Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(5):731-760. doi:10.7150/ijbs.4262 This issue Cite
Review
The Effect of Physiological Stimuli on Sarcopenia; Impact of Notch and Wnt Signaling on Impaired Aged Skeletal Muscle Repair
Susan Tsivitse Arthur ![]() , Ian D. Cooley
, Ian D. Cooley
Department of Kinesiology, Laboratory of Systems Physiology, University North Carolina - Charlotte, Charlotte, NC 28223, USA.
Received 2012-2-21; Accepted 2012-4-6; Published 2012-5-23
Abstract
The age-related loss of skeletal muscle mass and function that is associated with sarcopenia can result in ultimate consequences such as decreased quality of life. The causes of sarcopenia are multifactorial and include environmental and biological factors. The purpose of this review is to synthesize what the literature reveals in regards to the cellular regulation of sarcopenia, including impaired muscle regenerative capacity in the aged, and to discuss if physiological stimuli have the potential to slow the loss of myogenic potential that is associated with sarcopenia. In addition, this review article will discuss the effect of aging on Notch and Wnt signaling, and whether physiological stimuli have the ability to restore Notch and Wnt signaling resulting in rejuvenated aged muscle repair. The intention of this summary is to bring awareness to the benefits of consistent physiological stimulus (exercise) to combating sarcopenia as well as proclaiming the usefulness of contraction-induced injury models to studying the effects of local and systemic influences on aged myogenic capability.
Keywords: sarcopenia, physiological stimuli
Introduction
With aging there is a decline in the function of multiple organ systems including heart, brain, nerves, blood, skin and skeletal muscle. Sarcopenia is the age-associated loss of muscle mass and function, which may appear as early as 50 years (y) in humans (1). The prevalence of sarcopenia greatly increases over the age of 80 y with reports of 50% or greater loss of muscle strength (2, 3).
The causes of sarcopenia appear to be multifactorial and include hormones, sedentary lifestyle, smoking, genetics, body size and composition. However, the cellular aspects of age-associated sarcopenia are not well elucidated. Because people are living longer, understanding the mechanisms underlying sarcopenia is critical to the development of therapeutic and prevention strategies that will decrease sarcopenia-associated morbidity and mortality in the aging population.
A contributing factor to sarcopenia is a diminished ability of aged muscle to repair itself following injury (4-8). There are multiple suggested mechanisms for the impaired muscle regenerative process in aged muscle including loss of satellite cell number or function, decreased myoblast proliferation or weakened differentiation states. However, more studies are needed before a definitive answer can be reached. Although satellite cell activity is rigorous during muscle formation in the young, with age there is an attrition in satellite cell function (5, 7, 9-11). The novel finding that impaired Notch signaling may be partly responsible for the loss of myogenic potential in aged muscle is intriguing, and provides a potential clue into the mechanisms underlying sarcopenia (7, 12, 13). In addition, there may be a dysfunctional orchestration of the signaling pathways, Notch and Wnt that result in impaired muscle repair in the aged (14-17). The coordination of these signaling pathways during postnatal myogenesis is not well defined, nor is their contribution to impaired myogenic potential in aged muscle.
Physiological stimuli (exercise, injurious muscle contractions, hypertrophy models) are known to increase Notch and Wnt signaling in young muscle (18-23). However, little is known about the influence of physiological stimuli on the expression and coordination of Notch and Wnt signaling in repairing aged muscle. Studying the effect of physiological stimuli on Notch and Wnt coordination during the regenerative process of aged muscle may lead to an effective interventional strategy for sarcopenia. Consequently, such studies deserve further exploration. The purpose of this review is to examine the cellular regulation of sarcopenia, including impaired muscle regenerative capacity in the aged, and to discuss if physiological stimuli have the potential to slow the loss of myogenic potential that is associated with sarcopenia. In addition, this review article will discuss the effect of aging on Notch and Wnt signaling, and whether physiological stimuli have the ability to restore Notch and Wnt signaling resulting in rejuvenated aged muscle repair. A better understanding of these issues may lead to novel therapeutic strategies to prevent the loss of muscle quality seen in sarcopenia.
Impact of sarcopenia
Sarcopenia (also called senile muscle atrophy) is an age-related loss of skeletal muscle mass and function that is often determined by measuring the skeletal mass index (24). The percentage of skeletal muscle index is calculated by dividing muscle mass by the squared height, or by dividing muscle mass by body mass (24, 25). Sarcopenia is experienced primarily in the lower extremities, which may be related to reduced physical activity with age, or to greater loss of motor units in the legs than arms (25, 26). Evidence suggests that the age-related loss of muscle mass is the cause of poor strength, yet the impairment of muscle strength and power (dynapenia) is more profound than the loss of muscle mass (27).
There is a dichotomy in the relationship between age-associated mortality and sarcopenia. The negative attributes associated with aging including poor nutrition, sedentary lifestyle, pain associated with range of motion, as well as the degenerative cellular manifestations cause an inability to use the vital organ, skeletal muscle resulting in muscle atrophy. With the loss of muscle mass, there is an inability to perform activities of daily living which exacerbates the loss of strength, leading to disabilities, nursing home admissions and ultimately increased mortality (28). This review article will discuss the variety of manifestations associated with aging that cause sarcopenia. Sarcopenia exerts its effects on other systems and include, but are not limited to: cardiovascular, metabolic, and bone (1, 29, 30). Since skeletal muscle is a component of cardiorespiratory fitness, age-associated skeletal muscle atrophy results in poor oxyten uptake by skeletal muscle and sedentary lifestyle weakens cardiovascular function. In addition, there is impaired oxidative metabolism in atrophic skeletal muscle, resulting in poor glucose regulation and metabolic disease. Abnormal gait and disabilities ensues with muscle atrophy and sedentary lifestyle as well as the bone disease osteoporosis (muscle-associated force applied to the bone is needed for good health fitness) (1, 29, 30). Sarcopenia negatively influence other organs whose destructive outcomes result in loss of independence, and ultimately increased morbidity (1, 30).
Causes of sarcopenia
There are a variety of factors that contribute to sarcopenia, including both environmental and biological factors. For example, poor nutrition and a sedentary lifestyle are contributors to sarcopenia (2, 29). The loss of appetite that afflicts many elderly individuals leads to decreased food intake and Vitamin D and protein deficiencies. This may impair protein synthesis and ultimately decrease muscle mass (2, 29). Edstrom et al. suggest that dietary intervention may be a tool to combat sarcopenia (26). In addition to loss of appetite, many elderly individuals suffer from orthopedic pathologies, and associate movement with pain, resulting in decreased physical activity and ultimately loss of muscle mass and strength (30). However, elderly individuals who are highly active (such as Masters Athletes) also have significantly less muscle mass than their younger counterparts, suggesting that disuse may not be the sole contributor to sarcopenia (30, 31). Thus, other age-related causes of sarcopenia, besides an increased sedentary lifestyle, need to be delineated (25). Although lifestyle is an important player in the development of sarcopenia, the majority of contributors to age-related loss of muscle efficiency involve multiple biological systems such as: central and peripheral nervous systems, endocrine, immune, and metabolic systems (8, 25, 28-30, 32, 33). This review will discuss what are thought to be the major contributing factors underlying the physiological manifestations of sarcopenia.
I. Molecular and Cellular Manifestations associated with Sarcopenia
a. Increased intramuscular fibrosis and adipose tissue: The tissue composition of sarcopenic skeletal muscle is altered and consists predominately of connective and adipose tissue, a condition termed myosteatosis (25, 28). In obese aged individuals, this occurrence is termed “Sarcopenic Obesity” (25, 28). Increased fibrosis within the sarcopenic muscle may be related to elevated extracellular matrix protein (collagen) levels, as well as the accumulation of debris from impaired protein degradation (14, 26, 34). In addition, there is greater fibronectin expression in aged myofiber explants compared to young myofiber explants (14).
b. Increased pro-inflammatory cytokines TNFα and IL-6: Aging is associated with a state of chronic, low inflammation. There are many reports of increased levels of the pro-inflammatory cytokines tumor necrosis factor α (TNFα) and interleukin- 6 (IL-6) in the systemic circulation of the elderly (35-42). For example, there was a 2.8 fold increase in TNFα expression in skeletal muscle of aged (~ 70 y) male subjects compared to young (~20 y) male subjects (38). Phillips et al. also reported increased expression of TNFα in soleus and vastus lateralis (VL) of aged (26 month (mo)) rats relative to young (6 mo) rats (39). Furthermore, centurions were found to have significantly higher plasma TNFα levels than younger (18 - 30 y) controls with corresponding elevations of IL-6 (37). Studies report a link between elevated plasma IL-6 with age and increased mortality (40-42). Roubenoff et al. reported increased plasma levels of IL-6 in aged (~ 79 y) subjects relative to young (~ 39 y) controls. However, there was no difference in plasma TNFα levels between the age groups (42). High levels of IL-6 and TNFα are associated with a multitude of age-related diseases including obesity, cardiovascular diseases, type II diabetes and sarcopenia (35, 36, 43). It should be noted however, that some reports have not found differences in plasma and skeletal muscle TNFα or IL-6 levels between young and aged models; but rather suggest that the aged environment may be more sensitive to the effects of these pro-inflammatory cytokines (36).
Although the mechanism for the potential elevation of TNFα and IL-6 with age, and the relationship of these cytokines to sarcopenia are not well defined, they may be related to increased levels of adipose tissue in the elderly (1, 30). Adipocytes secrete IL-6 and TNFα as well as the adipokines leptin and adiponectin, which promote inflammation. Pro-inflammatory cytokines and adipokines deter muscle mass formation and promote fat mass accumulation (28, 29). Elevated TNFα in aged muscle is associated with decreased muscle force production (44, 45). TNFα is also linked to sarcopenia because this pro-inflammatory cytokine is known to be associated with other factors that contribute to sarcopenia including protein degradation, reactive oxygen species (ROS) accumulation and apoptosis (35, 46). In addition, TNFα may be associated with sarcopenia by promoting insulin resistance, delaying muscle repair, and exacerbating the pro-inflammatory response by up-regulating IL-6 (25, 43, 45-47).
Because IL-6 has both pro- and anti-inflammatory characteristics and has effects on muscle growth and atrophy, it is difficult to discern the role of IL-6 in the development of sarcopenia. There is a negative correlation between IL-6 and skeletal muscle strength in the elderly, and over-expression of IL-6 is associated with muscle atrophy (48, 49) IL-6 may contribute to insulin resistance and inhibit insulin-like growth factor-1 (IGF-1), which promotes protein degradation during sarcopenia (47, 50). Inhibiting IL-6 with an antibody or an anti-inflammatory reagent results in increased protein synthesis and a rescue of the loss of muscle mass (51, 52). Additional research is needed to delineate the relationship and contribution of TNFα and IL-6 to sarcopenia.
c. Decreased sex hormones: As one ages, there is a direct correlation between the levels of sex hormones and muscle mass suggesting that depletion of testosterone and estrogen may contribute to sarcopenia (1, 8). In addition, it is suggested that the age-associated decline in estrogen and testosterone are related to increases in levels of the pro-inflammatory cytokines IL-6 and TNFα, which may accelerate the loss of muscle mass during sarcopenia (8, 53, 54). With aging, there is also a correlation between decreased sex hormone levels and a decline in the growth factors of growth hormone (GH) and IGF-1, which may contribute to sarcopenia (54, 55). Postmenopausal (58-70 y) women possess lower GH levels than premenopausal (45-51 y) women, and the lack of GH is known to promote intramuscular fat accumulation and loss of muscle mass (8, 55). Ferrando et al. report that aged (> 60 y) men who were administered testosterone therapy showed increased IGF-1 protein levels (56).
Between the age ranges of young (20 - 29 y) and aged (70 - 84 y), bioavailable testosterone drops ~ 4.2 fold and the testosterone precursor dehydroepiandrosterone (DHEA) falls ~ 67 fold (57). Between the ages of 70 and 102 years, testosterone levels decrease with a corresponding decline in muscle strength, and with testosterone supplementation, there is a rescue of muscle mass and function (56-60). Ferrando et al. reported that aged (> 60 y) male subjects who were administered testosterone supplementation for 6 mo experienced gains in leg and arm muscle strength as well as increased muscle protein synthesis (56). Sinha-Hikim et al. reported similar findings in which aged (60-75 y) males who were administered testosterone enanthate for 20 weeks (wk) displayed increased cross sectional area of VL muscle and increased expression of markers of myogenicity (58). Furthermore, aged (22 mo) mice given a testosterone implant for 2 mo experienced diminished oxidative stress and myostatin levels, as well as increased myogenicity (60). These data show that testosterone correlates with sarcopenia, muscle mass and function as well protein synthesis function. Consequently, restoring testosterone levels in aged skeletal muscle may be favorable for the prevention or reversal of sarcopenia.
The decline in expression of estrogen in women that contributes to menopause (average age of onset is 51.4 y) (57) is associated with sarcopenia (8, 61). Loss of estrogen may promote body composition changes, including a loss of muscle mass, but an increase in adipose tissue as well as a redistribution of body fat to the visceral region (54, 61, 62). Maltais et al. suggests that estrogen may prevent fat accumulation within skeletal muscle and may have a direct relationship with lipoprotein lipase (which catalyzes triglyceride utilization). Thus, increased intramuscular fat in postmenopausal women may be related in part to deceased estrogen levels. Decreased muscle strength correlates with the age-associated loss of estrogen (54). Replacing estrogen levels with hormone replacement therapy (HRT) to increase muscle mass and strength is controversial (61, 63). Some reports show that HRT increases muscle mass and strength (61, 62). Taking HRT for one year resulted in increased quadriceps cross sectional area (6.3%) and decreased quadriceps intramuscular fat (4.9%) (61). However, there are studies that suggest HRT may not be an ideal tool for sarcopenia as there were no changes in muscle mass or strength with HRT and there are reports of significant negative consequences associated with HRT such as increased risks of cancer and cardiovascular diseases including stroke (54, 63-65). Rossouw et al. report a 26% and 37% increase in invasive breast cancer and colorectal cancer respectively (65). It is interesting to note that women over the age of 65 who were on estrogen replacement therapy for three years did not experience any changes in body fat percentage or physical performance (64). The age-associated loss of sex hormones contributes to sarcopenia directly, but also influences other factors that exacerbate the loss of muscle mass and function and the accumulation of intramuscular adipose tissue. Thus although the decline of sex hormone levels with aging contributes to sarcopenia, straightforward replacement of under-produced estrogen or testosterone may not be the most successful treatment strategy because these hormones also interact with other systems (inflammation, growth factors).
d. Alterations to muscle fibers and motor units: Both muscle fiber loss and decrease in skeletal muscle cross-sectional area are associated with sarcopenia (66). There is a switch of muscle fiber types from type II to type I, and a greater loss of type II muscle fibers than type I in aged skeletal muscle (25, 31, 67, 68). The VL of aged (~ 68 y) men had a higher percentage of fibers expressing Myosin heavy chain (MHC) type I (~ 20%) relative to VL of young (~ 28 y) subjects (~ 8%) (31). In addition to the loss of muscle fibers is an age-associated remodeling of the muscle architecture in which the muscle fiber fascicle length and the insertion angle decreases and there is a loss of sarcomeres (69). Multiple mechanisms appear to underlie the structural alteration of the skeletal muscle architecture, including a decreased rate of protein synthesis within the aged muscle. These changes ultimately result in decreased aged muscle force production (25).
Motor unit alterations may be a contributing factor to the age-associated loss of Type II muscle fibers (70). These motor unit alterations include a decrease in firing frequency and motor unit recruitment, loss of Type II motor units and a reduced number of motoneurons innervating muscle fibers, especially Type II muscle fibers which are then more susceptible to muscle atrophy and possibly sarcopenia (66, 71-73). Accelerated denervation may be related to elevated oxidative stress and accumulation of dysfunctional protein machinery at the neuromuscular junction within aged skeletal muscle (74). Using electron microscopy images to study the neuromuscular junction (NMJ) structure, superoxide dismutase knockout transgenic mice displayed greater NMJ degeneration with dysfunctional mitochondrial activity in close proximity as well as decreased contractile force production relative to wild type, suggesting impaired neuromuscular junction contributes to sarcopenia (75). In addition, there is decreased presence of ciliary neurotrophic factor and vascular endothelial growth factor, both of which are important for motorneuron stability (25). Remaining denervated muscle fibers attempt to compensate by re-innervating the surviving motor units, which consequently change the fiber type to slow-twitch (Type I). However the accelerated rate of denervation is faster than the decreased rate of reinnervation, resulting in loss of muscle function (25, 30, 66, 73, 75). Some suggest that degeneration within the neuromuscular system may be the greatest contributing factor to sarcopenia, while others contend that the muscle fiber denervation plays only a minor role (26, 76). Edstrom et al. reports that there is insignificant motorneuron loss during aging and suggests that impaired reinnervation of the myofiber is a cause of poor muscle regenerative response and sarcopenia rather than motorneuron denervation (Edstrom #26). Although correlations of motorneuron degeneration and sarcopenia are reported, more research is needed to provide compelling evidence that motorneuron degeneration is a major player in the development of sarcopenia. Potential studies could include testing if reinnervation is the limiting factor by comparing markers of sarcopenia in denervated muscle that was exposed to the microenvironment that promotes reinnervation or a microniche that inhibits reinnervation.
e. Protein synthesis/degradation imbalance: A disruption of the balance between protein synthesis and protein degradation, resulting in the accumulation of damaged proteins, is also associated with aging skeletal muscle and sarcopenia (1, 77, 78). There is increased protein modification with aging possibly related to oxidative stress (78). Exacerbating the problem is the fact that the primary pathways used to remove modified proteins (proteasome system, autophagy and lysosomal degradation) are less active in aged muscle (1, 78, 79). For example, in the lysosomal pathway in aged muscle, there is diminished delivery of membrane proteins or organelles to the lysosome and fusion of vacuoles with lysosomes (79). The attrition of protein degradation pathways results in accumulation of damaged proteins.
The rate of protein synthesis in aged muscle is reduced, with reports of decreased myofibrillar protein content and MHC expression (77, 78). There are multiple causes of decreased protein synthesis in aged skeletal muscle, such as underproduction of circulating and tissue-associated GH and IGF-1 isoforms (80, 81). Low levels of IGF-1 in aged muscle is related to high expression of muscle growth inhibitors including the pro-inflammatory cytokines TNFα and IL-6 (2, 38, 73, 82). In skeletal muscle of aged (70 y) subjects, there is a 45% decrease in growth hormone receptor protein (GHR) and IGF-1 mRNA, along with a 2.8 fold increase in TNFα mRNA relative to young (20 y) subjects (38). Aged women with lower IGF-1 levels and greater IL-6 expression relative to younger counterparts, demonstrate aspects of disability and increased mortality (28, 83). This opposing relationship between IGF-1 and the cytokines TNFα and IL-6 are considered indicators for sarcopenia (83).
f. Decreased IGF-1 and mRNA translation: The decreased presence of IGF-1 in aged skeletal muscle may contribute to sarcopenia by severely impacting protein synthesis. IGF-1 has anabolic effects on muscle protein content by inhibiting protein degradation and promoting myogenesis. Thus, IGF-1 attrition in aged skeletal muscle is associated with less protein synthesis and muscle growth (84, 85). In the presence of IGF-1, aged (25 mo) rats increased total protein content by 27% in gastrconemius muscle relative to control (84). In addition, there was increased muscle mass and force production in mdx mice (mouse model that represents Duchenne muscular dystrophy) that were treated with IGF-1 (86, 87). Injection of an adeno-virus with overexpressed IGF-IEa into aged (27 mo) mice resulted in increased muscle mass and strength (88). IGF-1 administration to the aged skeletal muscle may negate the impaired protein synthesis observed in sarcopenia.
