisite phosphorylation of the TAD of p53 which mediates the binding with p300/CBP. Rather than a switch-like behavior, an additive manner of phosphorylations of S15, S20, T18, S46, S33, S37 and T55 was demonstrated [45]. In this scenario, the electrostatic forces generated by negative charges of phosphate and the positive charge of CBP contribute to the interaction. In addition, p53 transcriptional activity can exhibit both on/off switch and graded response after genotoxic stress [132]. An extension of this fact is that the seeming redundancy of the N-terminal phosphorylation can contribute to the graded response. The redundancy may also play critical roles in sensing the nature and the severity of cellular stresses, whereby prolonged or severe genotoxic stress leads to phosphorylation of additional sites and gradual increase in the affinity for CBP/p300. Apart from the TAD, modifications of lysines on CTD, especially the acetylation of the six lysines in proximity, may also neutralize the positive charge on CTD [19]. Other models of multisite acetylation functioning as charged patches have already been established, including histone acetylation and p300 autoacetylation [133, 134]. These models further increase the possibility of the acetylation on p53's CTD to function as electrostatic regulator.
The bulk electrostatic mechanism explains the graded response of p53 and confers a quantitative feature on p53 response. However, since various modifications share the same electric property, this mechanism is not precise enough. Hence, structure of different kinds of moieties is necessary in distinguishing different modifications.
The three mechanisms presented above function cooperatively. The conformational switch regulates the rigid backbone of p53's globular regions and exposes the docking motifs for interaction proteins. Docking motifs, in turn, directly coordinate the binding of p53 with its partners through interacting with specific modification recognizing modules. Electrostatic forces increase the binding efficiency by specifically utilizing the flexibility of the unstructured regions. Henceforth, modifications inducing conformational changes behave as 'on/off' switches toggling between different subsets of p53 events through exposure of different groups of docking motifs. Specific docking motifs serve as combinatorial signals to recruit binding partner. Modifications regulating the electrostatic attraction yield a rheostat behavior of p53 to quantitatively coordinate the events like a sensor or blocker. In consequence, these mechanisms form a complex and precise coordination network for p53.
Deciphering the 'p53 code'
Spatial and sequential interplay of p53 modifications provides it with vast indexing potential and expands its functional spectrum. A semiotic view, such as 'code', 'barcode' or 'cassette', is adopted to define the complex network among the modifications of both histones and non-histone proteins [40, 135]. In this semiotic system, several elements, including code, regulator and the 'meaning' of the code are indispensable. Regulators can be classified into 'reader', 'writer' and 'eraser' (see Figure 4). 'Reader' refers to binding partner like chromatin, non-histone protein and DNA that interacts specifically with the modification marks and initiates specific effects. These include conformational changes, catalytically activation and transcriptional activation. Protein adding chemical groups to specific sites is defined as 'writer'; by contrast, 'eraser' is responsible for the removal of chemical group from specific sites. However, there are no strict divisions between the definitions of these regulators; a protein can have multiple properties such that the process of reading and writing can be completed by the same protein. These regulators in the semiotic system are coordinated either at protein level or, more precisely, at the posttranslational level. Specific modification at particular sites can distinguish between different forms of a protein and guarantee the functional specificity of the regulators. Basic to p53 degradation is Mdm2, of which the most relevant modifications are autoubiquitination [136, 137], phosphorylation [138-142], SUMOylation [143, 144] and acetylation [145]. Similar to Mdm2, other ubiquitin ligases including Pirh2 [146] and constitutively photomorphogenic 1 (COP1) are also subject to the regulation by PTMs [147].
Diagram of the p53 'code system'. At homeostasis, p53 is mainly presented in two forms: chromatin-bound (b) form and unbound form (c). Under stress, p53 is modified combinatorially by various enzymes. Thus, the 'code' is written (a). Specific 'code' on chromatin-bound p53 can recruit either histone modifying enzymes (d), histone remodelers (e) or other regulatory proteins (f) to the vicinity of the response element p53 is bound to. As for the unbound form, DNA with p53-binding sites (g) and other enzymes (h) recognize the code. Different 'readers' lead to distinct outcomes. Modifications of histones, remodeling of chromatin, directly activation or repression of transcription, conformational changes as well as extensive modifications of p53 are the effects of histone modifying enzymes, histone remodelers, additional regulatory proteins, specific DNA sequences and other enzymes, respectively.
