Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(8):1097-1108. doi:10.7150/ijbs.4524 This issue Cite
Research Paper
Molecular Characterization of an Ice Nucleation Protein Variant (InaQ) from Pseudomonas syringae and the Analysis of Its Transmembrane Transport Activity in Escherichia coli
Qianqian Li, Qi Yan, Jinsi Chen, Yan He, Jing Wang, Hongxing Zhang, Ziniu Yu, Lin Li ![]()
State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan 430070, Hubei, China.
Received 2012-4-25; Accepted 2012-8-27; Published 2012-9-1
Abstract
The ice nucleation protein (INP) of Pseudomonas syringae has gained scientific interest not only because of its pathogenicity of foliar necroses but also for its wide range of potential applications, such as in snow making, frozen food preparation, and surface-display system development. However, studies on the transport activity of INP remain lacking. In the present study, a newly identified INP-gene variant, inaQ, from a P. syringae MB03 strain was cloned. Its structural domains, signal sequences, and the hydrophilicity or hydrophobicity of each domain, were then characterized. The deduced amino acid sequence of InaQ shares similar protein domains with three P. syringae INPs, namely, InaK, InaZ, and InaV, which were identified as an N-terminal domain, a central repeating domain, and a C-terminal domain. The expression of the full-length InaQ and of various truncated variants was induced in Escherichia coli to analyze their transmembrane transport and surface-binding activities, while using the green fluorescence protein (GFP) as the fusion partner. With two transmembrane segments and a weak secretion signal, the N-terminal domain (InaQ-N) alone was found to be responsible for the transport process as well as for the binding to the outer membrane, whereas the C-terminal region was nonfunctional in protein transport. Increased membrane transport and surface-binding capacities were induced by a low isopropyl-β-D-thiogalactoside concentration (0.1 mmol/l) but not by culture temperatures (15 ºC to 37 ºC). Furthermore, by constructing the GFP-fused proteins with a single InaQ-N, as well as two and three tandemly aligned InaQ-N molecules, the transport and membrane-binding activities of these proteins were compared using Western blot analysis, immmunofluorescence microscopy, and assays of the GFP specific fluorescence intensity of subcellular fractions and flow cytometry, which showed that the increase of InaQ-N repeats resulted in a coordinated increase of the surface-immobilization efficiency. Therefore, the results of this study can serve as a molecular basis for improving the performance of INP-based cell surface-display systems.
Keywords: ice nucleation protein, Pseudomonas syringae, expression, transport, Escherichia coli.
Introduction
Pseudomonas syringae is a common pathogenic bacterium that causes foliar necroses in host plants and a hypersensitive response (HR) in nonhosts [1, 2]. This bacterium is also known as an ice nucleation-active (INA+) pathogen capable of synthesizing a secretory protein, the ice nucleation protein (INP), which confers on cells the ability to nucleate ice crystals at subfreezing temperatures (as high as -2 ºC to -3 ºC), thereby causing frost injury to plants [3]. This INP characteristic has received considerable interest not only for its application in multidisciplinary studies such as biosynthesis, regulation, and pathogenicity, but also for its potential applications in the production of frozen goods, snow making, and bacterial cell surface-display system development, with INP as the anchoring carrier [4, 5]. The potential applications has been expanded to include the development of live vaccines, the construction and screening of protein libraries, and the fabrication of environmental whole-cell biocatalysts, biosensors, and biosorbents, among others [6, 7].