A reduction in IGF-1 may also influence protein synthesis in aged skeletal muscle via inefficient mRNA translation (79, 89-92). The mammalian target of rapamycin (mTOR) signaling pathway is important for translation initiation and is therefore critical for muscle protein synthesis. One mechanism that activates mTOR signaling is the IGF-1/PI3k/serine/threonine kinase (Akt) pathway. Downstream effectors of mTOR signaling include 70-kilodalton ribosomal S6 protein kinase (p70s6k), eukaryotic translation initiation factor 4E binding protein 1 (4E-BP-1), eukaryotic translation initiation factor 4E (eIF-4E), and ribosomal protein S6 kinase (S6K1). Aged rodent muscles have reduced mTOR (Akt phosphorylation) and p70s6k signaling (89-93). Paturi et al. reported that aged (36 mo) mouse soleus had less Akt (and phosphorylated Akt), p70s6k and phosphorylated p70s6k), phosphorylated mTOR, S6 ribosomal protein, and AMP activated kinase (AMPK) than young (6 mo) mouse soleus (91). With underproduction of IGF-1 in aged skeletal muscle, there may be impaired mTOR signaling and mRNA translation, thereby imposing a barrier to protein synthesis in aged muscle.
g. Increased myostatin: Additionally, decreased protein synthesis during sarcopenia may be a result of elevated myostatin expression in aged skeletal muscle (32, 73, 94, 95). There is a two-fold increase in myostatin expression within skeletal muscle of aged (70 y) subjects relative to young (20 y) subjects (38). GH and IGF-1 may have an inhibitory effect on myostatin, therefore one potential cause of increased myostatin in aged skeletal muscle is the attrition of GH and IGF-1 expression (38, 73, 95, 96). Myostatin may inhibit muscle growth by preventing satellite cell activation, as well as promoting an adipogenic cell fate over myogenicity (95, 97). Inhibiting myostatin may be a key therapeutic target for sarcopenia. Administration of myostatin inhibitors to aged (18 mo) mice resulted in a 12% increase in fiber cross-sectional area and 35% increase in force production by the tibialis anterior (TA) (94). By understanding the mechanisms of the disruption in the pathways for protein degradation and protein synthesis during sarcopenia, therapeutic interventions can be devised to rejuvenate muscle growth and remove damaged muscle proteins in aged muscle.
h. Altered microRNA expression: MicroRNAs (miRNA; miR) are novel post-transcriptional regulators that may contribute to age-related impairments by dysregulating gene expression, either by cleaving mRNA or by inhibiting translation (98-100). Recent evidence suggests that miRNAs play a role in myogenesis (98, 101-107). During human muscle development there is increased expression of muscle specific miRNAs including miR-1,-133a, and -206 (103). During postnatal myogenesis, these specific miRs may play a role in skeletal muscle repair by influencing myoblast proliferation and differentiation (98, 101-108). One week after injecting a mixture of miR-1, - 206 and - 133 into injured muscle, there was accelerated muscle repair (104). miR-133a promotes myoblast proliferation by repressing factors required for muscle differentiation such as serum response factor (SRF), myogenin and MHC (107). miR-1 contributes to myoblast differentiation by inhibiting myocyte-specific enhancer factor 2C transcriptional repressor, histone deacetylast 4 (HDAC4). miR-206 also contributes to myoblast differentiation possibly by inhibiting Pax 7 (101, 106, 107). Inhibiting miR-1 and miR 206 delays myoblast differentiation and there is a concomitant increase in Pax 3 expression (102).
Because it has recently been determined that miRNA are important to myogenesis, it would be of interest to characterize miRNA in aged skeletal muscle. There is a correlation between miRNA expression and aging that may link miRNAs to sarcopenia (99, 109, 110). Drummond et al. profiled miRNAs in VL aged (~ 73 y) men relative to young (~ 31 y) male subjects and observed that miRNAs let-7b and let-7e (from the let-7 family) were elevated in aged muscle relative to young muscle. Let-7 family miRNAs are related to decreased cell cycle regulators during muscle repair, so the authors suggest that decreased muscle repair mechanisms in aged muscle may be a result of elevated Let-7 family miRNA in aged muscle (110). The expression of miRNAs associated with skeletal muscle repair (miR 221 and -181a) are decreased in aged (24 mo) mice quadriceps (109), suggesting that these skeletal muscle miRNAs may be associated with the decreased muscle growth in aged muscle. More research is needed to determine the influence of miRNAs on sarcopenia.
i. Apoptosis: Apoptosis of myofiber myonuclei and satellite cell nuclei within aging skeletal muscle may result in muscle mass attrition that is associated with sarcopenia (33, 111, 112). Apoptosis is a programmed death of a cell or nucleus, which is activated by either extrinsic or intrinsic cues such as activation of death receptors, reactive oxygen species (ROS) accumulation, endoplasmic reticulum stress or mitochondrial stress. Upon activation, components of apoptosis signaling are elevated including caspase 3, cytochrome c, BCL-2 associate-X protein (Bax), apoptosis-inducing factor (AIF), and/or apoptosis activating factors (APAF-1) (33, 111, 112). The end result of myonucleus or cellular apopotsis is DNA fragmentation and destruction of the cell, resulting in the formation of apoptotic bodies (33, 111-113).
Aged skeletal muscle has increased expression of pro-apoptotic proteins and caspases and DNA fragmentation, with a concomitant decrease in expression of anti-apoptotic proteins (112-119). There are elevations in APAF-1, Bax levels and caspase-3 activity (113, 117). Relative to adult (12 mo) rat gastrocnemius, aged (26 mo) rat gastrocnemius expressed higher levels of the pro-apoptotic protein AIF, with a concomitant decrease in the apoptotic inhibitor known as the apoptotic repressor with a caspase recruitment domain (ARC) (120). There are reports of increased anti-apoptotic proteins including XIAP in aged skeletal muscle, which are thought to assist in counteracting the pro-apoptotic environment within the aged muscle. However, the increased anti-apoptotic attempts may be futile because there are also elevations in a pro-apoptotic protein that inhibits XIAP (Smac/DIABLO) (33, 121). If apoptosis does indeed play a role in sarcopenia, then research directed towards decreasing DNA fragmentation and caspases and promoting anti-apoptotic protein expression in aged skeletal muscle is warranted.
j. Telomere shortening: With aging there is cellular senescence in multiple organs. If this process occurs in skeletal muscle, then it may contribute to sarcopenia (4, 122). The “Hayflick Limit”, defined as the limited number of cell replications prior to cell cycle arrest, may be a cause of cell senescence (123). Telomere instability may induce cellular arrest and limit cell replications (124). Telomeres are repeated DNA sequences that form a T loop cap at the end of a linear chromosome and protect the chromosome ends from degradation. Addition of DNA sequences to telomeres occurs in the presence of telomerase (4). With every round of cell division the telomeres shorten and telomerase activity decreases. With increasing age, cells reach the maximum number of cell divisions. At this point, telomere length reaches a critical length and the chromosome may become uncapped. This in turn may result in disruption of chromosome integrity because uncapped telomeres activate signaling pathways that correlate with DNA damage and apoptosis (Figure 1) (124).
Telomere function and attrition with aging.
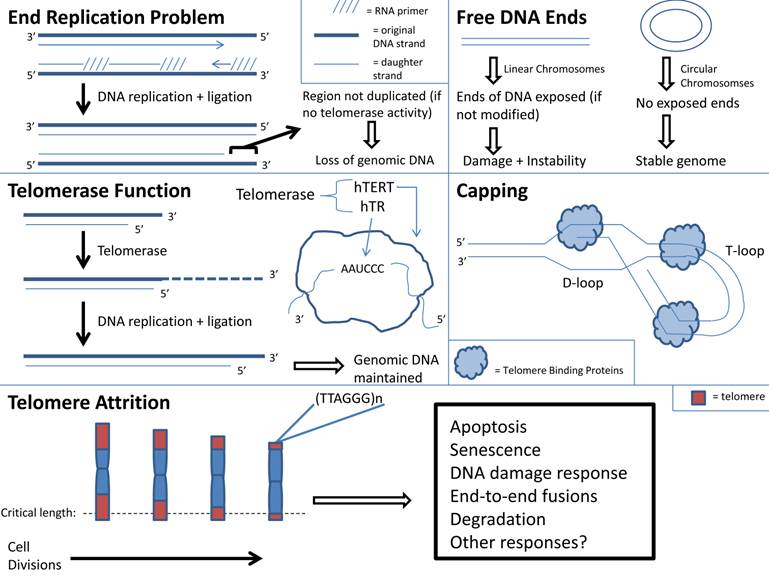
Telomere shortening is used as a marker for aging, and it is recognized that telomere shortening induces premature aging and renders an organism susceptible to disease (122, 125). Mice with deficient telomerase activity exhibit shortened telomeres and have a decreased lifespan (126, 127). There is also a relationship between telomere shortening and senescence of aged stem cells (128, 129). Shortened telomeres within hematopoietic stem cells (HSCs) result in dysfunctional HSCs and increased HSC senescence (128, 129). There is impaired activation and proliferation of HSCs with shortened telomeres (128, 129). Since telomere shortening has been shown to impair stem cell function, it is plausible to consider that telomere shortening occurs within cells of aged skeletal muscle, including the adult skeletal muscle stem cells termed satellite cells. The destructive outcome of telomere shortening in aged skeletal muscle cells may contribute to sarcopenia, but more research is needed. However, it should be noted that investigators have found that satellite cells from aged skeletal muscle maintained their telomerase activity although to a lesser degree than those from young skeletal muscle. This suggests that aged skeletal muscle stem cells have the ability to maintain their myogenic potential (130).
k. Oxidative Stress: Accumulation of reactive oxygen species (ROS), known as oxidative stress within aging skeletal muscle contributes to sarcopenia (1, 4, 131, 132). There are multiple sources of ROS production within aged skeletal muscle (30, 132-134). Lifelong accumulation of mitochondrial DNA (mtDNA) mutations in tissues that undergo high oxidative phosphorylation such as skeletal muscle, results in decreased electron transport chain activity, impaired oxidative phosphorylation and ROS accumulation (30, 132, 133). Pro-inflammatory cytokines such as TNFα, which have been shown to be elevated in aging skeletal muscle, also promote ROS accumulation (134).
ROS (as well as reactive nitrogen species) accumulation within aged skeletal muscle causes tissue degradation, skeletal muscle atrophy, decreased muscle function and increased presence of fibrotic tissue (1, 4, 30, 131-134). Oxidative stress may also induce DNA damage and impair the ability of DNA polymerases to copy strands, resulting in telomere shortening (122). The end result of oxidative stress is decreased longevity, and this is exacerbated by age-associated attrition of antioxidants and heat shock protein levels (8, 30, 131). It would seem reasonable that inhibiting ROS accumulation would increase longevity (135). In transgenic mice that over-express the antioxidant catalase, there is decreased production of ROS and hydrogen peroxide and fewer mitochondrial deletions resulting in a 21% increase in lifespan relative to control (135). However, many researchers report that inhibiting ROS does not prolong lifespan (123, 136, 137). Transgenic mice that over-express the antioxidant, superoxide dismutase did not have an extended lifespan (137). Furthermore, transgenic mice that over-expressed two antioxidants; superoxide dismutase and catalase, did not display an extended lifespan (136). Because of the age-associated accumulation of mtDNA mutations, elevated pro-inflammatory levels and depressed antioxidant expressions, there is a robust presence of oxidative stress within aged skeletal muscle. The propensity for ROS accumulation within aged skeletal muscle may induce DNA damage, deplete energy stores and exacerbate apoptosis, thereby contributing to sarecopenia (111, 132). Devising methods to decrease ROS accumulation within aged skeletal muscle may be a promising interventional strategy for sarcopenia.
There are multiple contributors to sarcopenia that are associated with DNA damage, apoptosis and protein degradation. More research is needed to further delineate these molecular manifestations of aging, and to devise strategies to counteract these deleterious changes. Another contributing factor to sarcopenia is an impaired ability of aged skeletal muscle to regenerate following exposure to injury.
II. Impaired Muscle Regeneration as a Contributor to Sarcopenia
With aging there is a delay in healing of tissue when exposed to injury as evidenced by poor skin healing, delayed bone repair, inability to re-myelinate damaged axons, impaired angiogenesis and delayed colonic mucosal repair (138-140). There is also a deficient regenerative response in aged skeletal muscle following exposure to injury (4-8). While there was complete recovery of muscle mass at 21 days post chemical-induced muscle injury in 3 mo old rat tibialis anterior muscle, there was 40% loss of muscle mass in 31 mo old rat tibialis anterior muscle at the same time point (6). The ability to reinnervate and revascularize is reduced in aged muscle compared to young muscle (141-143). There was significant motor denervation, including loss of motor axon terminals in the gastrocnemius of aged (32 mo) mice (142). Using an in situ blood vessel growth protocol (corneal micropocket assay), Smythe et al. reported that aged (21 mo) rat muscle exhibited less blood vessel growth than young (3 mo) rat muscle (141). In addition to poor reinnervation and revascularization, the muscle contraction apparatus is affected during recovery of aged muscle (144) Lorenzon et al. reported no excitation-contraction (e-c) coupling mechanism present in primary culture myoblasts obtained from aged human muscle biopsies, while young primary culture myoblasts demonstrated e-c coupling at day 6 of differentiation (144). The delay in skeletal muscle repair following injury (chemical-induced, exercise-induced) may be a contributing factor to sarcopenia. Below is a brief discussion of causes of impaired repair of aged skeletal muscle.
a. Cause of impaired aged skeletal muscle repair - resident satellite cell number: A dysregulation of satellite cells could result in impaired repair of aged muscle. Muscle regeneration was compared between young (20 y) and aged (70 y) human subjects who were exposed to 2 wk muscle cast immobilization followed by 3 day or 4 week re-loading (cast removal) (5). During re-loading there was a ~ four - fold decrease in satellite cell activation in aged muscle relative to young muscle, suggesting that aged muscle may possess dysfunctional satellite cells (5). It is unclear if the contributing role of satellite cells to delinquent aged muscle regeneration is related to changes in resident satellite cell numbers, or to their ability to function. It is difficult to determine if the overall number of satellite cells in aged muscle is a contributing factor to poor repair because there is confounding evidence regarding changes in satellite cell number with age. Some investigators report a decrease in satellite cell number with age, while others suggest no change or an increase in resident satellite cell numbers in aged skeletal muscle (5, 7-9, 32, 73, 139, 145-149). Using Pax 7, neural cell adhesion marker (NCAM) and MCadherin satellite cell markers, Carlson et al. reported a decrease in satellite cell number in resting aged (~ 70 y) relative to young (20 y) human muscle (5). Using a mouse model, Shefer et al. showed a 60% decrease in Pax 7 positive-cells of aged (19 - 25 mo) relative to young (3 - 6 mo) EDL myofibers (9). A plausible explanation for the perceived decrease of resident satellite cell number with age is that satellite cells may have reached their “Hayflick limit” in which they have reached the maximum threshold of cell divisions. Although there is a preponderance of evidence supporting the concept of decreased satellite cell number with aging, it should be noted, however, that there are reports stating no differences in satellite cell number with age (11, 150). Using electron microscopy, dystrophin-laminin immunohistochemistry, and MyoD immunohistochemistry techniques for quantifying satellite cells, Brooks et al. did not observe any significant differences in satellite cell numbers between young (5 mo) and aged (24 mo) Fisher rat soleus muscles (150).
b. Cause of impaired aged skeletal muscle repair - altered myogenic program: Independent of the contribution of resident satellite cell number, it is well known that there is a problem with satellite cell function in the myogenic program of aged muscle (7, 9-11, 151). However, exactly what aspect of the myogenic program is defective in aged muscle remains controversial. Some investigators state that myoblast proliferation is the limiting factor for effective age muscle repair, while others suggest that the impairment is due to diminished myotube formation (7, 9-11, 13, 30, 151, 152). Shefer et al. reported a reduced proliferation in aged myoblasts because there was only a 10 fold increase in proliferation of myogenic cells from cultured aged (28 - 33 mo) mouse myofibers (at 6th day of plating relative to 4th day of plating), while myoblast proliferation of young mouse (3 - 4 mo) myofiber cultures increased 15 fold at the same time point. Additionally, there was a 24 h delay in myotube formation in the aged group, but no morphological differences in myotube formation between the young and aged groups were observed (9). Similarly, Conboy et al. reported fewer proliferating myoblasts and myotubes from myofiber explants of aged (23 - 24 mo) relative to young (2 -3 mo) mouse muscle (7). These authors suggest that aged satellite cells retain their myogenic potential but have an impaired ability to activate and proliferate resulting in a delay of myotube formation (7, 9).
However, there is evidence that the differentiation component of the myogenic program is defective in aged muscle (10, 11, 13, 30, 151, 153). Although there was no significant difference in the percentage of desmin-positive cells between myoblasts obtained from young (~ 30y) and aged (~83y) human myofiber explants, there was a significant decrease in the fusion index of aged myotubes relative to young myotubes (11). The authors also reported that the aged myotubes had fewer myonuclei than young myotubes, and that the aged myotubes morphology appeared meager (11). Lees et al. reported that although there was no difference in the profile of proliferating proteins from cultured myogenic precursor cells (mpcs) between young (3 mo) and aged (32 mo) rat muscle, the aged mpcs expressed 50% less of the differentiation proteins myogenin and creatine kinase relative to young mpcs. In addition, there was less expression of p27kip1, and FOXO1 (proteins essential for myoblast differentiation) in the aged relative to young mpcs (151).
Altered expression of myogenic regulatory factors (MRFs) and myostatin may play a role in inhibiting proliferation or differentiation in aged regenerating muscle. Expression of MyoD, myf5, myogenin and MHC are decreased in aged muscle (30, 73). In addition, there may be decreased protein turnover in aged muscle resulting in impaired myosin replacement (30). Furthermore, the presence of free radicals in aged muscle renders the myosin molecule susceptible to oxidation and weakens cross bridge formation (30, 154). There is also decreased expression of E-proteins, which are critical co-regulators of MRFS, but increased expression of Id, an inhibitor of differentiation (30, 155). Id promotes apopotosis and muscle atrophy by inhibiting E-protein/MRF dimerization or by preventing MRFS binding to DNA (which prevents muscle growth) (155-157).
As previously discussed, the muscle growth inhibitor myostatin may contribute to impaired muscle repair (32, 73, 158, 159). Administration of recombinant myostatin to C2C12 cells (a skeletal muscle cell line) inhibited myoblast differentiation and decreased expression of MyoD, myf5, myogenin and p21 (159). The authors suggested that myostatin may inhibit MyoD through increased Smad 3 signaling (a mediator of myostatin signaling that inhibits myogenesis). MyoD expression was recovered when Smad 3 molecule was inhibited (through the expression of dominant-negative Smad 3 in C2C12 cells) (159). There may be a positive-feedback mechanism in which myostatin may inhibit MRF expression, and the loss of MyoD and myogenin may further up-regulate myostatin expression (73, 159).