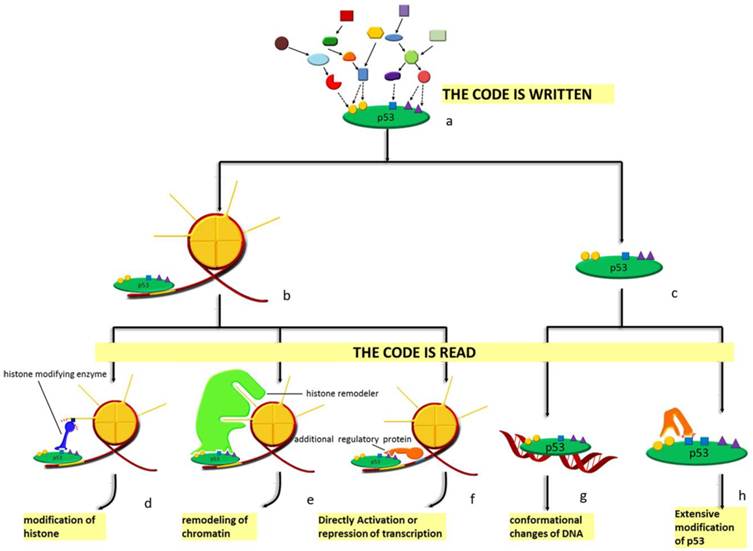
histone acetyltransferase (HAT) and histone deacetylase (HDAC) regulate acetylation of both histones and transcription factors, yielding a connection between chromatin accessibility and transcription activity. p300 and CBP are coactivators for a variety of transcription factors, whose activation is mainly regulated by autoacetylation [134, 148]. Similarly, we and other groups also found that class III HDACs sirtuins, especially SIRT1 and SIRT7 participating the deacetylation of p53, are phosphorylated and methylated [149-151]. Notably, different proteins modified by the same enzyme can function either synergistically to enhance the overall effects, or antagonistically to create a delicate balance [152]. This well explains the inconsistencies in the in vitro and in vivo experimental results: the in vitro methods used may always disturb the stoichiometry between different targets subject to same modifications.
Another critical element within this semiotic system of p53 is the interpretation of the code. 'PTM code' of non-histone proteins [153] is originally extrapolated from the 'histone code', [77, 78] which is interpreted as 'transcription starts or stops at a specific time and place' [154]. Although there are still arguments against the code's generality considering the context-dependent meaning and the weak predictability of modifications themselves [113], extensive combinations of modifications strengthen the specificity and more clearly define the concepts of 'code'. The meaning of the 'code' of p53 modification can be interpreted as the functions of interaction partners—including non-histone proteins, histones as well as DNA—encoded combinatorially by the modifications (Figure 4). PTMs on the regulators can be translated into anti-code matching with specific code on p53. In this way, only the properly modified regulators can recognize a specific form of p53. More recently, as revealed by the interdependence between p53 modifications and histone H3 modifications [155, 156], a 'p53-histone' code-to-code model connecting the histone and non-histone modifications has been raised.
With respect to this semiotic system, a stress-specific performance of p53 can be explained by the differences of modification marks induced by distinct stresses [157]. Likewise, cell- and tissue-type dependency of p53 behavior can be attributed to specific combinations of p53 modifications introduced by intrinsically distinct regulator pools in different types of cells or tissues [158]. Furthermore, a modification cascade can serve as a driving force for p53 pathway to progress spontaneously, which indicates a more general regulating rationale for other signaling pathways.