INP has been found in more than 10 bacterial species, typically Gram-negative phyllospheric bacteria that include P. syringae, Erwinia herbicola, Xanthomonas campestris, and Pseudomonas fluorescens [5, 8]. The genes corresponding to the INA+ phenotype have been identified in more than 10 different strains. Three of them, inaK [9], inaV [10], and inaZ [11], were previously cloned and characterized from P. syringae strains, which exhibit high similarities in sequences and in primary organization. All INPs (1200 aa to 1500 aa) encoded by these three genes comprise of three distinct structural domains: (1) the N-terminal domain (approximately 15% of the total sequence), which is relatively hydrophobic and contains no signal peptide sequences. This region is rich in asparagine, serine, and threonine, which are potentially capable of being coupled to the mannan-phosphatidylinositol group in the outer membrane through N-glycan (Asp) or O-glycan (Ser, Thr) linkages [12, 13]; (2) the C-terminal domain (approximately 4%), which is a relatively hydrophilic terminus; and (3) the central repeating domain (CRD) (approximately 81%), which constitutes contiguous repeats given by 16-residue (or 48-residue) periodicities with a consensus octapeptide (Ala-Gly-Tyr-Gly-Ser-Thr-Leu-Thr) [8, 14]. This domain presumably acts as a template for ice crystal formation. Neither the C-terminal domain nor the CRD of these INPs harbors the signal peptide sequences.
However, the intracellularly synthesized INPs can be exported to both the plant tissue in natural habitats and to the medium when grown under pure culture conditions [3]. In addition, the heterologous expression of INPs as a result of recombinant DNA techniques has been previously performed in various developed bacterial cell-surface display systems including Escherichia coli [15-17], Zymomonas mobilis [9], Salmonella sp. [18], Vibrio anguillarum [19], Pseudomonas putida [16, 20], and a cyanobacterium [21]. These systems have demonstrated that the INPs can transport extracellularly and are then primarily bound to the surface of the target cells. Although these results clearly indicated that INP must be exported via some other signal peptide-independent pathway, little is known of the secretion mechanism and of the functional segment responsible for the transmembrane transport across the parent cell membrane. To date, limited effort has been exerted to promote the membrane-binding capacity of the INP anchoring motifs.
The INP-based anchor is currently considered a logical choice for the construction of cell-surface display systems in Gram-negative bacteria because of its stable expression, feasible transmembrane transport, and adjustable CRD length, which are helpful in ensuring the appropriate protein fusion length [6, 22]. In addition, a number of investigations had reported on other advantages of this anchor protein, such as its negligible negative effect on either the membrane integrity or the growth rate of host cells as well as the feasibility of directing a number of large proteins [17]. Unfortunately, while these reports describe the successful application of the INP-anchored functional systems, some previous reports had noted that only a limited amount of intracellularly expressed INP-fused proteins were effectively transported to the surface of host cells, thereby conferring insufficient surface display activities [15, 16, 23]. Therefore, the development of a more efficient INP-mediated surface display system remains urgent.
In the present study, a newly identified INP-gene variant from a highly active P. syringae bacterium MB03 through an in site hybridization, inaQ, was compared both the sequence similarity and the structural organization with those of three other P. syringae-harbored INP genes. The membrane transport activities of the full-length InaQ, as well as those of the truncated variants lacking partial N-terminal sequences, and those lacking whole N-terminal and CRD domain, were determined using the model protein green fluorescence protein (GFP) as a reporter protein in E. coli cells. After the investigation of the effects of the expression level and the culture temperatures on the transport and membrane-binding activities, the surface display efficiencies of the fusion proteins with 1-3 tandemly aligned InaQ N-terminal domain anchors were compared using several assays. This study aims to improve the understanding of the membrane transport of InaQ and to develop activity-promoted surface-immobilization systems.
Materials and Methods
Bacterial strains and culture conditions
E. coli DH5α cells (TaKaRa Bio, Inc.) were used to construct various recombinant plasmids. E. coli JM109 [F′ traD36, proAB+ lacIq. lacZΔM15/Δ(lac-proAB) glnV44 e14- gyrA96 recA1 relA1 endA1 thi hsdR17] (TaKaRa Bio, Inc.) was used as the host strain for the expression of the target proteins and for surface-immobilization activity analysis. The wild-type P. syringae subsp. syringae MB03 (Microbial Genetic Stock Center, Wuhan, China) was used as the parent strain for the inaQ gene cloning. Recombinant E. coli strains harboring various recombinant plasmids were grown in Luria-Bertani (LB) medium containing 100 μg/ml of ampicillin (Amp) at 37 ºC. For the expression of the target proteins in E. coli, when the OD600 of the cell suspensions reached 0.6, isopropyl-β-D-thiogalactoside (IPTG) was added at a final concentration of 0.1 mmol/l, unless otherwise specified.