While there preponderance of evidence shows decreased MRF and elevated myostatin in aged skeletal muscle, it should be acknowledged that there are reports stating either no change in MRF or myostatin expression, or elevations of MRF expression in aged muscle (6, 142, 160-162). Myogenin protein levels were ~ 5.5-fold higher in gastrconemius of aged (32 mo) rats relative to young (4 mo) rats (142). Aged (31 mo) rats exposed to chemical injury displayed elevated myogenin mRNA levels at 21 days post-injury, while myogenin mRNA returned to normal at that time point in 3 mo old rats (6). Myostatin expression in mouse gastrocnemius and plantaris increased from birth until 11 wks of age (161). Although there are studies that suggest aging does not negatively affect myogenic protein and myostatin expression, the authors' support the notion of decreased MRF in aged skeletal muscle that affect muscle repair. The plethora of evidence offer viable mechanisms for impaired MRF expression which occur in aging tissue such as decreased protein synthesis, increased oxidative stress, as well as evidence of increased MRF inhibitor, myostatin.
c. Cause of impaired aged skeletal muscle repair - intrinsic factors: The intrinsic alterations within satellite cells that occur during aging negatively affect satellite cell integrity. With aging, satellite cells may have reached their “Hayflick limit” and reached their cell division threshold, which would result in decreased satellite number and ultimately impaired regeneration (8). In addition, there is increased apoptosis in aged satellite cells and myonuclei, which further depletes the satellite cell pool as judged by differences between aged (31 mo) and young (3 mo) rat EDL satellite cells (33, 163). Cultured aged myofibers with depleted Pax7+ satellite cells became apoptotic resulting in few differentiated myotubes (164).
Cell size regulation is another intrinsic factor that may impact satellite cells (165). Brack et al. suggest that nuclear domain size is maintained with age because the age-associated loss of myonuclei, including loss of satellite cells, causes the cytoplasm to diminish and the size of the muscle fiber decreases. Whether the loss of myofiber nuclei causes muscle atrophy, or if muscle atrophy causes the loss of myonuclei is controversial. Brack et al. suggest that impaired satellite cell function or quantity may contribute to the age-associated loss of myonuclei. Although there is controversy about the ability of aged muscle to regulate nuclear domain size, Brack et al. propose that effective satellite cell function may be critical for maintaining nuclear domain and cell size control, and that impaired satellite cell function in aged muscle may contribute to the loss of myonuclei and age-associated muscle atrophy (165).
Another intrinsic factor that contributes to aging-associated impaired satellite cell function is alteration of RNA pathways, including dysfunctional pre-RNA processing within satellite cell nuclei that impacts gene expression (166). In addition, there are alterations of ribonucleopreotein (RNP)-containing structures within aged muscle (166, 167). In aged rat satellite cell nuclei, there was accumulation of rnRNPS (involved with pre-mRNA splicing), which signified dysfunctional intranuclear transport of transcripts (166). Malatesta et al. also found decreased pre-mRNA transcription rate and decreased nucleoplasmic splicing factors in myonuclei of aged myofibers (167). The authors suggest that mRNA processing is less efficient in aged than young muscle, and that impaired mRNA function may contribute to the impaired satellite cell activity reported in aged muscle (166).
Cell fate choice is another intrinsic factor influencing aged satellite cell function (14, 149). The increased presence of adipose tissue (such as adipocyte differentiation marker C/EBPα) in aged muscle suggests that satellite cells may prefer an altered cell fate. Instead of choosing myogenic cells, the satellite cells commit to non-myogenic cell fates including adipogenic or fibrogenic cells (149). There was greater expression of adipogenic genes (fatty acid binding protein aP2) in myoblasts cultured from aged (23 mo) relative to young (8 mo) mouse hindlimb muscles (168). The change in cell fate decision away from myogenesis in aged skeletal muscle may be associated with decreased myogenic capacity and increased adipogenesis (168). Aged satellite cells may also choose fibrogenesis over myogenic cell fate (14). Aged myofiber explant cultures had increased fibronectin expression relative to young myofiber explants cultures(14). A satellite cell's preference for adipogenic or fibrogenic cell fate would result in an impairment in the regenerative potential of aged skeletal muscle.
d. Cause of impaired aged skeletal muscle repair - microenvironment mileu: Another reason for the delay in the aged myogenic program may be the composition of the environmental mileu surrounding the satellite cells (9, 13, 139, 145, 169-172). Young muscle transplantation into aged tissue milieu results in myogenic delay (141, 170, 172). In addition, there is decreased satellite cell proliferation and expression of myoblast markers desmin and myf5 in satellite cells exposed to serum from aged muscle (170). Furthermore, when young (2 mo) or aged (15 - 21 mo) mouse EDL muscle was transplanted into an aged host mouse, there was a delay in the inflammatory response and myoblast differentiation (141). These results suggest that the microenvironment of aged skeletal muscle suppresses myogenic capability. However, positioning aged skeletal muscle in a young muscle tissue environmental milieu accelerates its repair (9, 13, 139, 145, 169, 170, 172, 173). Heterochronic transplantation studies and parabiotically paired mice demonstrate that in the presence of young environment, both aged and young satellite cells are able to fully activate and the muscle is completely repaired (13, 173, 174). EDL muscle of aged (32 mo) rats that were transplanted into young (4 mo) rat muscle achieved regenerative capacity and force production on par with that of young to young muscle transplantation. This finding suggests that extrinsic factors within the muscle microenvironment may govern a muscle's capacity for regeneration (169). In addition, there is decreased collagen replacement of aged muscle fibers during young and aged parabosis experiments, suggesting that factor(s) within the young circulation prevent non-myogenic cell fate, and also that factor(s) in aged circulation promotes fibrosis and loss of myogenesis (14). These data suggest that both young and aged satellite cells have myogenic potential, and that it is the tissue environment that may govern the success of myogenic repair.
Determining the difference in composition between the young and aged muscle environment may result in the identification of factors present in each respective milieu that promote or inhibit myogenesis. Satellite cells are physically located next to the myofiber (sarcolemma, basal lamina) and are also adjacent to capillaries, rendering them susceptible to exposure to cells from the systemic environment (interstitial cells, endothelial cells, inflammatory cells, adipocytes, fibroblasts) (171, 175, 176). Neighboring cells may physically interact with satellite cells to influence satellite cell function, and any disruption in the relationship could impact the ability of the satellite cells to activate (145, 171, 176-182)Satellite cells physically connect with laminin, a major protein of the basal lamina, which accelerates myoblast movement and proliferation (177). The cooperation of satellite cells and endothelial cells is also critical for muscle growth (171, 175, 179, 181). There is increased myoblast proliferation in co-culture experiments using isolated human myogenic cells and human microvascular endothelial cells compared to controls (myogenic cells co-cultured with other myogenic cells or fibroblasts) (179). Inflammatory cells such as macrophages also interact with myoblasts to promote proliferation and inhibit apoptosis as demonstrated by co-culture experiments (179-181). Furthermore, myoblasts and connective tissue may interact to facilitate muscle repair (178, 182). Muscle-connective tissue fibroblasts (tcf4+) are physically situated near Pax 7+ satellite cells and regenerating myofibers (embryonic MHC+). Murphy et al. found a concomitant increase in expression of tcf4+, Pax 7+ and embryonic myosin heavy chain (eMHC +) cells following muscle injury. When Pax 7+ - cells were ablated there was a 52% decrease in tcf4+ fibroblast expression, and following the ablation of tcf4+ cells, there was a 51% decrease in Pax 7+- satellite cell expression and impaired myoblast proliferation. This relationship between satellite cells and tcf4+- muscle connective tissue fibroblasts suggests that these cells communicate to orchestrate muscle repair (178).
Although not well known, one potential contributing factor to impaired muscle regeneration in aged skeletal muscle is a dysfunction in the interaction of satellite cells with neighboring cells. Age-associated accumulation of damaged proteins may affect the physical interaction of aged satellite cells with their neighboring cells (145). In addition, there are dysfunctional monocytes and decreased phagocytosis in aged skeletal muscle (172, 174). Because the relationship between macrophages and satellite cells is important for muscle repair, the impaired phagocytic activity in injured aged skeletal muscle could be one cause of poor muscle repair in aged skeletal muscle. Also, age-associated nerve denervation and decreased blood supply, including impaired neo-vascularization within the aged skeletal muscle, may result in reduced physical contact between satellite cells and nerves or endothelial cells in aged skeletal muscle which may negatively impact muscle repair (172, 183). Little is known about the physical connections between aged satellite cells and neighboring cells, but because it is apparent that the interactions of neighboring cells and satellite cells are critical for muscle repair, it would appear logical that a contributing factor to impaired regeneration of aged skeletal muscle could be the altered relationships between aged satellite cells and neighboring cells (145).
In addition to the physical interaction with neighboring cells, satellite cells are exposed to released factors that could have positive or negative influences on satellite cell integrity, depending on the age of the microenvironment (145, 176, 181, 184-187). The literature is robust in describing the sources of growth factors within the satellite cell niche, and their importance for postnatal myogenesis in young skeletal muscle (175, 178, 181, 184). However, within the aged muscle microenvironment, there is an increased presence of inhibitory factors that may contribute to impaired regenerative capability (13, 145, 173, 181, 185, 188). As discussed in the previous sections of this review, in aged organisms there is a decline in systemic factors such as IGF-1, which are known to be important for postnatal myogenesis (188, 189). Aged transgenic mice with over-expressed IGF-1 did not demonstrate the classic muscle atrophy signs typically associated with aged wild type mice suggesting that IGF-1 may protect against sarcopenia (88, 188). There is also attrition of other skeletal muscle repair growth factors including Vascular Endothelial Growth Factor (VEGF) within aged skeletal muscle. Aged (22 mo) mouse myofibers expressed less VEGF relative to young (2.5 mo) controls (181, 187). In addition, there is increased endothelial cell apoptosis in aged tissue, which may result in a reduction in the secretion of endothelial-derived factors that activate satellite cells (179, 187, 190). Because endothelial cells and their secreted VEGF are critical for myogenic regulation, it is plausible to suggest that fewer endothelial cells, and decreased VEGF expression, may contribute to aged-associated impairment of muscle regeneration.
While some growth factors are suppressed in aged skeletal muscle, other growth factors within the satellite cell niche are elevated, and these growth factors, including transforming growth factor (TGF) α and β1 function to inhibit muscle repair (5, 12, 181). Relative to young (20 yr) human vastus lateralis myofibers, aged (70 yr) human myofibers showed greater expression of TGFβ/pSmad and subsequent decreased myogenic proliferation (5). Administration of TGFβ to aged human satellite cells resulted in decreased expression of desmin and MyoD in proliferating myoblasts, which suggests that TGFβ inhibits muscle repair (5). TGFβ increases the expression of cyclin-dependent kinase (CDK) inhibitors and prevents cMyc expression, which results in attenuated myogenic proliferation (5, 12).
Depressed levels of steroid hormones in aged skeletal muscle may indirectly elevate pro-inflammatory cytokines IL-6 and TNFα levels in the satellite cell niche (8, 53, 54, 191). IL-6 has conflicting roles during muscle repair because it is known to be essential for satellite cell activation (192), but, when acting as a pro-inflammatory cytokine, IL-6 is also known to be associated with decreased satellite cell number and a loss of proliferative capacity (8). As described above, TNFα expression levels increase in aged muscle and TNFα has antagonist effects on aged muscle regeneration (29, 30, 38, 45, 193). Transgenic mice over-expressing TNFα in muscle tissue show impaired satellite cell proliferation and differentiation following reloading of atrophic muscle (193). TNFα suppresses MRFs and myoblast differentiation, resulting in impaired muscle repair processes (29, 30). Based on the evidence of the deleterious effects of elevated IL-6 and TNFα on aged skeletal muscle repair, it is interesting to speculate that developing therapeutic tools that target increased pro-inflammatory mediators may assist in combating sarcopenia. It is well identified that the host environment plays an essential role in the success of myogenic processes, and that this microniche is altered in aged skeletal muscle, thereby negatively impacting myogenic potential.
There are many potential mechanisms that may account for, or contribute to, the impaired muscle repair that is associated with aged skeletal muscle including alterations in extrinsic or intrinsic factors. The effect of aging on signaling pathways known to be critical for muscle repair also need to be considered as a contributing factor to impaired muscle repair and sarcopenia.
III. The Role of Notch and Wnt on Impaired Muscle Repair in Aged Muscle
Understanding the differences in cell signaling pathways between young and aged skeletal muscle may help identify mechanisms underlying impaired cellular regeneration in aged skeletal muscle. Notch and Wnt signaling are known to be important for muscle repair and appear to be dysfunctional in aged muscle (7, 12, 13, 194).
a. Notch signaling in aged skeletal muscle repair: The literature is robust in discussion of Notch signaling and its importance to myogenesis (23, 195). In summary, Delta ligand interacts with full length Notch receptor which is cleaved, and translocated to the nucleus where it up-regulates transcription factors (Hes1, Hey1) resulting in myoblast proliferation. Notch signaling is impaired in regenerating aged skeletal muscle (7, 12, 13, 195). There is less Notch1 receptor and Delta1 ligand present in aged activated satellite cells than in young activated satellite cells (7, 12). Insufficient activation of Notch contributes to the diminished regenerative capacity of aged muscle (7, 13). In satellite cells isolated from aged mouse skeletal muscle, decreased Delta ligand expression on activated aged satellite cells corresponded with less myoblast proliferation compared to young satellite cells (7). Force-activating Notch in injured aged muscle increased satellite cell activation and myoblast proliferation (7).
b. Influence of circulating factors on Notch signaling: Using the parabiotic model, Conboy et al. determined that Delta ligand expression can be rescued in aged skeletal muscle (13). In parabiotically paired young and aged mice, the circulatory systems between the animals are shared. Consequently, the satellite cells of the aged mouse muscle are exposed to factors circulating within the young mouse. This pairing model permits the investigation of the effect of serum from young mice on the repair of aged skeletal muscle. The extent of aged skeletal muscle repair when aged mice are paired with young mice is compared to that observed in aged mice when two aged mice are parabiotically paired. When young and aged mice are paired and the TA of the aged mouse muscle is injured, there is an approximate 5-fold increase in Delta ligand expression in activated satellite cells from the injured aged mouse relative to aged-to-aged mouse pairing. In addition, the young-aged pairing restored aged muscle regeneration capability (13). These data suggest that extrinsic factors within the young serum may rescue the Notch signaling deficit, and contribute to the reversal of the poor regenerative capacity in aged muscle (13). In addition, there was decreased muscle regenerative capacity in injured young mouse muscle that was paired with aged mice, suggesting the presence of muscle regenerative inhibitors in aged serum (7, 12).
c. Impairment of upstream factors that activate Notch signaling: The decreased Notch signaling in aged muscle may be associated with impaired expression of upstream factors that activate Notch signaling (5, 194). One potential Notch activator is the mitogein-activated protein kinase (MAPK)/phosphate extracellular signal-regulated kinase (pERK) pathway. MAPK/pERK is depressed in aged muscle (5). Aged (~ 70 y) human VL muscle exposed to cast immobilization followed by reloading-induced muscle injury expresses less MAPK/pERK than young (~ 20 y) muscle exposed to reloading of atrophic muscle (5). Carlson et al. showed that aged satellite cells treated with MAPK agonist increases Notch signaling (5). Developing a tactic to activate MAPK/pERK signaling in aged skeletal muscle may increase Notch signaling and rejuvenate impaired aged skeletal muscle repair.
The sex hormone, testosterone is known to increase Notch signaling. Because testosterone levels are depressed in aged skeletal muscle, one potential cause for impaired Notch signaling in aged skeletal muscle may be a reduction in testosterone levels (57). Young (7 - 8 wks) mice had endogenous testosterone levels depleted and were then administered a testosterone implant, which resulted in increased Notch 1, Notch 2 and Delta 1 expression (196). Application of a testosterone implant to aged (22mo) mice, also resulted in Notch 1 receptor expression in aged skeletal muscle (60). Thus, testosterone is a potential candidate that up-regulates Notch signaling and may be considered as a potential therapeutic treatment option for impaired aged skeletal muscle repair.
d. Increased Notch signaling inhibitors: Impaired Notch signaling in aged skeletal muscle may also be related to increased levels of inhibitors of Notch within the myogenic mileu (194, 195). As discussed in the preceding sections, aged skeletal muscle has increased levels of TNFα, which is thought to contribute to sarcopenia and impair myogenesis, possibly by inhibiting Notch signaling (197). There was decreased mRNA and protein expression of Notch 1 receptor in mononuclear cells isolated from mdx mouse muscle that was treated with TNFα (197). TGFβ may be another inhibitor of Notch signaling, and a source of reduced regenerative capacity in aged skeletal muscle (5). Aged (~ 71 y) human VL skeletal muscle expresses more TGFβ protein than young (~ 23 y) subjects, and when TGFβ was administered to satellite cells isolated from aged (~ 71 y) human skeletal muscle, there was decreased myogenic potential relative to young (~ 23 y) isolated human satellite cells (5). Research reports an imbalance between TGFβ and Notch signaling in human and rodent skeletal muscle models in which elevated TGFβ levels negatively correlate with decreased Notch signaling in aged skeletal muscle (5, 12, 195). In addition, forced activation of Notch inhibits TGFβ-induced up-regulation of CDK inhibitors in satellite cells (12). These two signaling pathways may be integrated during postnatal myogeneisis. Research is needed to delineate the mechanisms underlying the observation that an imbalance of TGFβ and Notch impairs aged skeletal muscle regeneration. In addition, inhibitors of Notch signaling that are present within the aged skeletal muscle mileu need be identified.
e. Wnt signaling and aged muscle repair: In addition to alterations in Notch signaling, Wnt signaling may contribute to impaired aged skeletal muscle repair. Activation of canonical Wnt/β-catenin signaling follows when soluble Wnt ligands interact with receptors such as Frizzled and low-density lipoproteins. Upon Wnt activation, GSK3β's phosphorylation of β-catenin is inhibited, which allows nuclear translocation of β-catenin. Within the nucleus, activated β-catenin binds to transcription factors such as T-cell factor-1 (TCF1) and lymphoid-enhancing factors -1 (LEF1), resulting in skeletal muscle repair (23, 198, 199).
The role of Wnt signaling in impaired aged skeletal muscle repair is controversial. Some suggest that there is an exacerbation of Wnt signaling in aged skeletal muscle. Although Wnt signaling is required for completion of muscle repair, the incongruous elevation of Wnt signaling in aged skeletal muscle may result in dysfunctional myogenic capability and acceleration of aging (14-17). Liu et al. used the klotho mouse model (klotho deficient mice have accelerated aging) and observed increased Wnt signaling that was associated with the observed cell senescence and decreased life span (200). There was increased Wnt signaling in mesenchymal stem cells that were incubated with aged (64 - 68 wks) rat serum (17). In the presence of Wnt inhibitors, Dickoppf (DKK1) or siRNA against β catenin, there is a reversal of the proliferation inhibition (17). Decreased GSK3β and increased β-catenin and Axin 2 (a downstream target of Wnt signaling) expression was observed in aged satellite cells relative to young satellite cells (14). In addition, Brack et al. also reported increased β-galactosidase activity in aged TOPGAL mice (which express the β-galactosidase reporter gene under the control of a promoter responsive to β-catenin) compared to young TOPGAL mice (14, 201). Scime et al. found decreased levels of Wnt antagonists (Secreted frizzled-related protein 2 [Sfrp2] and Wnt inhibitor factor 1 [Wif1]) in aged mouse skeletal muscle relative to young mouse muscle, suggesting increased Wnt activity with aging (15). The presence of Wnt signaling in the circulation was compared in young and aged serum (14). Brack et al. performed parabiotic experiments between young and aged mice and demonstrated that there was decreased Wnt signaling (increased GSK3β expression) in aged myogenic cells when exposed to young mouse serum, and increased Wnt signaling (decreased GSK3β expression) in young myogenic cells when paired with aged mice. In addition, using a fusion protein of Wnt receptor, Frizzled, (Frizzled-Fc), Brack et al. indirectly measured increased levels of Wnt signaling components in aged circulation. When Frizzled-Fc was incubated with serum from aged mice, there was decreased Wnt activity, however young serum incubated with Frizzled-Fc did not induce a change in the degree of Wnt signaling (14). These data suggests that there is an up-regulation of Wnt signaling components in aged systemic circulation relative to young circulation, and that up-regulation of Wnt signaling may promote aging.