Future perspectives
Promising as the 'code' model of p53 PTM is, the following questions remain open such as: what is the real basis for the redundancy of the individual modifications in vivo? What is the real mechanism that regulates the context-dependent behavior of p53? How exactly is p53 involved in the regulation of one specific biological effect? How general is the mechanism for the regulation of PTM and in what way do they really cooperate? With the emergence of the novel functions regulated by p53, such as metabolism and nutrient stress responses, is there a possibility to revise the demarcations between different phenotypic outcomes to a more subtle one? In order to tackle these problems, numerous further investigations are required: (i) more precise and subtle distinction of the effects of the 'code' in molecular level instead of phenotypic level; (ii) discrimination between the direct and indirect effects of specific modifications; (iii) identification of the combinatorial behaviors of the modifications using high-throughput testing method; (iv) in situ observation of the dynamic changes of the modifications marks using more reliable and direct time-resolved method. Although there is still a long way to go, it is believed that the final decipherment of the p53 code will arrive in the near future.
Acknowledgements
This review was supported by National Natural Science Foundation of China Grants 90919030, 31070691, 30921062 (to W.-G.Z); Ministry of Science and Technology of China Grants 2011CB504200 (to W.-G.Z.); “111 project” from the Ministry of Education of China; grants from the Ministry of Science and Technology of China (to W.-G.Z.). We appreciate Dr. Kate Morton to edit this manuscript. We also apologize for unable to include many valuable literatures regarding p53 post-translational modifications in this paper due to the space limitation.
Abbreviations
53BP1: p53 binding protein 1; CBP: CREB binding protein; Cln/CDK: cyclin-cyclin dependent kinase complex; COP1: constitutively photomorphogenic 1; CTD: C-terminal domain; DBD: DNA binding domain; FoxO: forkhead box protein O; HAT: histone acetyltransferase; HDAC: histone deacetylase; HDACi: histone deacetylase inhibitor; MEF: embryonic fibroblasts cells; Mdm2: mouse double minute 2; NES1: N-terminal nuclear export sequence; NES2: C-terminal nuclear export sequence; NLS: nuclear localization sequence; PARP-1: Poly (ADP-ribose) polymerase 1; PTM: post-translational modification; P-rich: proline rich domain; RE: response element; REG: C-terminal regulatory domain; TAD: transactivation domain; TD: tetramerization domain.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Linzer DIH, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43-52
2. DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proceedings of the National Academy of Sciences. 1979;76:2420
3. Lane D, Crawford L. T antigen is bound to a host protein in SY40-transformed cells. Nature. 1979;278:261-3
4. Horn H, Vousden K. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306-16
5. Lane D. Cancer. p53, guardian of the genome. Nature. 1992;358:15
6. Rajagopalan S, Huang F, Fersht AR. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Research. 2011;39:2294
7. Davison TS, Yin P, Nie E, Kay C, Arrowsmith CH. Characterization of the oligomerization defects of two p53 mutants found in families with Li-Fraumeni and Li-Fraumeni-like syndrome. Oncogene. 1998;17:651
8. Shieh SY, Taya Y, Prives C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. The EMBO Journal. 1999;18:1815-23
9. Itahana Y, Ke H, Zhang Y. p53 Oligomerization is essential for its C-terminal lysine acetylation. The Journal of biological chemistry. 2009;284:5158-64
10. Maki CG. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. Journal of Biological Chemistry. 1999;274:16531
11. Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299-303
12. Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. The EMBO Journal. 1999;18:1660-72
13. Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910
14. Tao W, Levine AJ. P19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proceedings of the National Academy of Sciences. 1999;96:6937
15. Arva NC, Gopen TR, Talbott KE, Campbell LE, Chicas A, White DE. et al. A chromatin-associated and transcriptionally inactive p53-Mdm2 complex occurs in mdm2 SNP309 homozygous cells. Journal of Biological Chemistry. 2005;280:26776