Gene cloning and plasmid construction
The plasmids and oligonucleotide primers used in this study as well as the 32P-labeled oligonucleotide probe for the in situ hybridization experiment are listed in Table 1. All routine DNA manipulations were performed using standard procedures [24]. To clone the inaQ gene, the P. syringae MB03 total DNA was digested with BglII/EcoRI and then separated via electrophoresis using 0.8% agarose gel. Bands with approximately 4 kb to 5 kb sizes were excised, purified from the gel, ligated into the BglII/EcoRI sites of the E. coli vector plasmid pRSET-B (Invitrogen), and then transformed into the E. coli DH5α. The Amp-resistant colonies were then transferred to a nylon membrane (Bio-Rad, USA) and further hybridized with the 32P-labeled probe (Table 1) using a previously described method [10]. The corresponding positive hybridization colonies were screened and their plasmid DNAs were prepared and then digested with BglII/EcoRI to allow the sequencing of the digested fragments of interest. A clone that harbors the full-length frame of an INP gene together with its cognate promoter sequence was identified and subsequently designated as inaQ. The corresponding plasmid harboring inaQ was designated as pMB101.
Plasmids, oligonucleotide primers, and probes used in this study
| Plasmid, primer, or probe | Phenotype or sequencea | Source or reference |
|---|---|---|
| Plasmids | ||
| pRSET-B | Ampr, E. coli expression vector, 2887 bp | Invitrogen |
| pTrcHis-C | Ampr, E. coli expression vector, 4412 bp | Invitrogen |
| pGFPuv | Ampr, E. coli expression vector encodes a variant of GFP, 3336 bp | CLONTECH Lab, Inc. |
| pMB101 | Ampr, a pRSET B derivative harboring inaQ and its cognate promoter, 7022 bp | This study |
| pMB102 | Ampr, a pTrcHis-C derivative harboring Ptrc, inaQ-N and gfp fusion gene (inaQ-N/gfp), 5543 bp | [16] |
| pMB107 | Ampr, a pMB102 derivative harboring Ptrc, inaQ-N′ (lacking the first 54 bp of inaQ-N) and gfp fusion gene, 5492 bp | This study |
| pMB109 | Ampr, a pMB102 derivative harboring fusion gene (inaQ-N)2/gfp, 6071 bp | This study |
| pMB110 | Ampr, a pMB102 derivative harboring fusion gene (inaQ-N)3/gfp, 6599 bp | This study |
| pMB117 | Ampr, a pTrcHis-C derivative harboring Ptrc, inaQ and gfp fusion gene, 8618 bp | This study |
| pMB118 | Ampr, a pTrcHis-C derivative harboring Ptrc, inaQ-C and gfp fusion gene (inaQ-C/gfp), 5168 bp | This study |
| Oligonucleotide primersb | ||
| inpN1 | 5′-TGCCATGGATCTCGACAAGGCGTTGGTGC-3′ (NcoI) | |
| inpN2 | 5′-TAAGATCTGGTCTGCAAATTCTGCGGCGTCG-3′ (BglII) | |
| inp2 | 5′-CGAAGATCTCTCGACCTCTATCCAGTC-3′ (BglII) | |
| gfpR | 5′-GGAATTCTTATTTGTAGAGCTCATCCATGCC-3′ (EcoRI) | |
| inpC1 | 5′-TTTCCATGGTCAGGCTCTGGGACG-3′ (NcoI) | |
| PND1 | 5′-TTTCCATGGATTGCGGCCTTATATGGCC-3′ (NcoI) | |
| PN1 | 5′-TTAGGATCCGATCTCGACAAGGCGTTGGTG-3′ (BamHI) | |
| Oligonucleotide probe | 5′-GCNGGNTAYGGNAGYACNCARAC-3′ | [10] |
aAmpr, ampicillin resistance; inaQ, ice nucleation protein gene from P. syringae MB03; inaQ-N, N-terminal domain of inaQ; inaQ-N′, truncated inaQ-N lacking the first 54 bp; inaQ-C, C-terminal domain of inaQ; gfp, green fluorescent protein gene; Ptrc, pTrcHis-C promoter.