Increased Wnt signaling in aged skeletal muscle may inhibit myogenicity (14). Brack et al. reported that relative to satellite cells isolated from young mice, satellite cells isolated from aged (24 - 26 mo) mice expressed less myogenic marker Pax 7. Brack et al. also reported that satellite cells isolated from aged (24 - 26 mo) mice treated with the canonical Wnt inhibitor, Sfrp3, resulted in an increased number of desmin+/BrdU+ cells and rejuvenation of myogenicity (14). These data suggest that inappropriate increases in Wnt signaling within aged skeletal muscle may impair myogenic capability. It is plausible that untimely elevation of Wnt in the circulation may be considered a myogenic inhibitor in the aged muscle micro-environment
Wnt may also contribute to impaired muscle regenerative potential by promoting the age-associated transition of aged skeletal muscle to fibrogenic tissue, thereby accelerating aging (14, 26, 34, 200, 202-204). Wnt signaling is known to promote fibrosis in pathologies such as pulmonary and renal fibrosis, cirrhosis and colorectal carcinoma (205-209). There is increased active ß-catenin expression in hepatocytes of cirrhosis patients, as well as within the fibroblasts of proliferating bronchiole lesions in patients with pulmonary fibrosis (206, 207). Injections of Wnt4 into renal capsule of mice produced fibroblastic lesions, while administration of Wnt inhibitor, sFRP4 resulted in decreased renal fibrosis formation (208, 209). In regards to aged skeletal muscle, Brack et al. injected soluble Wnt3a into young muscle and observed increased Gomori stain (a connective tissue marker). Brack et al. also reported a greater propensity for aged satellite cells to differentiate into fibroblasts than satellite cells isolated from young mice (14). When young (6 wk) mdx mice were injected with Wnt inhibitor DKK1, there was a decrease in fibrosis (203). These data provide evidence that a potential mechanism by which Wnt signaling may impair aged skeletal muscle regeneration is the promotion of fibrogenesis in aged skeletal muscle.
A shift in the equilibrium between Notch and Wnt signaling may be another contributor to impaired regeneration of aged skeletal muscle. Wnt signaling orchestrates with Notch during post-natal myogenesis for complete muscle repair (23, 210, 211). During post-natal myogenesis, Notch signaling is critical for satellite cell activation and myoblast proliferation. At this point, Notch is down-regulated and Wnt signaling completes the muscle repair process by advancing myoblast differentiation and fusion to ultimately form myofibers (23, 210, 211). A suggested imbalance of these signaling pathways during aged skeletal muscle repair may occur as a result of an attenuation of Notch signaling and exacerbation of Wnt signaling resulting in dysfunctional repair of aged skeletal muscle (12-14, 195, 200, 202, 204). There is evidence for Wnt's negative influence on aging skeletal muscle repair, which supports the need for further investigations to ascertain Wnt's role in impaired aged muscle repair and sarcopenia.
Evidence suggesting that Wnt signaling does not accelerate aging, or play a critical role in inhibiting muscle repair in aged muscle should be recognized (185, 202). Carlson et al. did not find any differences in Wnt3a expression in serum from aged humans or mice relative to young human or mouse serum. These authors also did not observe rejuvenation of myogenic proliferation (desmin+/BrdU+) in aged myofibers explants that were incubated with aged serum treated with Wnt inhibitor, Sfrp3. Furthermore, these authors suggest that Wnt activity may be greater in young muscle because there is less active GSK3β in young satellite cells treated with young serum compared to aged satellite cells treated with aged serum (185). Research suggests that Wnt signaling may impede the aging process (202, 212, 213). Inhibition of GSK-3ß and activation of Wnt signaling may delay the onset of age-related diseases such as Alzheimer's Disease (202, 212, 213). Ye et al. showed an inverse correlation between cell senescence and Wnt signaling in aged human cells in which there was decreased expression of Wnt signaling components (elevated GSK-3ß and decreased ß-catenin activities) in cultured human fibroblasts that underwent 58 population doublings and were characterized as senescent (213).
Although there are studies that suggest Wnt signaling does not influence aging, the review of literature reports compelling evidence that Wnt signaling contributes to aging. The reports of increased Wnt signaling in the mouse model of accelerated aging (Klotho) (200), presence of Wnt induced-fibrosis in aged skeletal muscle (14) and increased Wnt signaling in aged TOPGAL mice (14, 201) provide confirms the concept that elevated Wnt contributes to aging and may affect repair of aged muscle. The inconsistencies in the results of studies designed to explore the role of Wnt in impaired aged skeletal muscle repair may be related to the models utilized to study the effect of Wnt signaling on aged skeletal muscle repair. Alternatively, the differences may be associated with the pleitropic roles of Wnt, which depend on a variety of signals. More work is needed is ascertain Wnt's contribution to impaired aged skeletal muscle repair. For example, therapeutic modalities (such as exercise) that take advantage of the benefits of circulatory factors that promote the proper orchestration of Notch and Wnt signaling and rejuvenation of impaired aged skeletal muscle repair need to be explored.
IV. The Effect of Physiological Stimuli on the Molecular and Cellular Manifestations of Sarcopenia
To obtain a more complete understanding of the factors that may underlie the pathological manifestations of sarcopenia (Table 1), studies exploring the responses of aged skeletal muscle to physiological stress (such as exercise) should be included. Important information regarding the deleterious effects of aging on skeletal muscle can be obtained by performing experiments in which the muscle is exposed to an acute bout of physiological stimuli. Furthermore, an examination of the adaptive mechanisms of aged skeletal muscle that is exposed to repeated bouts of physiological stimuli (exercise training) may assist in determining potential counter measures to sarcopenia.
Strength of evidence for factor contribution to sarcopenia.
| Factor suggested to contribute to sarcopenia | Extent of evidence | References |
|---|---|---|
| ↑ Intramuscular fibrosis and adipose tissue | Strong | 14,25,26,28,34 |
| ↑ TNFα and IL-6 | Strong | 35-42,44,45 |
| ↓ Sex hormone | Strong | 1,8,56-60,61 |
| ↓IGF-1 and mRNA translation | Strong | 79,84-92 |
| ↑ Myostatin | Strong | 32,73,94,95 |
| ↓ MicroRNA | More research needed | 99,109,110 |
| ↑ Apoptosis | More research needed | 33,111,112 |
| ↑ Telomere shortening in satellite cells | More research needed | 124 |
| ↑ oxidative stress | Strong | 1,4,30,131-134 |
| Impaired muscle regeneration | Strong | 4-8,141-143 |
| ↓ satellite cells in aged muscle | Controversy | ↓ satellite cells in aged muscle: 5,7-9,32,73,139,145-149 No change in satellite cell in aged muscle: 11,150 |
| Impaired myoblast proliferation vs. myotube formation to cause impaired muscle repair | Controversy | Impaired myoblast proliferation: 79,151 Impaired myotube formation: 10,11,13,30,151,153 |
| ↓ MRF | Controversy | ↓ MRF: 30,73,154,155 ↑/no change MRF: 6,142,160-162 |
| ↑ Myostatin | Controversy | ↑ Myostatin: 32,73,158,159 ↑/no change Myostatin: 161 |
| ↓ Notch signaling | Strong | 7,12,13,195 |
| ↑ Wnt signaling | Controversy | ↑ Wnt signaling: 14-17,200,201 ↓ Wnt signaling:185,202,212,213 |
a. Intramuscular fibrosis and adipose tissue: With the prevalence of fibrosis and adiposity in aged skeletal muscle, it is of interest to determine if physiological stimuli reduce fibrosis and intramuscular fat (25, 28, 214). In aged skeletal muscle of both human and rodent models, exercise training decreases intermuscular adipose tissue (214-216). There was a 7% increase in lean thigh tissue and 11% decrease in intermuscular adipose tissue in aged (> 55 yrs) subjects who performed a 12 wk resistance training program (214). Aged (23 mo) rats that performed life-long exercise had a 50% reduced muscle triglyceride count than sedentary controls (215). Aged (~ 76 y) subjects who participated in a complete exercise program (aerobic, muscle strength, flexibility and balance) did not report any increases in intramuscular fat content, while subjects who did not exercise had an 18.4% increase in muscle fat infiltration (216). Exercise training has the potential to prevent the age-associated accumulation of fibrosis and adipogenicity in skeletal muscle.
b. Pro-inflammatory cytokines TNFα and IL-6: Although contentious, reports suggest that age-associated elevations in the pro-inflammatory cytokines TNFα and IL-6 within aged skeletal muscle may contribute to sarcopenia by inducing muscle atrophy and promoting protein degradation and ROS accumulation. Anti-inflammatory medications are one countermeasure employed to combat the chronic inflammatory response that accompanies sarcopenia. Little is known about the benefits of exercise as a non-pharmacologial tactic against the pro-inflammatory cytokines TNFα and IL-6 (35). An acute bout of exercise increases TNFα and IL-6 levels in young and aged skeletal muscle (46, 47, 217, 218). For example, serum IL-6 levels can increase 100-fold during exercise (217). Following an acute bout of exercise, there is elevation of pro-inflammatory cytokines in aged subjects, but to a lesser degree than in young subjects (47, 218). Young subjects had greater increases in plasma IL-6 than aged subjects up to four days after performing heavy eccentric resistance exercises (218). At 72h post-downhill running, TNFα mRNA in VL was elevated 2.8 fold for young (23 - 35 y) subjects, while TNFα mRNA in aged subjects (66 - 78 y) VL was elevated only 1.8 fold (47).
The exercise-induced elevation of pro-inflammatory cytokines in aged muscle in an already inflamed environment suggests that an acute bout of physical activity is not beneficial to sarcopenic patients. The exercise-induced release of TNFα may exacerbate muscle injury (219). Inhibiting TNFα with cV1q antibody in dystrophic mice (mdx mice) resulted in a decrease in muscle necrosis at 24h following wheeling running exercise (219). However, it should be noted that there are inconsistencies in the data on the effect of age and exercise on TNFα and IL-6 s more research is needed to better understand their interaction following an acute bout of exercise (36).
It is interesting to report that, contrary to what is seen following a single bout of exercise, consistent exercise training may elicit a decrease in pro-inflammatory cytokines (220-224). Epidemiological studies report that aged (70 - 79y) subjects with higher levels of daily physical activity had lower levels of plasma TNFα and IL-6 (220). Exercise-induced attrition of TNFα and IL-6 within aged skeletal muscle may prevent their deleterious effects, resulting in preservation of skeletal muscle mass and function. It is plausible to suggest that daily physical activity (aerobic, strength, flexibility) may be a good countermeasure to sarcopenia and age-associated chronic inflammation (220). A cycle ergometry exercise program decreased TNFα in aged subjects (223). A year of physical activity resulted in an 8.5% decrease in plasma IL-6 levels in aged (70 - 89y) subjects (224). A 12-week cardiac rehabilitation program resulted in a decrease in IL-6 and an increase in anti-inflammatory marker IL-10 in aged (64 y) subjects (221). An exercise training program consisting of both aerobics and strength training resulted in a 50% decrease in TNFα and IL-6 mRNA in aged (~ 69 y) subjects (222). Because TNFα and IL-6 may be associated with sarcopenia, the data supporting a reduction in TNFα and IL-6 levels associated with long-term exercise training demonstrates the potential of physical activity as a therapeutic strategy in the treatment of sarcopenia. Further research is needed to determine the relationship between exercise and TNFα and IL-6, and their roles in sarcopenia.
c. Sex hormones: Sex hormones independently and in orchestration with other factors, play important roles in maintaining skeletal muscle mass integrity, and preventing the accumulation of intramuscular fat (1, 8, 53, 54). With aging, there is a decrease in sex hormone levels which increases intramuscular adipose tissue accumulation and decreases skeletal muscle mass, thereby contributing to sarcopenia (1, 8, 53, 54, 191). Treatment with sex hormone supplements has not consistently demonstrated recovery or delay of sarcopenia, therefore other tactics to replenish testosterone and estrogen levels in aged skeletal muscle need to be considered (54, 63, 64). One potential strategy to increase sex hormone levels in aged skeletal muscle is physiological stimuli (225, 226). Total testosterone increased in aged (62 y) men who performed a 10 wk power-training program (226). 17β-estradiol levels increased in overectomized rats that performed 12 wks of treadmill training relative to overectomized controls (225). Training programs have also been shown to counteract the negative effects of decreased sex hormone levels in aged skeletal muscle, both in the presence and absence of hormone replacement therapy (HRT) (62, 227). Middle-aged women (50 - 57 years) who performed a one-year resistance training program in conjunction with HRT saw positive results in skeletal muscle performance (62). Hormone therapy treatment may not be essential for negating the detrimental effects of depleted sex hormones and sarcopenia. Postmenopausal women between the ages of 55 - 75 y who were not on HRT performed a 3 mo resistance training program and found a decrease in percent body fat, an increase in lean body mass and an increase in physical performance (227). Physiological stimuli, which may take the form of endurance or resistance training, can replenish depleted sex hormones in aged skeletal muscle, and may be considered an effective intervention in the treatment of sarcopenia.
d. Skeletal muscle function: It is known that with sarcopenia, there is a decrease in aged muscle function that is often observed following exposure to physiological stimuli(228). Aged (~78 y) subjects exposed to a injurious plantar flexion exercises experienced a 26% drop in isometric plantar flexion maximal voluntary contraction (MVC) torque, and produced 38% less peak power compared to young (~ 24 y) subjects (229). In addition, aged (~ 66 y) women who performed an acute bout of knee extensions experienced a 27% decrease in eccentric 1 repetition maximum (1 RM) compared to a 10% decrease in 1 RM in young (~ 23 y) women (230). Using a mouse model, one bout of an in situ lengthening contraction protocol resulted in greater force deficits in aged (27-34 mo) muscle relative to young (5-6 mo) muscle (231). Likewise, following two bouts of in vitro lengthening contractions of the extensor digitalis longus (EDL) muscle, there was a greater force deficit reported in aged (24 - 25 mo) mouse muscle (~ 27.4%) than young (3 - 4 mo) mouse muscle (~ 21.5%) (232).
In addition to loss of function, aged muscle also experiences a delay in recovery of force production following injurious physiological stimuli (233, 234). Young mouse muscle exposed to a bout of injurious in situ lengthening contractions had complete restoration of muscle force production 28 days post-exercise, while aged mice muscle demonstrated only 84% of force production 28 and 60 days post-exercise (233). Following injurious in situ lengthening contractions, adult (6 mo) rat TA muscle contractile properties recovered in 5 days, while aged (32 mo) rat TA did not recover until 14 days post-exercise (234).
A single bout of physiological stimuli is not beneficial in improving age-associated loss of muscle force production, but multiple exercise bouts are considered to have beneficial effects on sarcopenia, including recovery of aged muscle force production. There was a 174% increase in muscle strength, a 9% increase in mid-thigh cross sectional area and a 48% increase in gait speed in aged (~ 90 y) subjects who performed an 8 week high-intensity resistance training program (235). Following a 24 wk resistance training program, quadricep muscle strength and cross sectional area increased by 29% and 7% respectively in aged men (62 - 77 y) (236). Similar muscle strength findings were reported by Verdijk et al. who found that aged (~72 y) male subjects experienced a 25% - 30% increase in quadriceps muscle strength, and a 6% - 9% increase in quadricep muscle mass following a 12 wk resistance training program (237).
Repeated bouts of physiological stimuli also produced protection against muscle injury and expediated recovery of force production (230, 234, 238). A training effect was observed in aged (32 mo) rat TA muscle that was exposed to two bouts of injurious in situ lengthening contractions. There was less force deficit after the second bout of lengthening contractions compared to that experienced after the first bout of lengthening contractions (234). Henwood et al. assessed muscle function recovery following training, de-training and re-training of muscles. Aged (65 - 84 y) subjects experienced 24 wks of training of either high velocity power training (HV) or strength training (ST) followed by 24 wks of de-training and then a subsequent 12 wks re-training protocol. Isometric and dynamic muscle strength and muscle power increased after the initial 24 wk HV and ST training programs. There was a minimal loss of muscle function (strength and power) following the de-training period, suggesting that the initial training protocol offered adaptation and protection of muscle strength (238). Likewise, aged (~ 66 y) women who performed an injurious bout of knee extension exercise following a 12 wk knee extension resistance training program experienced no significant edema (muscle injury marker) relative to young (~ 23 y) women, suggesting that the resistance training produced protection against muscle injury (230).
Although physiological stimuli training improves muscle strength and adaptation in aged muscle, there is still a delay in recovery in aged muscle relative to young skeletal muscle (234, 239, 240). Young (21 - 27 y) and aged (61 - 74 y) male subjects were exposed to whole leg immobilization for two wks and subsequently performed lower body resistance exercises for 4 wks. After 4 wks of re-training, young subjects showed a significant increase in their quadricep muscle volume (~ 8.2%) while aged quadriceps muscle volume increased only ~ 3.8% (240). In rodent models, reports show that there is a delay in muscle force recovery in aged muscle relative to young muscle, but that aged muscle does indeed adapt (234, 239). Brooks et al. exposed young (7 mo) and aged (22 mo) mice dorsi flexors to a 4 wk in situ muscle lengthening training protocol, and found that while young mice experienced adaptation and no significant force deficit, aged mice had a delay in adaptation such that it took 6 wks for force recovery (239).
It should be noted that some investigators report that training has deleterious effects on aged muscle function (228, 241, 242). Cutlip et al. exposed young (~ 6 wks) and aged rats (~ 30 mo) to a 4.5 wk training protocol of low-velocity intermittent in vivo stretch-shortening muscle contractions of the dorsi flexors. The training program increased isometric force production 25% in the young TA, but there was a 34% decrease in TA isometric force production in the aged TA. In addition, while the exercise protocol significantly increased the wet muscle mass of the young TA, there was no change in wet mass of the aged TA (228). Although there may be differing reports on the effects of training on function of aged muscle, the majority of evidence suggests that multiple bouts of physiological stimuli increase force production and recovery of force in aged muscle, and are beneficial in the prevention and treatment of sarcopenia.
e. Muscle fibers and motor units: With sarcopenia there is a preferential loss of Type II muscle fibers (25, 31, 66, 67, 243). Physiological stimuli may have the potential to prevent age-associated loss of muscle fibers, specifically Type II muscle fibers (31, 237, 244, 245). Verdijk et al. found a ~ 28% increase in Type II muscle fibers in aged male subjects (~72 y) who performed a 12 wk resistance training program (237). Type IIA plantaris muscle fibers increased in aged (24 mo) rats that performed a 12 wk endurance treadmill program (244). Resistance training may be a more effective physiological stimulus model than endurance training to prevent the loss of Type II muscle fibers (31, 245). Klitgaard et al. found that aged subjects who strength trained for 12 - 17 years possessed muscle fiber morphology more similar to young sedentary subjects than aged subjects who were swimmers or runners (31). Strength training may be a physiological stimulus that protects aged skeletal muscle from sarcopenia (31, 67, 237, 245).