16. Minsky N, Oren M. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Molecular Cell. 2004;16:631-9
17. Chen L, Li Z, Zwolinska AK, Smith MA, Cross B, Koomen J. et al. MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. The EMBO Journal. 2010;29:2538-52
18. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609-22
19. Gu W, Roeder RG. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell. 1997;90:595-606
20. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P. et al. p53 has a direct apoptogenic role at the mitochondria. Molecular Cell. 2003;11:577-90
21. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M. et al. Regulation of autophagy by cytoplasmic p53. Nature Cell Biology. 2008;10:676-87
22. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127-30
23. Midgley CA, Owens B, Briscoe CV, Thomas DB, Lane DP, Hall PA. Coupling between gamma irradiation, p53 induction and the apoptotic response depends upon cell type in vivo. Journal of Cell Science. 1995;108:1843-8
24. MacCallum DEea. The p53 response to ionising radiation in adult and developing murine tissues. Oncogene. 1996;13:2575-8
25. Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or lonizing radiation: Defects in chromosome instability syndromes? Cell. 1993;75:765-78
26. Alvarez S. A comprehensive study of p53 transcriptional activity in thymus and spleen of gamma irradiated mouse: high sensitivity of genes involved in the two main apoptotic pathways. International Journal of Radiation Biology. 2006;82:7612-770
27. Zhang Y, Ma WY, Kaji A, Bode AM, Dong Z. Requirement of ATM in UVA-induced signaling and apoptosis. Journal of Biological Chemistry. 2002;277:3124
28. Amundson SA, Do KT, Vinikoor L, Koch-Paiz CA, Bittner ML, Trent JM. et al. Stress-specific signatures: expression profiling of p53 wild-type and-null human cells. Oncogene. 2005;24:4572-9
29. Espinosa JI. p53 functions through stress-and promoter-specific recruitment of transcription initiation components before and after DNA damage. Molecular cell. 2003;12:1015-27
30. Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH. et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. GENES & DEVELOPMENT. 2000;14:981-93
31. Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827-39
32. Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, Lu X. et al. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nature Structural & Molecular Biology. 2007;14:912-20
33. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS. et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular cell. 2006;24:841-51
34. Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD. et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Molecular Cell. 2001;8:1243-54
35. Roy S, Tenniswood M. Site-specific acetylation of p53 directs selective transcription complex assembly. Journal of Biological Chemistry. 2007;282:4765
36. Saito S, Yamaguchi H, Higashimoto Y, Chao C, Xu Y, Fornace Jr AJ. et al. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. Journal of Biological Chemistry. 2003;278:37536-44
37. Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death & Differentiation. 2003;10:400-3
38. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nature Reviews Cancer. 2004;4:793-805
39. Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T. et al. Acetylation of Mouse p53 at Lysine 317 Negatively Regulates p53 Apoptotic Activities after DNA Damage. Mol Cell Biol. 2006;26:6859-69
40. Knights CD. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. The Journal of Cell Biology. 2006;173:533-44
41. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T. et al. p53AIP1, a Potential Mediator of p53-Dependent Apoptosis, and Its Regulation by Ser-46-Phosphorylated p53. Cell. 2000;102:849-62
42. D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S. et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nature Cell Biology. 2001;4:11-9
43. Feng L, Hollstein M, Xu Y. Ser46 Phosphorylation Regulates p53-Dependent Apoptosis and Replicative Senescence. Cell Cycle. 2006;5:2812-9
44. Dumaz N, Meek DW. Serine 15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. The EMBO Journal. 1999;18:7002-10
45. Chul Won Lee JCF. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proceedings of the National Academy of Sciences. 2010;107:19290-5