bThe underlined sequences indicate the restriction enzyme sites.
To construct the recombinant plasmid expressing the fusion gene inaQ-gfp under the control of the regulative promoter Ptrc, the primer pair inpN1/inp2 was used for the polymerase chain reaction (PCR), with plasmid pMB101 DNA as the template. The amplified 3603 bp fragment was digested with NcoI/BglII and then ligated into the NcoI/BglII sites of the E. coli expression vector plasmid pTrcHis-C (Invitrogen). A 743 bp BglII/EcoRI-digested fragment of a previous plasmid pMB102 [16] harboring the N-terminal domain of the inaQ (inaQ-N) and gfp fusion genes was then inserted into the BglII/EcoRI sites, thereby yielding the pMB117 plasmid.
The pMB102 plasmid was then used to construct several recombinant plasmids harboring genes fused with different inaQ and gfp domains. Of these plasmids, the truncated inaQ-N lacking the first 54 bp (designated as inaQ-N′) was amplified using primers PND1 and inpN2 and then digested with NcoI and BglII. The resulting fragment was then inserted into the NcoI/BglII sites of pMB102 to obtain plasmid pMB107. The inaQ-N/gfp fusion gene was amplified using primers PN1 and gfpR from pMB102 and then digested with BamHI and EcoRI. The resulting fragment was then inserted into the BglII/EcoRI sites of pMB102, thereby yielding plasmid pMB109, which harbor the fusion gene with two tandemly aligned inaQ-N and gfp [(inaQ-N)2/gfp]. In a similar manner, this digested fragment was further inserted into the BglII/EcoRI sites of pMB109 to generate plasmid pMB110, which harbor the (inaQ-N)3/gfp fusion gene with three tandemly aligned inaQ-N. To construct the pMB118 plasmid harboring the C-terminal domain of inaQ (inaQ-C) and the gfp fusion gene (inaQ-C/gfp), the inaQ-C fragment was amplified using primers inpC1 and inp2 from plasmid pMB101 and then digested with NcoI and BglII. The resulting fragment was then inserted into plasmid pMB102.
GFP fluorescence intensity assay
Prior to the GFP fluorescence intensity determination, the cells were harvested and diluted to the unit cell density (OD600 = 1.0) using phosphate-buffered saline (PBS) (pH 7.0). The specific GFP fluorescence intensities of the whole-cell and/or cell fractions were measured using a fluorescence spectrophotometer (RF-5103PC, Shimadzu, Japan) at an excitation wavelength of 395 nm and an emission wavelength of 509 nm.
Cell fractionation
Cell suspensions were passaged twice through a French pressure cell (Thermo, USA) at 13,000 psi. The suspension was then fractionated following previously described procedures [15].
Western blot analysis
The whole-cell InaQ proteins as well as the various truncated InaQ-based fusion proteins were separated in 12.0% gels via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were then transferred to Hybond-PVDF membranes (Amersham, UK). Western blot analysis was performed to verify the expression of GFP-fused proteins with the full-length InaQ and various truncated InaQ variants using monoclonal anti-GFP antibodies (Chemicon, USA) as the primary antibody and the horseradish peroxidase-conjugated goat anti-mice IgG (Chemicon, USA) as the secondary antibody, following previously described procedures [16].