Another variable that contributes to the loss of muscle fibers with age is alterations in motor units. There are decreases in motorneuron number, fast twitch motor unit number and motor unit recruitment (66, 70, 73). Endurance training may be beneficial in counteracting the deleterious effect of motor unit adaptation that is associated with aging (246). Aged (~ 65 y) male runners possessed comparable TA motor unit quantity to young (~25 y) controls and significantly more motor units than aged sedentary subjects (246). Strength training has also been shown to improve motor unit function in aging muscle (72, 247). Leong et al. reports increased motor unit discharge in the rectus femoris of aged (67-79 y) men who resistance trained (247). Similarly, a 6 wk resistance training program resulted in a 40% increase in motor unit discharge of the VL of aged (~77 y) adults (72). Chronic physiological stimuli appear to rejuvenate impaired motor unit function in aged skeletal muscle.
f. Protein synthesis/degradation balance: Physiological stimuli consisting of a variety of exercise modes may be a form of treatment that can counteract the protein imbalance in aging skeletal muscle (77, 93, 248-255). An acute bout of leg extension resistance exercise increases the fractional synthesis rate (FSR) of mixed muscle protein and myofibrillar protein, as well as amino acid transporter expression (LAT1/SLC7A5, CD98/SLC3A2, SNAT2/SLC38A2, PAT1/SLC36A1, and CAT1/SLC7A1) in the VL of aged men (248, 251, 253). Exercise training programs also increase protein synthesis in aged muscle (77, 249, 250, 252, 254, 255). Using leucine incorporation as a marker for protein synthesis, leg extension training also increases protein synthesis in VL of aged male subjects (77, 255). Also, training programs consisting of endurance, strength, flexibility and balance exercises increase mixed muscle protein FSR in aged subjects by 50% (254). (Furthermore, novel techniques of administrating either amino acid supplementation or blood flow restriction (reducing blood flow to the exercising muscle) in conjunction with resistance exercise have also been shown to increase protein synthesis in aged muscle (93, 249, 250, 252). During the three hours post-blood flow restriction and exercise combination treatment, there was a 56% increase in muscle protein synthesis in the VL of aged men (mean 70 ys) (250).
g. IGF-1 and mRNA translation: The finding that aged muscle responds to physiological stimuli by increasing protein synthesis supports the notion that aged muscle possesses the capability to manufacture protein in response to certain stimuli. There is a strong correlation between loss of muscle mass and attrition of IGF-1 and GH levels in aged skeletal muscle (80, 81, 85). Physiological stimuli may be an effective strategy to increase IGF-1 and GH levels in sarcopenic muscle (256-259). Resistance training increases IGF-1 expression (including the isoforms IGF-1Ea, mechano-growth factor [MGF]) in aged subjects (256-259). There was a 500% increase in IGF-1 expression in aged (~ 87 y) men exposed to a 10 wk resistance training program (257). Hameed et al. examined the effect of GH administration with and without resistance training on expression of IGF-1 isoforms (MGF and IGF-IEa) in aged (74 y) men. Resistance training alone increased mRNA levels of MGF (163%), IGF-IEa (68%) and IGF-IEb (75%). Resistance training in combination with GH administration resulted in greater increase of MGF mRNA (456%) and IGF-IEa mRNA (167%) (258). Hameed's results indicate that physiological stimuli alone can increase IGF-1 isoforms, but administration of GH further elevates IGF-1 isoforms. Hameed's group also examined the effect of aerobic exercise on IGF-1 isoforms and found that eccentric cycling (reverse pedaling) increases MGF mRNA in both young (20-27 y) and aged (67 - 75 y) quadriceps muscles (260).
Since insulin is known to up-regulate IGF-1 activity, there may be an age-associated correlation between insulin and IGF-1 in which a decrease in insulin sensitivity is associated with a decrease in IGF-1 activity. It is plausible to suggest that treating insulin insensitivity in aging muscle with physiological stimuli could re-establish IGF-1 levels (261). Insulin signaling and sensitivity was improved in aged rats that performed an acute bout of swimming exercise (262). Resistance training also improves insulin-dependent glucose uptake in skeletal muscle of aged subjects (263). These findings suggest that exercise may be effective in restoring IGF-1 isoforms in aged skeletal muscle. Because IGF-1 is critical to muscle growth and protein synthesis, IGF-1 may prove to be an effective countermeasure to sarcopenia.
Studies have examined the relevance of physiological stimuli to the improvement of translational efficiency in aged skeletal muscle (249-251). mTOR signaling components including mTOR phosphorylation and phosphorylation of S6K1 are elevated in aged muscle exposed to leg resistance exercise (249). The treatment combination of blood flow restriction and exercise also increases mTOR components including mTOR phosphorylation, phosphorylation of S6K1 and increased S6 ribosomal protein in aged muscle (250). This treatment combination may be beneficial in counteracting sarcopenia because it induces muscle protein synthesis and mTOR signaling (249, 250).
Although aged skeletal muscle has the ability to generate proteins and activate mTOR signaling, this potential is diminished relative to young skeletal muscle (245, 251, 264, 265). There was less leucine incorporation and phosphorylation of p70rS6k and 4EBP1 in aged (~ 70 y) men relative to young men who performed one bout of resistance exercise (251). Mayhew et al. report a 60% increase in phosphorylated Akt in young muscle compared with 39% in aged muscle following a single bout of resistance exercise (245). After 28 days of plantaris overload, mTOR phosphorylation was elevated by 44% and 35% in young (6 mo) rats and aged (30 mo) rat muscle respectively. In addition, ribosomal protein S6 phosphorylation increased 114% and 24% in young and aged plantaris, respectively, after overload (265). The accumulation of studies support the notion that physiological stimuli such as overloading elevates mTOR signaling and improves translational efficiency in aged skeletal muscle.
h. Myostatin: Physiological stimuli may further enhance protein synthesis by inhibiting myostatin, an inhibitor of protein synthesis (67, 266-270). An acute bout of exercise decreases myostatin levels (67, 267-269). Myostatin mRNA in the VL decreased in aged (60-75 y) subjects who performed a single bout of resistance exercise (268). Training exercises also decrease myostatin levels (266, 270). Myostatin mRNA in aged VL muscle decreased 37% following 9 wks of leg extensions training (270). Following three bouts of resistance training, myostatin mRNA decreased and myostatin-inhibitor follistatin -like mRNA increased in aged (68 y) subjects relative to young (28 y) subjects (266). In summary, physiological stimuli influence proteins and signaling pathways that promote protein synthesis in the aged muscle, and may be a useful strategy to combat sarcopenia.
i.MicroRNA: There is evidence to suggest that the novel post-transcriptional inhibitors, muscle-specific microRNA (miRNAs) are critical for muscle repair. Given the altered expression of certain muscle-specific miRNAs in aged skeletal muscle, there is an emerging concept that miRNA levels may predict sarcopenia (Table 2) (109, 110, 271). Research is ongoing to determine the effect of physiological stimuli on young and aged skeletal muscle miRNA expression (98, 108, 110, 272, 273). Drummond et al. reported discrepancies in the changes in expression of primary miRNA (pri-miRNA) and mature miRNA (miR) between young (~ 29 y) and aged (~ 70y) VL of subjects that performed a single bout of leg-extension exercise. pri-miRNA -206 increased in both young and aged VL muscle, however pri-miRNA - 1-2,-133a-1, and -133a-2 and miR-1 decreased in young muscle only (108). Using a rodent plantaris overload model, McCarthy et al. reported similar findings to Drummond et al. in regards to physiological stimuli inducing a decrease in miR-1 and an increase in pri-mRNA 206 expression in young muscle (98, 108). Conversely, McCarthy's group reported that plantaris overloading increased pri-miRNA 1-2 and 133a-2 in muscle and Drummond's group reported that resistance exercise decreases these factors. In addition, while McCarthy et al. reported that plantaris overload did not alter pri-miRNA 133a-1 expression in muscle, Drummond et al. saw a decrease in pri-miRNA 133a-1 in muscle following resistance exercise (98, 108).
The effect of physiological stimuli on micro-RNA/pri- RNA expression.
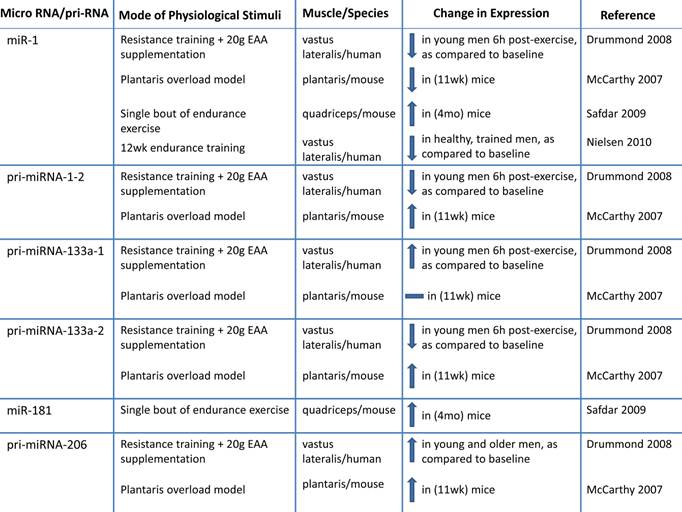
There are also differing reports of miR expression following endurance exercise (272, 273). While MCarthy et al. and Drummond et al. found that both resistance exercise and overloading models decreased miR-1 in muscle, Safdar et al. found that a bout of endurance exercise increased miR-1 expression (as well as miR 187) in mouse quadriceps muscle (98, 108, 273). However, Nielson et al. found a decrease in miR 1 expression in VL of subjects who performed a 12 wk endurance training program (272). There are numerous conflicting reports on the effect of physiological stimuli on miR and pri-miRNA expression in skeletal muscle. One potential explanation for the differences in expression of muscle pri-miRNAs or miRs following physiological stimuli is the assortment of physiological stimuli models used (overload hypertrophy model, resistance exercise, endurance exercise) which may induce varying degrees of miR or pri-miRNA up-regulation. These conflicting reports underscore the need for more research on the effect of various modes of physiological stimuli on miRNA and pri-miRNA expression in young and aged skeletal muscle. Such studies will generate a more complete understanding of the importance of miR in aging skeletal muscle.
j. Apoptosis: Because sedentary conditions are known to up-regulate skeletal muscle apoptosis, it appears worthwhile to study the effect of exercise on apoptotic-associated loss of muscle mass. An acute bout of exercise in rodent skeletal muscle resulted in increased DNA fragmentation and apoptotic nuclei (274, 275). However, in human models, young subjects who performed a bout of cycle ergometry did not display any alterations in pro-apoptotic proteins, caspase activity, DNA fragmentation or anti-apoptotic protein activity (276). The differences in apoptotic responses following a single bout of physiological stimuli could be related to species, the capacity of the specific stimulus to incur or a variety of other reasons. Consequently, more research is needed to determine the effect of an acute bout of exercise on apoptosis in both young and aged skeletal muscle.
Multiple bouts of physiological stimuli suppress apoptosis in both young and aged skeletal muscle (33, 114, 275, 277). In addition to increasing the expression of apoptosis inhibitor ARC by 38%, an 8 wk treadmill-training program increased anti-apoptotic protein XIAP and decreased DNA fragmentation in young rodent skeletal muscle (277). A 12 wk treadmill-training program in aged (24 mo) rats decreased DNA fragmentation and Bax expression and decreased caspase-3 activity by 95% (114). In addition, there was a decrease in DNA fragmentation and Bax levels in the patagialis wing muscle of aged (~ 48 mo) Japanese quail exposed to a loading training protocol (278). These findings suggest that an appropriate exercise-training program may attenuate age-related skeletal muscle apoptosis, and could be considered a countermeasure for sarcopenia.
k. Telomere length: Telomere shortening is a marker for age-associated cellular senescence and may be associated with sarcopenia (124, 126). Chronic physiological stimuli such as life-long physical activity may delay telomere shortening (279-282). A study that compared leukocyte telomere length between physically active and sedentary twins showed a direct correlation between physical activity and leukocyte telomere length. Leukocyte telomere lengths of the most active were ~ 200 nucleotides longer than the leukocyte telomere lengths of the least active subjects (280). Lullow et al. found that aged (50 - 70 y) subjects with high exercise energy expenditure displayed longer telomere lengths than subjects with low exercise energy expenditure (282). Ponsot et al. reported no differences in minimum telomere length in TA satellite cell DNA between young (~ 25 y) and aged (~ 75 y) recreationally active subjects (279). Similarly, endurance-trained aged subjects (55 - 72 y) had leukocyte telomeres ~ 900 base pairs longer than sedentary age-matched controls, and were not significantly different in length from those of young (18 - 32 years) exercised subjects (281). These data suggest that moderate physical activity may have a protective effect against age-associated telomere shortening, and may have therapeutic potential for the treatment of sarcopenia.
l. Oxidative stress: Certain types of physiological stimuli have the potential to decrease ROS-producing sources and may therefore be countermeasures to sarcopenia. Exercise training (either aerobic- or resistance-type exercises) appears to decrease oxidative stress in aged skeletal muscle, possibly by elevating mitochondrial content and function (283-285). A 12 wk endurance-training program increased mitochondrial content and activity (succinate oxidase activity and NADH oxidase activity) in aged (~67y) subjects (283). In mtDNA-mutated mice that express progeroid aging (characterized by age-associated mitochondrial dysfunction), a five-mo endurance training program resulted in increased systemic mitochondrial biogenesis and oxidative capacity (285). There is decreased lipid peroxidation in serum of aged (~ 68y) subjects following a six-mo high resistance training program (286). A 14 wk resistance training program increased mitochondrial ETC complex IV activity and decreased the oxidative stress marker 8-hydroxydeoxyguanosine (8-OHdG) in aged (~ 68y) subjects (284). Physiological stimuli also increase antioxidant activity and heat shock protein levels (284, 287). Parise et al. reported a 75% and 82% increase in copper zinc superoxide dismutase (CuZnSOD) and catalase respectively in aged (~ 71y) subjects who performed a 12 wk resistance training program (288). Lastly, Naito et al. reported elevations in heat shock protein 72 in aged (23 mo) rats that performed a 10 wk treadmill training program (287). These studies suggest that physiological stimuli may be a beneficial countermeasure to decrease oxidative stress and elevate antioxidant expression in sarcopenic muscle.
V. Effect of Physiological Stimuli on Muscle Regeneration and Notch and Wnt Signaling in Sarcopenic Muscle
One potential effective interventional strategy for impaired repair in sarcopenic muscle is physiological stimulation, which is known to up-regulate regenerative pathways in young muscle (289-294). Both voluntary free wheel and resistance wheel training increased satellite cell number in young (7 - 10 wks) rats (294). There was a 5-fold increase in embryonic MHC in young (5 - 8 wks) mice that performed a bout of downhill running (293). Since physiological stimuli activate the pathways needed for repair in young muscle, it would be meaningful to determine if exercise has the ability to positively impact muscle repair mechanisms within aged muscle. For example, the effects of exercise on dysfunctional satellite cell regulation could be determined.
a. Muscle regeneration: There is a wealth of evidence suggesting that physiological stimuli rescues aspects of impaired muscle regeneration in aged skeletal muscle (67, 125, 289-292, 295-299). Treadmill exercise training increases satellite cell numbers in aged rats (289, 291). Following 13 wks of treadmill running, aged (15 - 17 mo) rats expressed increased MyoD-positive and Pax-7-positive satellite cells (289) Aerobic training also increases satellite cell numbers and MRFs in aged subjects (300). Following a 12 wk aerobic training protocol, the percentage of MHC I mRNA expression increased from ~ 54% (at rest) to ~ 61% (post training) in aged (~70 yrs) women subjects (300). Resistance training exercises elevated satellite cell numbers, MHC, MyoD, and myogenin in aged subjects (67, 292, 295, 298, 299). Mackey et al. reported that following 12 wks of resistance training, there was a 27% increase in NCAM + cells in VL of aged (~76 yrs) subjects, suggesting that aged muscle has myogenic potential (298). Furthermore, using the stretch overload model in the latissimus dorsi (ADL) of aged (90 day) Japanese quail, Carson et al. reported there was an increase in number of BrdU- positive cells in myofibers relative to un-stretched controls (297).
Although physiological stimulation is effective in reviving regeneration of aged skeletal muscle, there is a delay in the achievement of repair, and the magnitude of repair is not as robust as seen in young muscle (290, 291). While young (~6 wks) rats completely recovered by one week after an acute bout of treadmill running, aged (16 wks) rats continued to show markers of muscle injury and repair at 2 weeks post-exercise (291). At 24h post injurious eccentric knee extensor exercises, young (25 - 35 yrs) males showed a greater elevation of NCAM+-cells than aged (60 - 75 yrs) males. However, it should be noted that the injurious exercise increased satellite cell number in the aged subjects (290).
The overwhelming evidence that physiological stimuli induce muscle repair mechanisms in aged muscle provides strong support for the idea that exercise should be considered a practical intervention strategy to up-regulate the impaired cellular and molecular mechanisms of aged muscle repair. Physiological stimuli may release systemic factors that are otherwise not present in aged muscle, and induce a rejuvenation of muscle repair in sarcopenic muscle (67, 125, 296). For example, exercise increases VEGF, which is known to be important for muscle repair, and is depleted in aged skeletal muscle (296). More research is needed to investigate the effect of physiological stimuli on the delivery of factors that influence the satellite cell niche and enhance repair of aged skeletal muscle.
b. Notch and Wnt signaling: Studying the influence of physiological stimuli on Notch and Wnt signaling could be a novel therapeutic tactic to tackle impaired aged skeletal muscle repair that is associated with sarcopenia. As described above, Notch and Wnt are critical to muscle repair, and there is evidence suggesting that these pathways may be dysfunctional in aged muscle (7, 12-17, 195). As described in the preceding sections of this review article, physiological stimuli, through a variety of forms of exercise and contraction-induced injury models, release factors that promote a pro-myogenic environment, and the rescue of dysfunctional repair of aged skeletal muscle. Physiological stimuli increases Notch and Wnt signaling in young muscle models (Figure 2) (18-23). Downhill running and overload hypertrophy models increase components of Notch signaling in young skeletal muscle (18, 19). Tsivitse et al. reported increased expression of Notch1 and Delta1 in mouse hindlimb muscles exposed to an acute bout of injurious downhill running (18). Treadmill running and overload hypertrophy models decreased GSK3β activity and increased canonical Wnt signaling in young skeletal muscle (20-22). Following plantaris overload, β-catenin, Frizzled 1 and Lef-1 mRNA levels increased suggesting that muscle growth stimuli activate Wnt signaling (21).
The effect of aging & physiological stimuli on Notch and Wnt signaling during aged muscle repair.
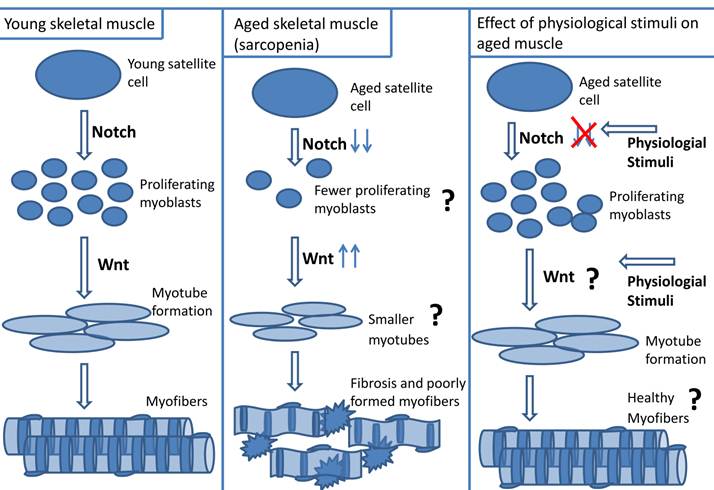
It is clear that research looking at physiological stimuli and regulation of Notch and Wnt signaling during skeletal muscle repair is promising in terms of therapeutic potential for the treatment of sarcopenia.