46. Teufel DP, Bycroft M, Fersht AR. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene. 2009;28:2112-8
47. Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y. et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21Waf1/Cip1. Molecular and Cellular Biology. 2006;26:2782
48. Sykes SM, Stanek TJ, Frank A, Murphy ME, McMahon SB. Acetylation of the DNA Binding Domain Regulates Transcription-independent Apoptosis by p53. Journal of Biological Chemistry. 2009;284:20197-205
49. Blaydes JP, Luciani MG, Pospisilova S, Ball HM-L, Vojtesek B, Hupp TR. Stoichiometric Phosphorylation of Human p53 at Ser315Stimulates p53-dependent Transcription. Journal of Biological Chemistry. 2001;276:4699-708
50. Fogal V, Hsieh JK, Royer C, Zhong S, Lu X. Cell cycle-dependent nuclear retention of p53 by E2F1 requires phosphorylation of p53 at Ser315. The EMBO Journal. 2005;24:2768-82
51. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F. et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nature Genetics. 2003;36:55-62
52. Buschmann T, Adler V, Matusevich E, Fuchs SY, Ronai Ze. p53 Phosphorylation and Association with Murine Double Minute 2, c-Jun NH2-Terminal Kinase, p14ARF, and p300/CBP during the Cell Cycle and after Exposure to Ultraviolet Irradiation. Cancer Research. 2000;60:896-900
53. Sakaguchi K, Saito Si, Higashimoto Y, Roy S, Anderson CW, Appella E. Damage-mediated Phosphorylation of Human p53 Threonine 18 through a Cascade Mediated by a Casein 1-like Kinase. Journal of Biological Chemistry. 2000;275:9278-83
54. Ou Y-H, Chung P-H, Sun T-P, Shieh S-Y. p53 C-Terminal Phosphorylation by CHK1 and CHK2 Participates in the Regulation of DNA-Damage-induced C-Terminal Acetylation. Mol Biol Cell. 2005;16:1684-95
55. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS. et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nature Cell Biology. 2006;8:1074-83
56. Watanabe F, Fukazawa H, Masutani M, Suzuki H, Teraoka H, Mizutani S. et al. Poly(ADP-ribose) polymerase-1 inhibits ATM kinase activity in DNA damage response. Biochemical and Biophysical Research Communications. 2004;319:596-602
57. Valenzuela MT, Guerrero R, Nunez MI, Ruiz de Almodovar JM, Sarker M, de Murcia G. et al. PARP-1 modifies the effectiveness of p 53-mediated DNA damage response. Oncogene. 2002;21:1108-16
58. Sakaguchi K, Herrera JE, Saito Si, Miki T, Bustin M, Vassilev A. et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. GENES & DEVELOPMENT. 1998;12:2831-41
59. Zhu W-G, Hileman T, Ke Y, Wang P, Lu S, Duan W. et al. 5-Aza-2'-deoxycytidine Activates the p53/p21Waf1/Cip1 Pathway to Inhibit Cell Proliferation. Journal of Biological Chemistry. 2004;279:15161-6
60. Wang H, Zhao Y, Li L, McNutt MA, Wu L, Lu S. et al. An ATM- and Rad3-related (ATR) Signaling Pathway and a Phosphorylation-Acetylation Cascade Are Involved in Activation of p53/p21Waf1/Cip1 in Response to 5-Aza-2'-deoxycytidine Treatment. Journal of Biological Chemistry. 2008;283:2564-74
61. Wang H, Zhou W, Zheng Z, Zhang P, Tu B, He Q. et al. The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA Repair. 2011;11:146-56
62. Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proceedings of the National Academy of Sciences. 2009;106:6591-6
63. Chernov MV, Bean LJH, Lerner N, Stark GR. Regulation of ubiquitination and degradation of p53 in unstressed cells through C-terminal phosphorylation. Journal of Biological Chemistry. 2001;276:31819
64. Topisirovic I, Gutierrez GJ, Chen M, Appella E, Borden KLB, Ronai ZA. Control of p53 multimerization by Ubc13 is JNK-regulated. Proceedings of the National Academy of Sciences. 2009;106:12676-81
65. Laine A, Topisirovic I, Zhai D, Reed JC, Borden KLB, Ronai Ze. Regulation of p53 Localization and Activity by Ubc13. Mol Cell Biol. 2006;26:8901-13
66. Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nature Cell Biology. 2007;9:428-35
67. Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H. et al. Methylation of p53 by Set7/9 Mediates p53 Acetylation and Activity In Vivo. Molecular Cell. 2008;29:392-400
68. Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F. et al. Methylation-Acetylation Interplay Activates p53 in Response to DNA Damage. Molecular and Cellular Biology. 2007;27:6756-69
69. Espinosa JM, Emerson BM. Transcriptional Regulation by p53 through Intrinsic DNA/Chromatin Binding and Site-Directed Cofactor Recruitment. Molecular Cell. 2001;8:57-69