Immunofluorescence microscopy and flow cytometry
Recombinant E. coli cells transporting and displaying fusion proteins with full-length or truncated InaQ and GFP were examined using immunofluorescence microscopy, as previously described [16]. The GFP-fluorescent as well as Cy5-labeled cells were quantified via flow cytometry [16] using a FACScan flow cytometer (Becton Dickinson, Oxnard, CA, USA) equipped with a dual laser system (Ar: 488 nm and He-Ne: 633 nm) for the simultaneous measurement of GFP and Cy5 fluorescence. Measurements are expressed as the percentage of total GFP-labeled cells relative to the total Cy5-fluorescent cells.
Pronase proteolysis assays, SDS and EDTA sensitivities
Recombinant E. coli JM109 cells expressing GFP-fused proteins were harvested by centrifugation, washed three times with PBS buffer (pH 7.0), and then diluted to OD600 1.0 suspensions. After the addition of 0.5 mg/ml pronase (5.2 units/mg, Sigma), 0.05% SDS (w/v), or 2 mmol/l EDTA, respectively, the cell suspensions were incubated at 37 ºC to determine the GFP fluorescence intensity at 1 h intervals. Relative GFP fluorescence intensity (%) denotes the percentage of the residual fluorescence intensity after the treatments relative to the total fluorescence intensity.
For determining the effect of different IPTG induction concentrations on cell-surface immobilization activities of the fusion proteins, when the OD600 of the cell suspensions reached 0.6, IPTG was added at the final concentrations of ranging from 0.1 mmol/l to 1.0 mmol/l, and allows a following 8 h culturing. The cells were harvested, washed, and diluted, and then treated by pronase at similar procedures as described above. The whole-cell GFP specific fluorescence intensity and residual GFP fluorescence after pronase proteolysis was determined.
For determining the effect of different temperatures on cell-surface immobilization activity of the fusion protein, 0.1 mmol/l IPTG was used to induce the expression of the fusion proteins under the given culture temperature conditions (15 ºC, 20 ºC, 28 ºC, and 37 ºC), the whole-cell and residual GFP fluorescence intensity was then measured following above procedures.
Database search and molecular modeling
The inaQ sequence was characterized by conducting BLASTN and BLASTP searches on the GenBank nucleotide and amino acid (aa) sequence database using the National Center for Biotechnology Information (NCBI) server (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The transmembrane sites were predicted using the HMMTOP (http://www.enzim.hu/hmmtop/). Mean hydropathy analysis was performed using the BioEdit software (http://www.mbio.ncsu.edu/bioedit/bioedit.html). The possible signal peptide sequences were analyzed using SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP/).
Results and Discussion
Cloning of inaQ gene from P. syringae MB03 and molecular characterization of InaQ
Sequence analysis showed that a 4197 bp BglII/EcoRI-digested fragment corresponding to a positive clone of in situ hybridization from the P. syringae subsp. syringae MB03 gene library was found to harbor a 3603 bp open reading frame (ORF), which is preceded by a 522 bp putative promoter region. The nucleotide alignment using the online BLASTN tool suggests that this ORF, designated as inaQ (GenBank accession no. EU360731.1), can be classified as an INP-encoding gene. The inaQ gene encodes a 1200 aa protein with a deduced molecular mass of 118.3 kDa and an isoelectric point of 4.59. The sequence alignments of the inaQ and three other genes (inaK, inaV, and inaZ) show similarities of at least 89% and 92% in the gene and aa sequences, respectively.