Muscle contraction-induced injury models may be used as therapeutic tools to rescue the attenuated Notch and potentially dysfunctional Wnt signaling in aged skeletal muscle. Carey et al. has demonstrated that both a single bout of resistance exercise, and a 12 week resistance training program increased Notch 1 and Hes 6 gene expression in VL of aged (60 - 75 y) males, suggesting that physiological stimuli may be a beneficial tool for rejuvenating attenuated Notch signaling (301). Little is known about the effects of physiological stimuli on Notch and Wnt signaling pathways in aged muscle, but because exercise is known to elevate circulating pro- myogenic factors, it appears reasonable to consider that appropriate physiological stimuli may release systemic factors that return the balance of Notch and Wnt signaling in aged skeletal muscle. Using “global” forms of muscle injury (exercise) that bring into play the influence of multiple biological systems to the regenerative process will help identify factors required for rejuvenation of impaired myogenic potential in aged muscle. For example, factors required for proper regulation of Notch and Wnt signaling during muscle repair may be identified. Further research will be required to determine if physiological stimuli have the ability to promote the proper orchestration of Notch and Wnt signaling for rejuvenating aged skeletal muscle repair.
Conclusion
It is well known that sarcopenia increases linearly with aging. It is also well known that the mechanisms underlying the physiological manifestations of sarcopenia are multifactorial, and are likely to be complex. The advancement of senescence in aged tissue has detrimental effects on skeletal muscle including loss of motor nerves and muscle fibers, apoptosis, telomere shortening, imbalance of protein synthesis and degradation. All of these factors can contribute to decreased muscle quality (2, 8, 25, 28-30, 32). Sarcopenia is also associated with increased intramuscular adipose tissue, low-grade inflammation and depleted sex hormone levels. Another contributing factor to sarcopenia is the impaired ability of aged skeletal muscle to regenerate following injury. Causes of loss of myogenic potential with age may be related to intrinsic dysfunction of satellite cells, or to extrinsic factors in which the environmental mileu within the aged skeletal muscle is anti-myogenic (4-8). Recent evidence suggests that Notch signaling is impaired and Wnt signaling may be dysregulated in aged skeletal muscle (7, 12-14, 16, 17). There is an abundance of evidence demonstrating that resistance and endurance training re-balances age-associated issues that cause sarcopenia including decreasing intramuscular adipose tissue, pro-inflammatory cytokine levels, oxidative stress and DNA fragmentation as well as delays/prevents telomere shortening and increases sex hormone levels, protein synthesis and growth factors. In addition, resistance training increases Type II muscle fiber expression in aged muscle and rejuvenates impaired motor units. Furthermore, exercise may positively influence the age-associated muscle repair deficiencies including enhancing postnatal myogenesis through increased satellite cell number, MRF expression, rescuing Notch signaling and regulating dysfunctional Wnt signaling. Consequently, physiological stimuli, consisting of a variety of exercise models, may be an effective therapeutic tool in the fight against sarcopenia (67, 77, 125, 269, 289-292, 295, 296, 298-300). Physiological stimuli release pro-myogenic systemic factors that may mimic a young skeletal muscle microenvironment. This beneficial aspect of exercise may rescue the age-associated lack of Notch and Wnt orchestration during aged skeletal muscle repair. More research is needed to determine what factors in the environment of aged skeletal muscle contribute to sarcopenia. A more complete understanding of the mechanisms underlying the physiological manifestations of sarcopenia will facilitate the development of effective treatment strategies to combat this debilitating disease.
Acknowledgements
The authors would like to thank Dr. Andrea Dulin for providing helpful input in the manuscript revisions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Di Iorio A, Abate M, Di Renzo D, Russolillo A, Battaglini C, Ripari P, Saggini R, Paganelli R, Abate G. Sarcopenia: age-related skeletal muscle changes from determinants to physical disability. Int J Immunopathol Pharmacol. 2006;4:703-19
2. Doherty T. Physiology of aging invited review: aging and sarcopenia. J Appl Physiol. 2003;95:1717-27
3. Taaffe D. Sarcopenia: exercise as a treatment strategy. Australian Family Physician. 2006;35(3):130-3
4. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130(2):223-33
5. Carlson ME, Suetta C, Conboy MJ, Aagaard P, Mackey A, Kjaer M, Conboy I. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med. 2009;1(8-9):381-91
6. Marsh DR, Criswell DS, Carson JA, Booth FW. Myogenic regulatory factors during regeneration of skeletal muscle in young, adult, and old rats. J Appl Physiol. 1997;83(4):1270-5
7. Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential aged muscle. Science. 2003;302:1575-7
8. Lee CE, McArdle A, Griffiths RD. The role of hormones, cytokines and heat shock proteins during age-related muscle loss. Clin Nutr. 2007;26(5):524-34
9. Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Developmental Biology. 2006;294:50-66
10. Renault V, Piron-Hamelin G, Forestier C, DiDonna S, Decary S, Hentati F, Saillant G, Butler-Browne GS, Mouly V. Skeletal muscle regeneration and the mitotic clock. Exp Gerontol. 2000;35(6-7):711-9
11. Beccafico S, Puglielli C, Pietrangelo T, Bellomo R, Fanò G, Fulle S. Age-dependent effects on functional aspects in human satellite cells. Ann N Y Acad Sci. 2007;1100:345-52
12. Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454(7203):528-32
13. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760-4
14. Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807-10
15. Scimè A, Desrosiers J, Trensz F, Palidwor GA, Caron AZ, Andrade-Navarro MA, Grenier G. Transcriptional profiling of skeletal muscle reveals factors that are necessary to maintain satellite cell integrity during ageing. Mech Ageing Dev. 2010;131(1):9-20
16. Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA, Ricci E, Mirabella M. Increased aging in primary muscle cultures of sporadic inclusion-body myositis. Neurobiol Aging. 2010;31(7):1205-14
17. Zhang DY, Wang HJ, Tan YZ. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS One. 2011;6(6):e21397
18. Tsivitse S, Peters MG, Stoy AL, Mundy JA, Bowen RS. The effect of downhill running on Notch signaling in regenerating skeletal muscle. Eur J Appl Physiol. 2009;106(5):759-67
19. Akiho M, Nakashima H, Sakata M, Yamasa Y, Yamaguchi A, Sakuma K. Expression profile of Notch-1 in mechanically overloaded plantaris muscle of mice. Life Sci. 2009;86(1-2):59-65
20. Aschenbach WG, Ho RC, Sakamoto K, Fujii N, Li Y, Kim YB, Hirshman MF, Goodyear LJ. Regulation of dishevelled and beta-catenin in rat skeletal muscle: an alternative exercise-induced GSK-3beta signaling pathway. Am J Physiol Endocrinol Metab. 2006;291(1):E152-8
21. Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005;289(4):C853-C9
22. Ishido M, Uda M, Masuhara M, Kami K. Alterations of M-cadherin, neural cell adhesion molecule and Beta-catenin expression in satellite cells during overload-induced skeletal muscle hypertrophy. Acta Physiol Scand. 2006;187:407-18
23. Tsivitse S. Notch and Wnt signaling, physiological stimuli and postnatal myogenesis. Int J Biol Sci. 2010;6(3):268-81
24. Janssen I, Heymsfield S, Ross R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J Am Geriatr Soc. 2002;50(5):889-96
25. Narici MV, Maffulli N. Sarcopenia: characteristics, mechanisms and functional significance. Br Med Bull. 2010;95:139-59
26. Edström E, Altun M, Bergman E, Johnson H, Kullberg S, Ramírez-León V, Ulfhake B. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol Behav. 2007;92(1-2):129-35
27. Frontera WR, Suh D, Krivickas LS, Hughes VA, Goldstein R, Roubenoff R. Skeletal muscle fiber quality in older men and women. Am J Physiol Cell Physiol. 2000;279(3):C611-C8
28. Roth SM, Metter EJ, Ling S, Ferrucci L. Inflammatory factors in age-related muscle wasting. Curr Opin Rheumatol. 2006;18(6):625-30
29. Buford TW, Anton SD, Judge AR, Marzetti E, Wohlgemuth SE, Carter CS, Leeuwenburgh C, Pahor M, Manini TM. Models of accelerated sarcopenia: critical pieces for solving the puzzle of age-related muscle atrophy. Ageing Res Rev. 2010;9(4):369-83
30. Degens H. Age-related skeletal muscle dysfunction: causes and mechanisms. J Musculoskelet Neuronal Interact. 2007;7(3):246-52
31. Klitgaard H, Mantoni M, Schiaffino S, Ausoni S, Gorza L, Laurent-Winter C, Schnohr P, Saltin B. Function, morphology and protein expression of ageing skeletal muscle: a cross-sectional study of elderly men with different training backgrounds. Acta Physiol Scand. 1990;140(1):41-54
32. Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology. 2008;9(4):213-28
33. Alway SE, Siu PM. Nuclear apoptosis contributes to sarcopenia. Exerc Sport Sci Rev. 2008;36(2):51-7
34. Serrano AL, Muñoz-Cánoves P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res. 2010;316(18):3050-8
35. Nicklas BJ, Brinkley TE. Exercise training as a treatment for chronic inflammation in the elderly. Exerc Sport Sci Rev. 2009;37(4):165-70
36. Peake J, Della Gatta P, Cameron-Smith D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am J Physio Regul Integr Comp Physiol. 2010;298(6):R1485-95
37. Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhøj P, Pedersen BK. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999;54(7):M357-64
38. Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11(1):163-75B
39. Phillips T, Leeuwenburgh C. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005;19(6):668-70
40. Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH Jr, Heimovitz H, Cohen HJ, Wallace R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106(5):506-12
41. Giovannini S, Onder G, Liperoti R, Russo A, Carter C, Capoluongo E, Pahor M, Bernabei R, Landi F. Interleukin-6, C-reactive protein, and tumor necrosis factor-alpha as predictors of mortality in frail, community-living elderly individuals. J Am Geriatr Soc. 2011;59(9):1679-85
42. Roubenoff R, Harris TB, Abad LW, Wilson PW, Dallal GE, Dinarello CA. Monocyte cytokine production in an elderly population: effect of age and inflammation. J Gerontol A Biol Sci Med Sci. 1998;53(1):M20-6
43. Pedersen BK, Bruunsgaard H. Possible beneficial role of exercise in modulating low-grade inflammation in the elderly. Scand J Med Sci Sports. 2003;13(1):56-62
44. Yende S, Waterer GW, Tolley EA, Newman AB, Bauer DC, Taaffe DR, Jensen R, Crapo R, Rubin S, Nevitt M, Simonsick EM, Satterfield S, Harris T, Kritchevsky SB. Inflammatory markers are associated with ventilatory limitation and muscle dysfunction in obstructive lung disease in well functioning elderly subjects. Thorax. 2006;61(1):10-6
45. Dirks AJ, Leeuwenburgh C. Tumor necrosis factor alpha signaling in skeletal muscle: effects of age and caloric restriction. J Nutr Biochem. 2006;17(8):501-8
46. Liao P, Zhou J, Ji LL, Zhang Y. Eccentric contraction induces inflammatory responses in rat skeletal muscle: role of tumor necrosis factor-alpha. Am J Physio Regul Integr Comp Physiol. 2010;298(3):R599-607
47. Hamada K, Vannier E, Sacheck JM, Witsell AL, Roubenoff R. Senescence of human skeletal muscle impairs the local inflammatory cytokine response to acute eccentric exercise. FASEB J. 2005;19(2):264-6
48. Bautmans I, Niemini R, Lambert M, Demanet C, Mets T. Circulating acute phase mediators and skeletal muscle performance in hospitalized geriatric patients. J Gerontol A Biol Sci Med Sci. 2005;60(3):361-7
49. Tsujinaka T, Ebisui C, Fujita J, Kishibuchi M, Morimoto T, Ogawa A, Katsume A, Ohsugi Y, Kominami E, Monden M. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem Biophys Res Commun. 1995;207(1):168-74
50. Pedersen BK, Akerström TC, Nielsen AR, Fischer CP. Role of myokines in exercise and metabolism. J Appl Physiol. 2007;103(3):2007
51. Rieu I, Magne H, Savary-Auzeloux I, Averous J, Bos C, Peyron MA, Combaret L, Dardevet D. Reduction of low grade inflammation restores blunting of postprandial muscle anabolism and limits sarcopenia in old rats. J Physiol. 2009;587(Pt 22):5483-92
52. Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H, Monden M. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. 1996;97(1):244-9
53. Maggio M, Basaria S, Ceda GP, Ble A, Ling SM, Bandinelli S, Valenti G, Ferrucci L. The relationship between testosterone and molecular markers of inflammation in older men. J Endocrinol Invest. 2005;28(11 Suppl Proceedings):116-9
54. Maltais ML, Desroches J, Dionne IJ. Changes in muscle mass and strength after menopause. J Musculoskelet Neuronal Interact. 2009;9(4):186-97
55. Kalleinen N, Polo-Kantola P, Irjala K, Porkka-Heiskanen T, Vahlberg T, Virkki A, Polo O. 24-hour serum levels of growth hormone, prolactin, and cortisol in pre- and postmenopausal women: the effect of combined estrogen and progestin treatment. J Clin Endocrinol Metab. 2008;93(5):1655-61
56. Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282(3):E601-7
57. Lamberts SW, van den Beld AW, van der Lely AJ. The endocrinology of aging. Science. 1997;278(5337):419-24
58. Sinha-Hikim I, Cornford M, Gaytan H, Lee ML, Bhasin S. Effects of testosterone supplementation on skeletal muscle fiber hypertrophy and satellite cells in community-dwelling older men. J Clin Endocrinol Metab. 2006;91(8):3024-33
59. Atkinson RA, Srinivas-Shankar U, Roberts SA, Connolly MJ, Adams JE, Oldham JA, Wu FC, Seynnes OR, Stewart CE, Maganaris CN, Narici MV. Effects of testosterone on skeletal muscle architecture in intermediate-frail and frail elderly men. J Gerontol A Biol Sci Med Sci. 2010;65(11):1215-9
60. Kovacheva EL, Hikim AP, Shen R, Sinha I, Sinha-Hikim I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology. 2010;151(2):628-38
61. Sipilä S, Taaffe D, Cheng S, Puolakka J, Toivanen J, Suominen H. Effects of hormone replacement therapy and high-impact physical exercise on skeletal muscle in post-menopausal women: a randomized placebo-controlled study. Clin Sci (Lond). 2001;101(2):147-57
62. Taaffe DR, Sipilä S, Cheng S, Puolakka J, Toivanen J, Suominen H. The effect of hormone replacement therapy and/or exercise on skeletal muscle attenuation in postmenopausal women: a yearlong intervention. Clin Physiol Funct Imaging. 2005;25(5):297-304
63. Tankó LB, Movsesyan L, Svendsen OL, Christiansen C. The effect of hormone replacement therapy on appendicular lean tissue mass in early postmenopausal women. Menopause. 2002;9(2):117-21
64. Kenny AM, Kleppinger A, Wang Y, Prestwood KM. Effects of ultra-low-dose estrogen therapy on muscle and physical function in older women. J Am Geriatr Soc. 2005;53(11):1973-7
65. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J; Writing Group for the Women's Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial[i]. JAMA. 2002;288(3):321-33
66. Lexell J, Downham DY, Larsson Y, Bruhn E, Morsing B. Heavy-resistance training in older Scandinavian men and women: short- and long-term effects on arm and leg muscles. Scand J Med Sci Sports. 1995;5(6):329-41
67. Snijders T, Verdijk LB, van Loon LJ. The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Research Reviews. 2009;8(4):328-38
68. Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007;292(1):E151-7
69. Narici MV, Maganaris CN, Reeves ND, Capodaglio P. Effect of aging on human muscle architecture. J Appl Physiol. 2003;95(6):2229-34
70. Faulkner JA, Larkin LM, Claflin DR, Brooks SV. Age-related changes in the structure and function of skeletal muscles. Clin Exp Pharmacol Physiol. 2007;34(11):1091-6
71. Kamen G, Sison SV, Du CC, Patten C. Motor unit discharge behavior in older adults during maximal-effort contractions. J Appl Physiol. 1995;79(6):1908-13
72. Kamen G, Knight CA. Training-related adaptations in motor unit discharge rate in young and older adults. J Gerontol A Biol Sci Med Sci. 2004;59(12):1334-8
73. Lang T, Streeper T, Cawthon P, Baldwin K, Taaffe DR, Harris TB. Sarcopenia: etiology, clinical consequences, intervention, and assessment. Osteoporos Int. 2010;21(4):543-9
74. Kullberg S, Ramírez-León V, Johnson H, Ulfhake B. Decreased axosomatic input to motoneurons and astrogliosis in the spinal cord of aged rats. J Gerontol A Biol Sci Med Sci. 1998;53(5):B369-79
75. Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24(5):1376-90
76. Ulfhake B, Bergman E, Edstrom E, Fundin BT, Johnson H, Kullberg S, Ming Y. Regulation of neurotrophin signaling in aging sensory and motoneurons: dissipation of target support? Mol Neurobiol. 2000;21(3):109-35
77. Hasten DL, Pak-Loduca J, Obert KA, Yarasheski KE. Resistance exercise acutely increases MHC and mixed muscle protein synthesis rates in 78-84 and 23-32 yr olds. Am J Physiol Endocrinol Metab. 2000;278(4):E620-6
78. Tavernarakis N. Ageing and the regulation of protein synthesis: a balancing act? Trends Cell Biol. 2008;18(5):228-35
79. Combaret L, Dardevet D, Béchet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Curr Opin Clin Nutr Metab Care. 2009;12(1):37-41
80. Kostek MC, Delmonico MJ, Reichel JB, Roth SM, Douglass L, Ferrell RE, Hurley BF. Muscle strength response to strength training is influenced by insulin-like growth factor 1 genotype in older adults. J Appl Physiol. 2005;98:2147-54
81. Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F. The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol. 2010;205(3):201-10
82. Marcell TJ, Harman SM, Urban RJ, Metz DD, Rodgers BD, Blackman MR. Comparison of GH, IGF-I, and testosterone with mRNA of receptors and myostatin in skeletal muscle in older men. Am J Physiol Endocrinol Metab. 2001;281(6):E1159-64
83. Payette H, Roubenoff R, Jacques PF, Dinarello CA, Wilson PW, Abad LW, Harris T. Insulin-like growth factor-1 and interleukin 6 predict sarcopenia in very old community-living men and women: the Framingham Heart Study. J Am Geriatr Soc. 2003;51(9):1237-43
84. Chakravarthy MV, Booth FW, Spangenburg EE. The molecular responses of skeletal muscle satellite cells to continuous expression of IGF-1: implications for the rescue of induced muscular atrophy in aged rats. Int J Sport Nutr Exerc Metab. 2001;11:S44-8