70. Wu S-Y, Chiang C-M. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. The EMBO Journal. 2009;28:1246-59
71. Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276-88
72. Zhao X, Gan L, Pan H, Kan D, Majeski M, Adam SA. et al. Multiple elements regulate nuclear/cytoplasmic shuttling of FOXO1: characterization of phosphorylation-and 14-3-3-dependent and-independent mechanisms. Biochemical Journal. 2004;378:839
73. Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR. et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. The EMBO Journal. 2002;21:2263-71
74. Perrot V, Rechler MM. Characterization of Insulin Inhibition of Transactivation by a C-terminal Fragment of the Forkhead Transcription Factor Foxo1 in Rat Hepatoma Cells. Journal of Biological Chemistry. 2003;278:26111-9
75. Verhey KJ, Gaertig J. The Tubulin Code. Cell Cycle. 2007;6:2152-60
76. Buratowski S. the CTD code. Nature Structural Biology. 2003;10:679-80
77. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074-80
78. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41-5
79. Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 Inhibits Its Ubiquitination by Mdm2. Journal of Biological Chemistry. 2002;277:50607-11
80. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M. et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105-8
81. Kachirskaia I, Shi X, Yamaguchi H, Tanoue K, Wen H, Wang EW. et al. Role for 53BP1 Tudor Domain Recognition of p53 Dimethylated at Lysine 382 in DNA Damage Signaling. Journal of Biological Chemistry. 2008;283:34660-6
82. Huang J, Dorsey J, Chuikov S, Zhang X, Jenuwein T, Reinberg D. et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. Journal of Biological Chemistry. 2010;285:9636
83. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- Versus Polyubiquitination: Differential Control of p53 Fate by Mdm2. Science. 2003;302:1972-5
84. Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA. et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629
85. Sakaguchi K, Sakamoto H, Lewis MS, Anderson CW, Erickson JW, Appella E. et al. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry. 1997;36:10117-24
86. Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak Peter M. A Mechanism for Cell-Cycle Regulation of MAP Kinase Signaling in a Yeast Differentiation Pathway. Cell. 2007;128:519-31
87. Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S. et al. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853-7
88. Li AG, Piluso LG, Cai X, Gadd BJ, Ladurner AG, Liu X. An acetylation switch in p53 mediates holo-TFIID recruitment. Molecular Cell. 2007;28:408-21
89. Rajagopalan S, Jaulent AM, Wells M, Veprintsev DB, Fersht AR. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Research. 2008;36:5983-91
90. Rajagopalan S, Sade RS, Townsley FM, Fersht AR. Mechanistic differences in the transcriptional activation of p53 by 14-3-3 isoforms. Nucleic Acids Research. 2010;38:893-906
91. Matthew J.F. Waterman ESS. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nature Genetics. 1998;19:175-8
92. Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nature Reviews Molecular Cell Biology. 2005;6:610-21
93. Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nature Cell Biology. 2003;5:461-6
94. Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J. et al. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. Journal of Biological Chemistry. 2003;278:21323
95. Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B. et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431-9
96. Walter W, Clynes D, Tang Y, Marmorstein R, Mellor J, Berger SL. 14-3-3 Interaction with Histone H3 Involves a Dual Modification Pattern of Phosphoacetylation. Mol Cell Biol. 2008;28:2840-9
97. Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. GENES & DEVELOPMENT. 2002;16:583
98. Derbyshire DJ, Basu BP, Serpell LC, Joo WS, Date T, Iwabuchi K. et al. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. The EMBO Journal. 2002;21:3863-72
99. Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT Repeats As Phosphopeptide-Binding Modules Involved in Protein Targeting. Science. 2003;302:636-9
100. Yu X, Chini CCS, He M, Mer G, Chen J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science. 2003;302:639-42
101. Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nature Reviews Molecular Cell Biology. 2006;7:473-83