In the molecular organization, the InaQ protein consists of three INP-characteristic domains, which can be structurally distinguished as an N-terminal domain (InaQ-N, 175 aa) comprising two transmembrane segments (TM) (TM1 from aa 6 to 30; TM2 from aa 44 to 62), a CRD (976 aa) constitutes of a series of tandemly aligned 16-residue repeats with the β-sheet confirmation, and a C-terminal domain (InaQ-C, 49 aa). InaQ-N is rich in hydrophobic aa residues (Supplementary Material: Fig. S1A), whereas the C-terminal domain InaQ-C is constituted mainly by hydrophilic residues such as Gly, Tyr, Thr, and Ser (Supplementary Material: Fig. S1B). Interestingly, although no clear signal sequences were found in both the InaQ-N and the InaQ-C domain via SignalP, the analysis of the C-score value used to predict the possible cleavage site of the signal peptide [25] indicates a weak signal between aa 18 and aa 19 of InaQ-N (Supplementary Material: Fig. S1C).
Transport activities of fusion proteins with full-length or truncated InaQ and GFP
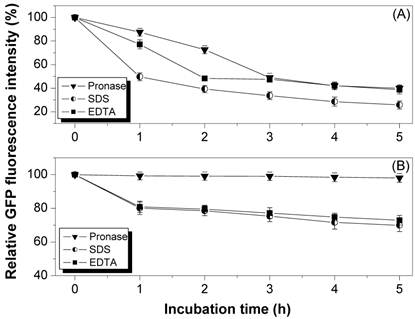
The reporter protein GFP was expressed in fusion form with the InaQ in E. coli JM109 to facilitate the surface localization assays of the different InaQ variant proteins. The surface localization of recombinant strains harboring the pMB117 and pGFPuv plasmids, which encode InaQ-GFP and alone GFP, respectively, were compared with those of their expressed products via the pronase accessibility assay as well as SDS and EDTA sensitivity determination (Fig. 1). GFP is considered sensitive to pronase but not to most common proteases [26]. In addition, only surface-immobilized GFP is subjected to proteolysis by externally added pronase. Therefore, the surface localization of InaQ-GFP in the transformed cells (Fig. 1) was theoretically confirmed by the observed 60% reduction in GFP fluorescence intensity during pronase treatment for 5 h (Fig. 1A). This finding is in contrast to the slight reduction (less than 10%) in the pronase-treated control cells expressing intracellular GFP (Fig. 1B). In addition, SDS and EDTA treatments were used to assay the membrane integrity in the transformed cells upon surface localization of fusion proteins. The cells with surface-expressed InaQ-GFP (Fig. 1A) were more sensitive to SDS and EDTA than the control cells (Fig. 1B), suggesting that the InaQ-GFP was localized on the surface of the transformed cells.
Pronase accessibility, SDS and EDTA sensitivity assays of (A) E. coli JM109/pMB117 cells expressing InaQ-GFP, and (B) the control strain E. coli JM109/pGFPuv expressing only cellular GFP. Relative values were based on the GFP fluorescence intensity at the initial incubation time.
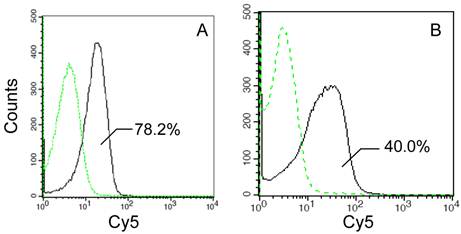
Several previous studies have described the alone N-terminal domain of an INP protein can direct surface immobilization of the foreign proteins [15, 17, 23]. However, it remained uncertain whether the truncated N-terminal domain maintained the full transmembrane transport activity of an INP. For this purpose, the transport and outer membrane-binding activities of the InaQ-GFP and InaQ-N/GFP were compared via flow cytometry assays. Compared with the transformed E. coli JM109/pMB102 strain expressing InaQ-N/GFP, the E. coli JM109/pMB117 cells expressing InaQ-GFP exhibited a pronounced Cy5 fluorescence intensity (Fig. 2), which suggests that the full-length InaQ is more efficient in targeting GFP than that of the InaQ-N. Therefore, the subdivisional domains responsible of transmembrane transport of the InaQ protein need to be further verified.