85. Hameed M, Harridge SD, Goldspink G. Sarcopenia and hypertrophy: a role for insulin-like growth factor-1 in aged muscle. Exerc Sport Sci Rev. 2002;30(1):15-9
86. Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157(1):137-48
87. Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27(2):195-200
88. Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci USA. 1998;95(26):15603-7
89. Flati V, Caliaro F, Speca S, Corsetti G, Cardile A, Nisoli E, Bottinelli R, D' Antona G. Essential amino acids improve insulin activation of AKT/MTOR signaling in soleus muscle of aged rats. Int J Immunopathol Pharmacol. 2010;23(1):81-9
90. Mouisel E, Vignaud A, Hourdé C, Butler-Browne G, Ferry A. Muscle weakness and atrophy are associated with decreased regenerative capacity and changes in mTOR signaling in skeletal muscles of venerable (18-24-month-old) dystrophic mdx mice. Muscle Nerve. 2010;41(6):809-18
91. Paturi S, Gutta AK, Katta A, Kakarla SK, Arvapalli RK, Gadde MK, Nalabotu SK, Rice KM, Wu M, Blough E. Effects of aging and gender on muscle mass and regulation of Akt-mTOR-p70s6k related signaling in the F344BN rat model. Mech Ageing Dev. 2010;131(3):202-9
92. Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3(2):90-101
93. Walker DK, Dickinson JM, Timmerman KL, Drummond MJ, Reidy PT, Fry CS, Gundermann DM, Rasmussen BB. Exercise, amino acids, and aging in the control of human muscle protein synthesis. Med Sci Sports Exerc. 2011;43(12):2249-58
94. Murphy KT, Koopman R, Naim T, Léger B, Trieu J, Ibebunjo C, Lynch GS. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24(11):4433-42
95. Solomon A, Bouloux PM. Modifying muscle mass-the endocrince perspective. Journal of Endocrinology. 2006;191:349-60
96. Liu W, Thomas SG, Asa SL, Gonzalez-Cadavid N, Bhasin S, Ezzat S. Myostatin is a skeletal muscle target of growth hormone anabolic action. J Clin Endocrinol Metab. 2003;88(11):5490-6
97. Artaza JN, Bhasin S, Magee TR, Reisz-Porszasz S, Shen R, Groome NP, Meerasahib MF, Gonzalez-Cadavid NF. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multipotent cells. Endocrinology. 2005;146(8):3547-57
98. McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007;102(1):306-13
99. Lanceta J, Prough RA, Liang R, Wang E. MicroRNA group disorganization in aging. Exp Gerontol. 2010;45(4):269-78
100. Wang D, Valdez MR, McAnally J, Richardson J, Olson EN. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development. 2001;128(22):4623-33
101. Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, Hijikata T. MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct. 2008;33(2):163-9
102. Goljanek-Whysall K, Sweetman D, Abu-Elmagd M, Chapnik E, Dalmay T, Hornstein E, Münsterberg A. MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc Natl Acad Sci USA. 2011;108(29):11936-41
103. Koutsoulidou A, Mastroyiannopoulos NP, Furling D, Uney JB, Phylactou LA. Expression of miR-1, miR-133a, miR-133b and miR-206 increases during development of human skeletal muscle. BMC Dev Biol. 2011;11:34
104. Nakasa T, Ishikawa M, Shi M, Shibuya H, Adachi N, Ochi M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J Cell Mol Med. 2010;14(10):2495-505
105. Ge Y, Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle. 2011;10(3):441-8
106. Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol. 2011;31(1):203-14
107. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38(2):228-33
108. Drummond MJ, McCarthy JJ, Fry CS, Esser KA, Rasmussen BB. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am J Physiol Endocrinol Metab. 2008;295(6):E1333-40
109. Hamrick MW, Herberg S, Arounleut P, He HZ, Shiver A, Qi RQ, Zhou L, Isales CM, Mi QS. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem Biophys Res Commun. 2010;400(3):379-83
110. Drummond MJ, McCarthy JJ, Sinha M, Spratt HM, Volpi E, Esser KA, Rasmussen BB. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol Genomics. 2011;43(10):595-603
111. Dirks A, Hofer T, Marzetti E, Pahor M, Leeuwenburgh C. Mitochondrial DNA mutations, energy metabolism and apoptosis in aging muscle. Ageing Research Reviews. 2006;5:179-95
112. Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and failty at old age. Experimental Gerontology. 2006;41:1234-8
113. Dirks A, Leeuwenburgh C. Apoptosis in skeletal muscle with aging. Am J Physiol Regul Integr Comp Physiol. 2002;282(2):R519-27
114. Song W, Kwak HB, Lawler JM. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid Redox Signal. 2006;8(3-4):517-28
115. Siu P. Muscle apoptotic response to denervation, disuse, and aging. Med Sci Sports Exerc. 2009;41(10):1876-86
116. Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2010;45(2):138-48
117. Pistilli EE, Siu PM, Alway SE. Molecular regulation of apoptosis in fast plantaris muscles of aged rats. J Gerontol A Biol Sci Med Sci. 2006;61(3):245-55
118. Alway SE, Degens H, Krishnamurthy G, Smith CA. Potential role for Id myogenic repressors in apoptosis and attenuation of hypertrophy in muscles of aged rats. Am J Physiol Cell Physiol. 2002;283(1):C66-76
119. Alway SE, Degens H, Krishnamurthy G, Chaudhrai A. Denervation stimulates apoptosis but not Id2 expression in hindlimb muscles of aged rats. J Gerontol A Biol Sci Med Sci. 2003;58(8):687-97
120. Dirks AJ, Leeuwenburgh C. Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis-inducing factor, X-linked inhibitor of apoptosis, caspase-3, and caspase-12. Free Radic Biol Med. 2004;36(1):27-39
121. Siu PM, Pistilli EE, Ryan MJ, Alway SE. Aging sustains the hypertrophy-associated elevation of apoptotic suppressor X-linked inhibitor of apoptosis protein (XIAP) in skeletal muscle during unloading. J Gerontol A Biol Sci Med Sci. 2005;60(8):976-83
122. von Zglinicki T, Martin-Ruiz CM. Telomeres as biomarkers for ageing and age-related diseases. Curr Mol Med. 2005;5(2):197-203
123. Blagosklonny M. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5(18):2087-102
124. von Zglinicki T, Saretzki G, Ladhoff J, d'Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126(1):111-7
125. Kadi F, Ponsot E. The biology of satellite cells and telomeres in human skeletal muscle: effects of aging and physical activity. Scand J Med Sci Sports. 2009;20(1):39-48
126. Blasco MA, Lee HW, Hande MP, Samper E, Landsorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25-34
127. Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569-74
128. Flores I, Benetti R, Blasco MA. Telomerase regulation and stem cell behaviour. Curr Opin Cell Biol. 2006;18(3):254-60
129. Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KE, Wüstefeld T, Blasco MA, Manns MP, Rudolph KL. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003;22(15):4003-13
130. O'Connor MS, Carlson ME, Conboy IM. Differentiation rather than aging of muscle stem cells abolishes their telomerase activity. Biotechnol Tech. 2009;25(4):1130-7
131. Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129(7-8):383-90
132. Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol. 2008;43(1):24-33
133. Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62(3):235-45
134. Reid MB, Li YP. Tumor necrosis factor-alpha and muscle wasting: a cellular perspective. Respir Res. 2001;2(5):269-72
135. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909-11
136. Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend lifespan of mice. Aging Cell. 2009;8(1):73-5
137. Huang T, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J Gerontol A Biol Sci Med Sci. 2000;55(1):B5-9
138. Strube P, Sentuerk U, Riha T, Kaspar K, Mueller M, Kasper G, Matziolis G, Duda GN, Perka C. Influence of age and mechanical stability on bone defect healing: age reverses mechanical effects. Bone. 2008;42(2):758-64
139. Conboy IM, Rando TA. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle. 2005;4(3):407-10
140. Sipos F, Leiszter K, Tulassay Z. Effect of ageing on colonic mucosal regeneration. World J Gastroenterol. 2011;17(25):2981-6
141. Smythe GM, Shavlakadze T, Roberts P, Davies MJ, McGeachie JK, Grounds MD. Age influences the early events of skeletal muscle regeneration: studies of whole muscle grafts transplanted between young (8 weeks) and old (13-21 months) mice. Exp Gerontol. 2008;43(6):550-62
142. Dedkov E, Kostrominova TY, Borisov AB, Carlson BM. MyoD and myogenin protein expression in skeletal muscles of senile rats. Cell Tissue Res. 2003;311:401-16
143. Mu X, Urso ML, Murray K, Fu F, Li Y. Relaxin regulates MMP expression and promotes satellite cell mobilization during muscle healing in both young and aged mice. Am J Pathol. 2010;177(5):2399-410
144. Lorenzon P, Bandi E, de Guarrini F, Pietrangelo T, Schäfer R, Zweyer M, Wernig A, Ruzzier F. Ageing affects the differentiation potential of human myoblasts. Experimental Gerontology. 2004;39:1545-54
145. Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7(4):590-8
146. Day K, Shefer G, Shearer A, Yablonka-Reuveni Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev Biol. 2010;340(2):330-43
147. Dedkov EI, Borisov AB, Wernig A, Carlson BM. Aging of skeletal muscle does not affect the response of satellite cells to denervation. J Histochem Cytochem. 2003;51(7):853-63
148. Dreyer HC, Blanco CE, Sattler FR, Schroeder ET, Wiswell RA. Satellite cell numbers in young and older men 24 hours after eccentric exercise. Muscle Nerve. 2006;33:242-53
149. Machida S, Narusawa M. The roles of satellite cells and hematopoietic stem cells in impaired regeneration of skeletal muscle in old rats. Annals New York Academy of Sciences. 2006;1067:349-53
150. Brooks NE, Schuenke MD, Hikida RS. No change in skeletal muscle satellite cells in young and aging rat soleus muscle. J Physiol Sci. 2009;59(6):465-71
151. Lees S, Rathbone CR, Booth FW. Age-associated decrease in muscle precursor cell differentiation. Am J Physiol Cell Physiol. 2006;290:C609-C15
152. Machida S, Booth FW. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Exp Gerontol. 2004;39(10):1521-5
153. Bigot A, Jacquemin V, Debacq-Chainiaux F, Butler-Browne GS, Toussaint O, Furling D, Mouly V. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biol Cell. 2008;100(3):189-99
154. Lowe DA, Warren GL, Snow LM, Thompson LV, Thomas DD. Muscle activity and aging affect myosin structural distribution and force generation in rat fibers. J Appl Physiol. 2004;96(2):498-506
155. Alway SE, Degens H, Lowe DA, Krishnamurthy G. Increased myogenic repressor Id mRNA and protein levels in hindlimb muscles of aged rats. Am J Physio Regul Integr Comp Physiol. 2002;282(2):R411-22
156. Reid M. Response of the ubiquitin-proteasome pathway to changes in muscle activity. Am J Physiol Regul Integr Comp Physiol. 2005;288(6):R1423-31
157. Yokota Y, Mori S. Role of Id family proteins in growth control. J Cell Physiol. 2002;190(1):21-8
158. Baumann AP, Ibebunjo C, Grasser WA, Paralkar VM. Myostatin expression in age and denervation-induced skeletal muscle atrophy. J Musculoskelet Neuronal Interact. 2003;3(1):8-16
159. Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831-40
160. Alway SE, Lowe DA, Chen KD. The effects of age and hindlimb suspension on the levels of expression of the myogenic regulatory factors MyoD and myogenin in rat fast and slow skeletal muscles. Exp Physiol. 2001;86(4):509-17
161. Kawada S, Tachi C, Ishii N. Content and localization of myostatin in mouse skeletal muscles during aging, mechanical unloading and reloading. J Muscle Res Cell Motil. 2001;22(8):627-33
162. Lowe DA, Lund T, Alway SE. Hypertrophy-stimulated myogenic regulatory factor mRNA increases are attenuated in fast muscle of aged quails. Am J Physiol. 1998;275(1 Pt 1):C155-62
163. Jejurikar SS, Henkelman EA, Cederna PS, Marcelo CL, Urbanchek MG, Kuzon WM Jr. Aging increases the susceptibility of skeletal muscle derived satellite cells to apoptosis. Exp Gerontol. 2006;41(9):828-36
164. Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25(4):885-94
165. Brack AS, Bildsoe H, Hughes SM. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci. 2005;118(Pt 20):4813-21
166. Malatesta M, Perdoni F, Muller S, Pellicciari C, Zancanaro C. Pre-mRNA processing is partially impaired in satellite cell nuclei from aged muscles. J Biomed Biotechnol. 2010;2010:410405
167. Malatesta M, Perdoni F, Muller S, Zancanaro C, Pellicciari C. Nuclei of aged myofibres undergo structural and functional changes suggesting impairment in RNA processing. Eur J Histochem. 2009;53(2):97-106
168. Taylor-Jones JM, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002;123(6):649-61
169. Carlson BM, Dedkov EI, Borisov AB, Faulkner JA. Skeletal muscle regeneration in very old rats. J Gerontol A Biol Sci Med Sci. 2001;56(5):B224-33
170. Carlson ME, Conboy IM. Loss of stem cell regenerative capacity within aged niches. Aging Cell. 2007;6(3):371-82
171. Mounier R, Chrétien F, Chazaud B. Blood vessels and the satellite cell niche. Curr Top Dev Biol. 2011;96:121-38
172. Shavlakadze T, McGeachie J, Grounds MD. Delayed but excellent myogenic stem cell response of regenerating geriatric skeletal muscles in mice. Biogerontology. 2010;11(3):363-76
173. Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol. 1989;256(6 Pt 1):C1262-6
174. Zacks SI, Sheff MF. Age-related impeded regeneration of mouse minced anterior tibial muscle. Muscle & Nerve. 1982;5(2):152-61
175. Abou-Khalil R, Mounier R, Chazaud B. Regulation of myogenic stem cell behavior by vessel cells: the "ménage à trois" of satellite cells, periendothelial cells and endothelial cells. Cell Cycle. 2010;9(5):892-6
176. Hall JK, Banks GB, Chamberlain JS, Olwin BB. Prevention of muscle aging by myofiber-associated satellite cell transplantation. Sci Transl Med. 2010;2(57):57ra83
177. Ocalan M, Goodman SL, Kühl U, Hauschka SD, von der Mark K. Laminin alters cell shape and stimulates motility and proliferation of murine skeletal myoblasts. Dev Biol. 1988;125(1):158-67
178. Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138(17):3625-37
179. Christov C, Chrétien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B, Gherardi RK. Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell. 2007;18(4):1397-409
180. Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, Gherardi RK. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J Cell Biol. 2003;163(5):1133-43
181. Ten Broek RW, Grefte S, Von den Hoff JW. Regulatory factors and cell populations involved in skeletal muscle regeneration. J Cell Physiol. 2010;224(1):7-16
182. Bi P, Kuang S. Meat Science and Muscle Biology Symposium: stem cell niche and postnatal muscle growth. J Anim Sci. 2011;90(3):924-35
183. Grounds M. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Annals New York Academy of Sciences. 1998;854:78-91
184. Bischoff R. Interaction between satellite cells and skeletal muscle fibers. Development. 1990;109(4):943-52
185. Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, Mikels AJ, Agrawal S, Schaffer DV, Conboy IM. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8(6):676-89
186. Conboy IM, Yousef H, Conboy MJ. Embryonic anti-aging niche. Aging (Albany NY). 2011;3(5):555-63
187. Wagatsuma A. Effect of aging on expression of angiogenesis-related factors in mouse skeletal muscle. Exp Gerontol. 2006;41(1):49-54
188. Carosio S, Berardinelli MG, Aucello M, Musarò A. Impact of ageing on muscle cell regeneration. Ageing Res Rev. 2011;10(1):35-42
189. Goldspink G, Harridge SD. Growth factors and muscle ageing. Exp Gerontol. 2004;39(10):1433-8
190. Kushner EJ, MacEneaney OJ, Weil BR, Greiner JJ, Stauffer BL, DeSouza CA. Aging is associated with a proapoptotic endothelial progenitor cell phenotype. J Vasc Res. 2011;48(5):408-14
191. Kramer PR, Kramer SF, Guan G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004;50(6):1967-75
192. Serrano AL, Baeza-Raja B, Perdiguero E, Jardí M, Muñoz-Cánoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7(1):33-44
193. Langen RC, Schols AM, Kelders MC, van der Velden JL, Wouters EF, Janssen-Heininger YM. Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol. 2006;35(6):689-96
194. Carlson M, Silva HS, Conboy IM. Aging of signal transduction pathways, and pathology. Exp Cell Res. 2008;314(9):1951-61
195. Buas MF, Kadesch T. Regulation of skeletal myogenesis by Notch. Exp Cell Res. 2010;316(18):3028-33
196. Brown D, Hikim AP, Kovacheva EL, Sinha-Hikim I. Mouse model of testosterone-induced muscle fiber hypertrophy: involvement of p38 mitogen-activated protein kinase-mediated Notch signaling. J Endocrinol. 2009;201(1):129-39
197. Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010;5(8):e12479
198. Rao TP, Kühl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106(12):1798-806
199. Tanaka S, Terada K, Nohno T. Canonical Wnt signaling is involved in switching from cell proliferation to myogenic differentiation of mouse myoblast cells. J Mol Signal. 2011;6:12
200. Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803-6
201. Al Alam D, Green M, Tabatabai Irani R, Parsa S, Danopoulos S, Sala FG, Branch J, El Agha E, Tiozzo C, Voswinckel R, Jesudason EC, Warburton D, Bellusci S. Contrasting expression of canonical Wnt signaling reporters TOPGAL, BATGAL and Axin2(LacZ) during murine lung development and repair. PLoS One. 2011;6(8):e23139
202. DeCarolis NA, Wharton KA Jr, Eisch AJ. Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays. 2008;30(2):102-6
203. Trensz F, Haroun S, Cloutier A, Richter MV, Grenier G. A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am J Physiol Cell Physiol. 2010;299(5):C939-47
204. White BD, Nguyen NK, Moon RT. Wnt signaling: it gets more humorous with age. Curr Biol. 2007;17(21):R923-5
205. Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162(5):1495-502
206. Königshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, Eickelberg O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One. 2008;3(5):e2142
207. Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Insights into the pathobiology of hepatitis C virus-associated cirrhosis: analysis of intrahepatic differential gene expression. Am J Pathol. 2002;160(2):641-54
208. Surendran K, McCaul SP, Simon TC. A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol. 2002;282(3):F431-41
209. Surendran K, Schiavi S, Hruska KA. Wnt-dependent beta-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J Am Soc Nephrol. 2005;16(8):2373-84
210. Brack A, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from Notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2:50-9