102. Barford D, Hu SH, Johnson L. Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP* 1,* 2. Journal of molecular biology. 1991;218:233-60
103. Turowski P, Fernandez A, Favre B, Lamb NJ, Hemmings BA. Differential methylation and altered conformation of cytoplasmic and nuclear forms of protein phosphatase 2A during cell cycle progression. The Journal of Cell Biology. 1995;129:397-410
104. Ulrich HD. SUMO modification: wrestling with protein conformation. Current Biology. 2005;15:R257-R9
105. Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G. et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849-53
106. Lippens G, Landrieu I, Smet C. Molecular mechanisms of the phospho-dependent prolyl cis/trans isomerase Pin1. FEBS Journal. 2007;274:5211-22
107. McKinney K, Mattia M, Gottifredi V, Prives C. p53 Linear Diffusion along DNA Requires Its C Terminus. Molecular Cell. 2004;16:413-24
108. Anderson ME, Woelker B, Reed M, Wang P, Tegtmeyer P. Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: implications for regulation. Molecular and Cellular Biology. 1997;17:6255
109. Hupp T, Meek D, Midgley C, Lane D. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875-86
110. Petty TJ, Emamzadah S, Costantino L, Petkova I, Stavridi ES, Saven JG. et al. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. The EMBO Journal. 2011;30:2167-76
111. Brent MM, Anand R, Marmorstein R. Structural Basis for DNA Recognition by FoxO1 and Its Regulation by Posttranslational Modification. Structure. 2008;16:1407-16
112. Finley D, Bartel B, Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394
113. Reinberg D, Sims RJ3rd. Is there a code embedded in proteins that is based on post-translational modifications? Nature Reviews Molecular Cell Biology. 2008;9:815-20
114. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou M-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491-6
115. Winston F, Allis CD. The bromodomain: a chromatin-targeting module? Nature Structural Biology. 1999;6:601-4
116. Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120-4
117. Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC. et al. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. The EMBO Journal. 2001;20:5232-41
118. Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O. et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100-3
119. Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T. et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96
120. Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J. et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86
121. Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends in Biochemical Sciences. 2006;31:35-40
122. Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L. et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397-403
123. Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and Function of a Human TAFII250 Double Bromodomain Module. Science. 2000;288:1422-5
124. Ahringer J. NuRD and SIN3: histone deacetylase complexes in development. Trends in Genetics. 2000;16:351-6
125. Tsai W-W, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S. et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927-32
126. Li B, Gogol M, Carey M, Lee D, Seidel C, Workman JL. Combined Action of PHD and Chromo Domains Directs the Rpd3S HDAC to Transcribed Chromatin. Science. 2007;316:1050-4
127. Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2004;24:1653-62
128. Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nature Reviews Molecular Cell Biology. 2005;6:197-208
129. Bell S, Klein C, Müller L, Hansen S, Buchner J. p53 contains large unstructured regions in its native state. Journal of molecular biology. 2002;322:917-27
130. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557-82
131. Serber Z, Ferrell Jr JE. Tuning bulk electrostatics to regulate protein function. Cell. 2007;128:441-4
132. Jõers A, Jaks V, Kase J, Maimets T. p53-dependent transcription can exhibit both on/off and graded response after genotoxic stress. Oncogene. 2004;23:6175-85
133. Cheung P, Allis CD, Sassone-Corsi P. Signaling to Chromatin through Review Histone Modifications. Cell. 2000;103:263-71
134. Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D. et al. Regulation of the p300 HAT domain via a novel activation loop. Nature Structural & Molecular Biology. 2004;11:308-15
135. Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nature Reviews Molecular Cell Biology. 2008;9:702-12
136. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. Journal of Biological Chemistry. 2000;275:8945
137. Okamoto K, Taya Y, Nakagama H. Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS letters. 2009;583:2710-4
138. Hay TJ, Meek DW. Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS letters. 2000;478:183-6
139. Mayo LD, Turchi JJ, Berberich SJ. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Research. 1997;57:5013
140. Maya R, Balass M, Kim ST, Shkedy D, Leal JFM, Shifman O. et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. GENES & DEVELOPMENT. 2001;15:1067
141. Winter M, Milne D, Dias S, Kulikov R, Knippschild U, Blattner C. et al. Protein kinase CK1 phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry. 2004;43:16356-64
142. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proceedings of the National Academy of Sciences. 2001;98:11598
143. Miyauchi Y, Yogosawa S, Honda R, Nishida T, Yasuda H. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. Journal of Biological Chemistry. 2002;277:50131
144. Buschmann T, Lerner D, Lee CG, Ronai Z. The Mdm-2 amino terminus is required for Mdm2 binding and SUMO-1 conjugation by the E2 SUMO-1 conjugating enzyme Ubc9. Journal of Biological Chemistry. 2001;276:40389
145. Wang X, Taplick J, Geva N, Oren M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS letters. 2004;561:195-201
146. Duan S, Yao Z, Hou D, Wu Z, Zhu W-g, Wu M. Phosphorylation of Pirh2 by Calmodulin-dependent kinase II impairs its ability to ubiquitinate p53. The EMBO Journal. 2007;26:3062-74
147. Dornan D, Shimizu H, Mah A, Dudhela T, Eby M, O'Rourke K. et al. ATM Engages Autodegradation of the E3 Ubiquitin Ligase COP1 After DNA Damage. Science. 2006;313:1122-6
148. Karanam B, Jiang L, Wang L, Kelleher NL, Cole PA. Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. Journal of Biological Chemistry. 2006;281:40292
149. Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT. et al. Phosphorylation regulates SIRT1 function. PLoS One. 2008;3:e4020
150. Ford J, Ahmed S, Allison S, Jiang M, Milner J. JNK2-dependent regulation of SIRT1 protein stability. Cell cycle (Georgetown, Tex). 2008;7:3091
151. Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L. et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proceedings of the National Academy of Sciences. 2011;108:1925-30
152. Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. European Journal of Biochemistry. 2001;268:2764-72
153. Benayoun BA, Veitia RA. A post-translational modification code for transcription factors: sorting through a sea of signals. Trends in Cell Biology. 2009;19:189-97
154. Turner BM. defining an epigenetic code. Nature Cell Biology. 2007;9:2-6
155. Allison SJ, Milner J. Loss of p53 Has Site-Specific Effects on Histone H3 Modification, Including Serine 10 Phosphorylation Important for Maintenance of Ploidy. Cancer Research. 2003;63:6674-9
156. Warnock LJ, Adamson R, Lynch CJ, Milner J. Crosstalk between site-specific modifications on p53 and histone H3. Oncogene. 2007;27:1639-44
157. Ljungman M. Dial 9-1-1 for p53: mechanisms of p53 activation by cellular stress. Neoplasia (New York, NY). 2000;2:208
158. Braithwaite AW, Royds JA, Jackson P. The p53 story: layers of complexity. Carcinogenesis. 2005;26:1161
159. Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Molecular Cell. 2004;13:251-63
160. Hannich JT, Lewis A, Kroetz MB, Li SJ, Heide H, Emili A. et al. Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. Journal of Biological Chemistry. 2005;280:4102
161. Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73α by SUMO-1. Journal of Biological Chemistry. 2000;275:36316
162. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains-from structures to functions. Nature Reviews Molecular Cell Biology. 2009;10:659-71
163. West LE, Roy S, Lachmi-Weiner K, Hayashi R, Shi X, Appella E. et al. The MBT Repeats of L3MBTL1 Link SET8-mediated p53 Methylation at Lysine 382 to Target Gene Repression. Journal of Biological Chemistry. 2010;285:37725
164. Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends in Molecular Medicine. 2010;16:528-36
165. Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell. 2008;133:930
Author contact
![]() Corresponding author: Tel: 86-1082202235. E-mail: zhuweiguoedu.cn.
Corresponding author: Tel: 86-1082202235. E-mail: zhuweiguoedu.cn.