Flow cytometric analysis of recombinant E. coli JM109/pMB117 and JM109/pMB102 cells expressing InaQ-GFP and InaQ-N/GFP, respectively. Cells were labeled with primary monoclonal anti-GFP antibodies, followed with secondary Cy5-conjugated IgG. The value of each histogram indicates the percentage of total Cy5-labeled fluorescent cells.
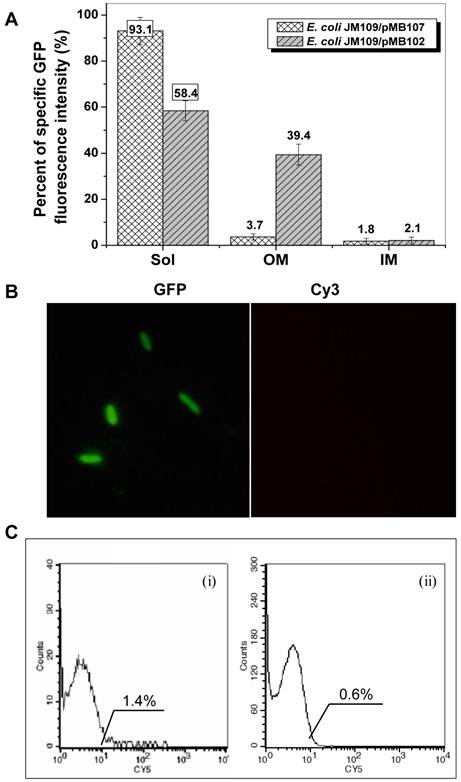
The presence of two transmembrane segments (TM1 and TM2, Fig. 1A) as well as a weak secretion signal (Supplementary Material: Fig. S1C, as indicated by the arrow) in InaQ-N region suggests that this domain is most likely responsible for the membrane transport activity. To investigate whether the leading region of InaQ-N is associated with the transport and membrane binding events, the truncated inaQ-N′ (which lacks the first 54 bp of inaQ-N) was fused with gfp to construct a hybrid inaQ-N′/gfp gene. The GFP-specific fluorescence intensity of the subcellular fractions of JM109/pMB107 and JM109/pMB102 cells expressing the InaQ-N′/GFP and InaQ-N/GFP, respectively, was comparatively determined. The total GFP fluorescence intensity of both transformed cells is comparable (by approximately 230 FI/OD600) upon similar IPTG inductions and culture conditions. However, as shown in Fig. 3A, approximately 93% of the total GFP fluorescence was maintained in the cell-soluble cytoplasmic fractions of JM109/pMB107 cells, whereas almost no GFP fluorescence was observed in the outer membrane fraction, which strongly contrasts with approximately 58% and 38% of the total GFP fluorescence occurred in the cytoplasmic and the outer membrane fractions of JM109/pMB102 cells, respectively. The nonoccurrence of surface localization of the InaQ-N′/GFP fusion protein on the corresponding JM109/pMB107 cells was further confirmed via immunofluorescence microscopy observation and flow cytometry assays (Figs. 3B and 3C, respectively). Therefore, these results suggest that the leading region in InaQ-N (consisting of the first 18 aa) is obligatorily necessary to the transmembrane transport of InaQ-N/GFP.
Expression profiles of E. coli JM109/pMB107 cells expressing InaQ-N′/GFP. (A) GFP fluorescence intensity measurements of subcellular fractions of JM109/pMB107 cells and the control strain JM109/pGFPuv expressing cellular GFP. Sol, soluble cytoplasmic fraction; OM, outer membrane fraction; IM, inner membrane fraction. Each value and error bar represents the mean of three independent experiments and its standard deviation, respectively. (B) Immunofluorescence micrographs of intact JM109/pMB107 cells. Prior to microscopic observation, the cells were treated with monoclonal anti-GFP antibodies, followed with Cy3-conjugated antibodies. (C) Flow cytometric assay of (i) JM109/pMB107 and (ii) JM109/pGFPuv (negative control). Cells were labeled with monoclonal anti-GFP primary antibodies followed with secondary Cy5-conjugated IgG. The value in each histogram indicates the percentage of total Cy5-labeled fluorescent cells.