211. Hurlbut G, Kankel MW, Lake RJ, Artavanis-Tsakonas S. Crossing paths with Notch in the hyper-network. Curr Opin Cell Biol. 2007;19(2):166-75
212. Inestrosa NC, Varela-Nallar L, Grabowski CP, Colombres M. Synaptotoxicity in Alzheimer's disease: the Wnt signaling pathway as a molecular target. IUBMB Life. 2007;59(4-5):316-21
213. Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007;27(2):183-96
214. Marcus R, Addison O, Kidde JP, Dibble LE, Lastayo PC. Skeletal muscle fat infiltration: impact of age, inactivity, and exercise. J Nutr Health Aging. 2010;14(5):362-6
215. Novelli M, Pocai A, Skalicky M, Viidik A, Bergamini E, Masiello P. Effects of life-long exercise on circulating free fatty acids and muscle triglyceride content in ageing rats. Exp Gerontol. 2004;39(9):1333-40
216. Goodpaster B, Chomentowski P, Ward BK, Rossi A, Glynn NW, Delmonico MJ, Kritchevsky SB, Pahor M, Newman AB. Effects of physical activity on strength and skeletal muscle fat infiltration in older adults: a randomized controlled trial. J Appl Physiol. 2008;105(5):1498-503
217. Ostrowski K, Rohde T, Asp A, Schjerling P, Pedersen BK. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol. 1999;515(Pt 1):287-91
218. Toft AD, Jensen LB, Bruunsgaard H, Ibfelt T, Halkjaer-Kristensen J, Febbraio M, Pedersen BK. Cytokine response to eccentric exercise in young and elderly humans. Am J Physiol Cell Physiol. 2002;283(1):C289-95
219. Radley HG, Davies MJ, Grounds MD. Reduced muscle necrosis and long-term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul Disord. 2008;18(3):227-38
220. Colbert LH, Visser M, Simonsick EM, Tracy RP, Newman AB, Kritchevsky SB, Pahor M, Taaffe DR, Brach J, Rubin S, Harris TB. Physical activity, exercise, and inflammatory markers in older adults: findings from the Health, Aging and Body Composition Study. J Am Geriatr Soc. 2004;52(7):1098-104
221. Goldhammer E, Tanchilevitch A, Maor I, Beniamini Y, Rosenschein U, Sagiv M. Exercise training modulates cytokines activity in coronary heart disease patients. Int J Cardiol. 2005;100(1):93-9
222. Lambert CP, Wright NR, Finck BN, Villareal DT. Exercise but not diet-induced weight loss decreases skeletal muscle inflammatory gene expression in frail obese elderly persons. J Appl Physiol. 2008;105(2):473-8
223. LeMaitre JP, Harris S, Fox KA, Denvir M. Change in circulating cytokines after 2 forms of exercise training in chronic stable heart failure. Am Heart J. 2004;147(1):100-5
224. Nicklas BJ, Hsu FC, Brinkley TJ, Church T, Goodpaster BH, Kritchevsky SB, Pahor M. Exercise training and plasma C-reactive protein and interleukin-6 in elderly people. J Am Geriatr Soc. 2008;56(11):2045-52
225. Hao L, Wang Y, Duan Y, Bu S. Effects of treadmill exercise training on liver fat accumulation and estrogen receptor alpha expression in intact and ovariectomized rats with or without estrogen replacement treatment. Eur J Appl Physiol. 2010;109(5):879-86
226. Kraemer WJ, Häkkinen K, Newton RU, Nindl BC, Volek JS, McCormick M, Gotshalk LA, Gordon SE, Fleck SJ, Campbell WW, Putukian M, Evans WJ. Effects of heavy-resistance training on hormonal response patterns in younger vs. older men. J Appl Physiol. 1999;87(3):982-92
227. Bouchard DR, Soucy L, Sénéchal M, Dionne IJ, Brochu M. Impact of resistance training with or without caloric restriction on physical capacity in obese older women. Menopause. 2009;16(1):66-72
228. Cutlip RG, Baker BA, Geronilla KB, Mercer RR, Kashon ML, Miller GR, Murlastis Z, Alway SE. Chronic exposure to stretch-shortening contractions results in skeletal muscle adaptation in young rats and maladaptation in old rats. Appl Physiol Nutr Metab. 2006;31(5):573-87
229. Dalton BH, Power GA, Vandervoort AA, Rice CL. Power loss is greater in old men than young men during fast plantar flexion contractions. Journal of Applied Physiology. 2010;109:1441-7
230. Ploutz-Snyder LL, Giamis EL, Formikell M, Rosenbaum AE. Resistance training reduces susceptibility to eccentric exercise-induced muscle dysfunction in older women. J Gerontol A Biol Sci Med Sci. 2001;56(9):384-90
231. Brooks SV, Faulkner JA. The magnitude of the initial injury induced by stretches of maximally activated muscle fibres of mice and rats increases in old age. Journal of Physiology. 1996;497(2):573-80
232. Lockhart NC, Brooks SV. Protection from contraction-induced injury provided to skeletal muscles of young and old mice by passive stretch is not due to a decrease in initial mechanical damage. J Gerontol A Biol Sci Med Sci. 2006;61A(6):527-33
233. Brooks SV, Faulkner JA. Contraction-induced injury: recovery of skeletal muscles in young and old mice. Am J Physiol Cell Physiol. 1990;258:C436-C42
234. McBride TA, Gorin FA, Carlsen RC. Prolonged recovery and reduced adaptation in aged rat muscle following eccentric exercise. Mechanisms of Ageing and Development. 1995;83:185-200
235. Fiatarone MA, Marks EC, Ryan ND, Meredith CN, Lipsitz LA, Evans WJ. High-intensity strength training in nonagenarians. Effects on skeletal muscle. JAMA. 1990;263(22):3029-34
236. Häkkinen K, Alen M, Kallinen M, Newton RU, Kraemer WJ. Neuromuscular adaptation during prolonged strength training, detraining and re-strength-training in middle-aged and elderly people. Eur J Appl Physiol. 2000;83(1):51-62
237. Verdijk LB, Gleeson BG, Jonkers RA, Meijer K, Savelberg HH, Dendale P, van Loon LJ. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J Gerontol A Biol Sci Med Sci. 2009;64A(3):332-9
238. Henwood TR, Taaffe DR. Detraining and retraining in older adults following long-term muscle power or muscle strength specific training. J Gerontol A Biol Sci Med Sci. 2008;63(7):751-8
239. Brooks SV, Opiteck JA, Faulkner JA. Conditioning of skeletal muscles in adult and old mice for protection from contraction-induced injury. J Gerontol A Biol Sci Med Sci. 2001;56(4):163-71
240. Suetta C, Hvid LG, Justesen L, Christensen U, Neergaard K, Simonsen L, Ortenblad N, Magnusson SP, Kjaer M, Aagaard P. Effects of aging on human skeletal muscle after immobilization and retraining. J Appl Physiol. 2009;107:1172-80
241. Baker BA, Cutlip RG. Skeletal muscle injury versus adaptation with aging: novel insights on perplexing paradigms. Exerc Sport Sci Rev. 2010;38(1):10-6
242. Raue U, Slivka D, Minchev K, Trappe S. Improvements in whole muscle and myocellular function are limited with high-intensity resistance training in octogenarian women. J Appl Physiol. 2009;106(5):1611-7
243. Verdijk L, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2006;292(1):E151-7
244. Deschenes MR, Roby MA, Glass EK. Aging influences adaptations of the neuromuscular junction to endurance training. Neuroscience. 2011;190:56-66
245. Mayhew DL, Kim JS, Cross JM, Ferrando AA, Bamman MM. Translational signaling responses preceding resistance training-mediated myofiber hypertrophy in young and old humans. J Appl Physiol. 2009;107(5):1655-62
246. Power GA, Dalton BH, Behm DG, Vandervoort AA, Doherty TJ, Rice CL. Motor unit number estimates in masters runners: use it or lose it? Med Sci Sports Exerc. 2010;42(9):1644-50
247. Leong B, Kamen G, Patten C, Burke JR. Maximal motor unit discharge rates in the quadriceps muscles of older weight lifters. Med Sci Sports Exerc. 1999;31(11):1638-44
248. Drummond MJ, Fry CS, Glynn EL, Timmerman KL, Dickinson JM, Walker DK, Gundermann DM, Volpi E, Rasmussen BB. Skeletal muscle amino acid transporter expression is increased in young and older adults following resistance exercise. J Appl Physiol. 2011Jul;111(1):135-42
249. Drummond MJ, Dreyer HC, Pennings B, Fry CS, Dhanani S, Dillon EL, Sheffield-Moore M, Volpi E, Rasmussen BB. Skeletal muscle protein anabolic response to resistance exercise and essential amino acids is delayed with aging. J Appl Physiol. 2008;104(5):1452-61
250. Fry CS, Glynn EL, Drummond MJ, Timmerman KL, Fujita S, Abe T, Dhanani S, Volpi E, Rasmussen BB. Blood flow restriction exercise stimulates mTORC1 signaling and muscle protein synthesis in older men. J Appl Physiol. 2010;108(5):1199-209
251. Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, Williams J, Smith K, Seynnes O, Hiscock N, Rennie MJ. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol. 2009;587(Pt 1):211-7
252. Fry CS, Rasmussen BB. Skeletal muscle protein balance and metabolism in the elderly. Curr Aging Sci. 2011;4(3):260-8
253. Sheffield-Moore M, Paddon-Jones D, Sanford AP, Rosenblatt JJ, Matlock AG, Cree MG, Wolfe RR. Mixed muscle and hepatic derived plasma protein metabolism is differently regulated in older and younger men following resistance exercise. Am J Physiol Endocrinol Metab. 2005;288(5):E922-9
254. Villareal DT, Smith GI, Sinacore DR, Shah K, Mittendorfer B. Regular multicomponent exercise increases physical fitness and muscle protein anabolism in frail, obese, older adults. Obesity (Silver Spring). 2011;19(2):312-8
255. Yarasheski KE, Pak-Loduca J, Hasten DL, Obert KA, Brown MB, Sinacore DR. Resistance exercise training increases mixed muscle protein synthesis rate in frail women and men >/=76 yr old. Am J Physiol. 1999;277(1 Pt 1):E118-25
256. Bamman MM, Petrella JK, Kim JS, Mayhew DL, Cross JM. Cluster analysis tests the importance of myogenic gene expression during myofiber hypertrophy in humans. J Appl Physiol. 2007;102(6):2232-9
257. Fiatarone Singh MA, Ding W, Manfredi TJ, Solares GS, O'neill EF, Clements KM, Ryan ND, Kehayias JJ, Fielding RA, Evans WJ. Insulin-like growth factor I in skeletal muscle after weight-lifting exercise in frail elders. Am J Physiol. 1999;277(1 Pt 1):E135-43
258. Hameed M, Lange KH, Andersen JL, Schjerling P, Kjaer M, Harridge SD, Goldspink G. The effect of recombinant human growth hormone and resistance training on IGF-I mRNA expression in the muscles of elderly men. J Physiol. 2004;555(Pt 1):231-40
259. Roberts MD, Dalbo VJ, Sunderland KL, Poole CN, Hassell CN, Hassell SE, Bemben D, Cramer J, Stout J, Kerksick CM. IGF-1 splice variant and IGF-1 peptide expression patterns in young and old human skeletal muscle prior to and following sequential exercise bouts. Eur J Appl Physiol. 2010;110(5):961-9
260. Hameed M, Toft AD, Pedersen BK, Harridge SD, Goldspink G. Effects of eccentric cycling exercise on IGF-I splice variant expression in the muscles of young and elderly people. Scand J Med Sci Sports. 2008;18(4):447-52
261. Giovannini S, Marzetti E, Borst SE, Leeuwenburgh C. Modulation of GH/IGF-1 axis: potential strategies to counteract sarcopenia in older adults. Mech Ageing Dev. 2008;129(10):593-601
262. Pauli JR, Ropelle ER, Cintra DE, De Souza CT, da Silva AS, Moraes JC, Prada PO, de Almeida Leme JA, Luciano E, Velloso LA, Carvalheira JB, Saad MJ. Acute exercise reverses aged-induced impairments in insulin signaling in rodent skeletal muscle. Mech Ageing Dev. 2010;131(5):323-9
263. Dela F, Kjaer M. Resistance training, insulin sensitivity and muscle function in the elderly. Essays Biochem. 2006;42:75-88
264. Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, Walker DK, Dhanani S, Volpi E, Rasmussen BB. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle. 2011;1(1):11
265. Chalé-Rush A, Morris EP, Kendall TL, Brooks NE, Fielding RA. Effects of chronic overload on muscle hypertrophy and mTOR signaling in young adult and aged rats. J Gerontol A Biol Sci Med Sci. 2009;64(12):1232-9
266. Dalbo VJ, Roberts MD, Sunderland KL, Poole CN, Stout JR, Beck TW, Bemben M, Kersick CM. Acute loading and aging effects on myostatin pathway biomarkers in human skeletal muscle after three sequential bouts of resistance exercise. J Gerontol A Biol Sci Med Sci. 2011;66(8):855-65
267. Hulmi JJ, Ahtiainen JP, Kaasalainen T, Pöllänen E, Häkkinen K, Alen M, Selänne H, Kovanen V, Mero AA. Postexercise myostatin and activin IIb mRNA levels: effects of strength training. Med Sci Sports Exerc. 2007;39(2):289-97
268. Kim JS, Cross JM, Bamman MM. Impact of resistance loading on myostatin expression and cell cycle regulation in young and older men and women. Am J Physiol Endocrinol Metab. 2005;288(6):E1110-9
269. Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Myogenic gene expression at rest and after a bout of resistance exercise in young (18-30 yr) and old (80-89 yr) women. J Appl Physiol. 2006;101(1):53-9
270. Roth SM, Martel GF, Ferrell RE, Metter EJ, Hurley BF, Rogers MA. Myostatin gene expression is reduced in humans with heavy-resistance strength training: a brief communication. Exp Biol Med (Maywood). 2003;228(6):706-9
271. Swan M. Meeting report: American Aging Association 40th Annual Meeting, Raleigh, North Carolina, June 3-6, 2011. Rejuvenation Res. 2011;14(4):449-55
272. Nielsen S, Scheele C, Yfanti C, Akerstrom T, Nielsen AR, Pedersen BK, Laye MJ. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J Physiol. 2010;588(Pt 20):4029-37
273. Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS One. 2009;4(5):e5610
274. Arslan S, Erdem S, Siyri A, Hascelik Z, Tan E. Exercise-induced apoptosis of rat skeletal muscle and the effect of meloxicam. Rheumatol Int. 2002;21(4):133-6
275. Marzetti E, Lawler JM, Hiona A, Manini T, Seo AY, Leeuwenburgh C. Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radic Biol Med. 2008;44(2):160-8
276. Quadrilatero J, Bombardier E, Norris SM, Talanian JL, Palmer MS, Logan HM, Tupling AR, Heigenhauser GJ, Spiret LL. Prolonged moderate-intensity aerobic exercise does not alter apoptotic signaling and DNA fragmentation in human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;298:E534-47
277. Siu PM, Bryner RW, Murlasits Z, Alway SE. Response of XIAP, ARC, and FLIP apoptotic suppressors to 8 wk of treadmill running in rat heart and skeletal muscle. J Appl Physiol. 2005;99(1):204-9
278. Siu PM, Alway SE. Age-related apoptotic responses to stretch-induced hypertrophy in quail slow-tonic skeletal muscle. Am J Physiol Cell Physiol. 2005;289(5):C1105-13
279. Ponsot E, Lexell J, Kadi F. Skeletal muscle telomere length is not impared in healthy physically active old women and men. Muscle Nerve. 2008;37(4):467-72
280. Cherkas LF, Hunkin JL, Kato BS, Richards JB, Gardner JP, Surdulescu GL, Kimura M, Lu X, Spector TD, Aviv A. The association between physical activity in leisure time and leukocyte telomere length. Arch Intern Med. 2008;168(2):154-8
281. LaRocca TJ, Seals DR, Pierce GL. Leukocyte telomere length is preserved with aging in endurance exercise-trained adults and related to maximal aerobic capacity. Mech Ageing Dev. 2010;131(2):165-7
282. Ludlow AT, Zimmerman JB, Witkowski S, Hearn JW, Hatfield BD, Roth SM. Relationship between physical activity level, telomere length, and telomerase activity. Med Sci Sports Exerc. 2008;40(10):1764-71
283. Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61(6):534-40
284. Parise G, Brose AN, Tarnopolsky MA. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Exp Gerontol. 2005;40(3):173-80
285. Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA. 2011;108(10):4135-40
286. Vincent KR, Vincent HK, Braith RW, Lennon SL, Lowenthal DT. Resistance exercise training attenuates exercise-induced lipid peroxidation in the elderly. Eur J Appl Physiol. 2002;87(4-5):416-23
287. Naito H, Powers SK, Demirel HA, Aoki J. Exercise training increases heat shock protein in skeletal muscles of old rats. Med Sci Sports Exerc. 2001;33(5):729-34
288. Parise G, Phillips SM, Kaczor JJ, Tarnopolsky MA. Antioxidant enzyme activity is up-regulated after unilateral resistance exercise training in older adults. Free Radic Biol Med. 2005;39(2):289-95
289. Shefer G, Rauner G, Yablonka-Reuveni Z, Benayahu D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS One. 2010;5(10):E13307
290. Dreyer HC, Blanco CE, Sattler FR, Schroeder ET, Wiswell RA. Satellite cell numbers in young and older men 24 hours after eccentric exercise. Muscle Nerve. 2006;33(2):242-53
291. Jacobs SC, Wokke JH, Bär PR, Bootsma AL. Satellite cell activation after muscle damage in young and adult rats. Anat Rec. 1995;242(3):329-36
292. Kosek DJ, Kim JS, Petrella JK, Cross JM, Bamman MM. Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. J Appl Physiol. 2006;101(2):531-44
293. Lueders TN, Zou K, Huntsman HD, Meador B, Mahmassani Z, Abel M, Valero MC, Huey KA, Boppart MD. The {alpha}7{beta}1 integrin accelerates fiber hypertrophy and myogenesis following a single bout of eccentric exercise. Am J Physiol Cell Physiol. 2011;301(4):C938-46
294. Smith HK, Merry TL. Voluntary resistance wheel exercise during postnatal growth in rats enhances skeletal muscle satellite cell and myonuclear content at adulthood. Acta Physiol (Oxf). 2011;204(3):393-402
295. Dieli-Conwright CM, Spektor TM, Rice JC, Sattler FR, Schroeder ET. Influence of hormone replacement therapy on eccentric exercise induced myogenic gene expression in postmenopausal women. J Appl Physiol. 2009;107(5):1381-8
296. Ambrosio F, Kadi F, Lexell J, Fitzgerald GK, Boninger ML, Huard J. The effect of muscle loading on skeletal muscle regenerative potential: an update of current research findings relating to aging and neuromuscular pathology. Am J Phys Med Rehabil. 2009;88(2):145-55
297. Carson JA, always SE. Stretch overload-induced satellite cell activation in slow tonic muscle from adult and aged Japanese quail. Am J Physiol. 1996;270(2 Pt 1):C578-84
298. Mackey AL, Esmarck B, Kadi F, Koskinen SO, Kongsgaard M, Sylvestersen A, Hansen JJ, Larsen G, Kjaer M. Enhanced satellite cell proliferation with resistance training in elderly men and women. Scand J Med Sci Sports. 2007;17(1):34-42
299. Verney J, Kadi F, Charifi N, Féasson L, Saafi MA, Castells J, Piehl-Aulin K, Denis C. Effects of combined lower body endurance and upper body resistance training on the satellite cell pool in elderly subjects. Muscle Nerve. 2008;38(3):1147-54
300. Konopka AR, Traape TA, Jemiolo B, Trappe SW, Harber MP. Myosin heavy chain plasticity in aging skeletal muscle with aerobic exercise training. J Gerontol A Biol Sci Med Sci. 2011;66(8):835-41
301. Carey K, Farnfield MM, Tarquinio SD, Cameron-Smith D. Impaired expression of notch signaling genes in aged human skeletal muscle. J Gerontoloty Biol Sci A. 2007;62A(1):9-17
Author contact
![]() Corresponding author: Dr. Susan Tsivitse Arthur, Department of Kinesiology, UNC Charlotte, 9201 University City Blvd., Charlotte, NC 28223. Telephone: 704-687-0856 FAX: 704-687-0930 Email: sarthur8edu.
Corresponding author: Dr. Susan Tsivitse Arthur, Department of Kinesiology, UNC Charlotte, 9201 University City Blvd., Charlotte, NC 28223. Telephone: 704-687-0856 FAX: 704-687-0930 Email: sarthur8edu.