The functionality of the alone C-terminal domain was also analyzed by determining the expression and surface localization of the fusion proteins with InaQ-C and GFP via flow cytometry assay (Fig. 4). In contrast to the over 90% GFP-associated fluorescent cells, the Cy5-associated cells were very limited, indicating that the expressed InaQ-C/GFP fusion protein was almost exclusively maintained in the cell cytoplasm. The results show that InaQ-C was unable to direct the transmembrane transport.
Flow cytometry assay of E. coli JM109/pMB118 cells expressing InaQ-C/GFP. Prior to the assay, the cells were treated with monoclonal anti-GFP primary antibodies, followed with Cy5-conjugated secondary IgG. The value in each histogram indicates the percentage of total Cy5-labeled fluorescent cells.
Effect of IPTG concentrations and culture temperatures on the transport and outer membrane binding activities of InaQ-N/GFP
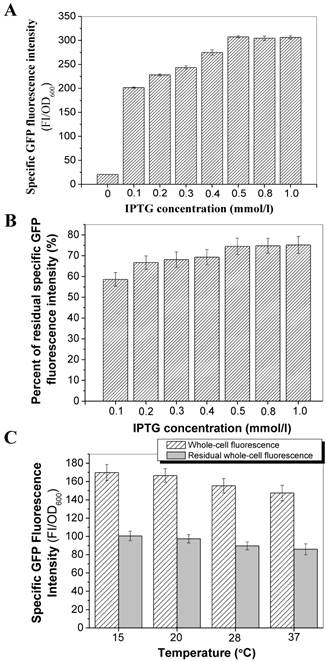
The transmembrane transport and surface immobilization of the fusion protein InaQ-N/GFP was investigated upon the induction of different IPTG concentrations. A steadily increasing expression pattern of E. coli JM109/pMB102 whole-cell GFP fluorescence intensity was observed at the 0.1 mmol/l to 0.5 mmol/l IPTG concentration until a threshold concentration (0.5 mmol/l), at which no increase in fluorescence was detected with increasing IPTG concentration, was reached (Fig. 5A). Interestingly, a reverse pattern was observed in the GFP fluorescence intensity of intact cells subjected to pronase proteolysis, as was shown of the percentage of the residual GFP fluorescence in Fig. 5B, suggests that low IPTG concentrations (especially at 0.1 mmol/l) resulted in increased membrane transport and surface immobilization efficiencies, which is presumable that the cellular transcription and secretion could be more coordinated under such induction conditions. Moreover, the effects of different culture temperatures on the transport and surface immobilization of InaQ-N/GFP in E. coli JM109/pMB102 cells upon induction by 0.1 mmol/l IPTG and with or without pronase treatment was also investigated. As was shown in Fig. 5C, only a slight difference in the activities of both whole-cell and residual whole-cell GFP fluorescence was observed within the 15 ºC to 37 ºC temperature range, suggested the effect of the culture temperature on transport and surface immobilization of the fusion protein was weak.
Effect of IPTG concentrations and culture temperatures on the transport and surface-binding activities of InaQ-N/GFP. (A) Correlation between IPTG concentration and GFP fluorescence intensity of E. coli JM109/pMB102 expressing the InaQ-N/GFP fusion protein. (B) Residual GFP fluorescence intensity of intact cells after pronase proteolysis. (C) Whole-cell and residual GFP fluorescence intensity of the outer membrane fractions of JM109/pMB102 cells cultured at different temperatures and without/with pronase proteolysis.
Effects on GFP transport and surface-display efficiency using tandemly aligned InaQ-N anchors
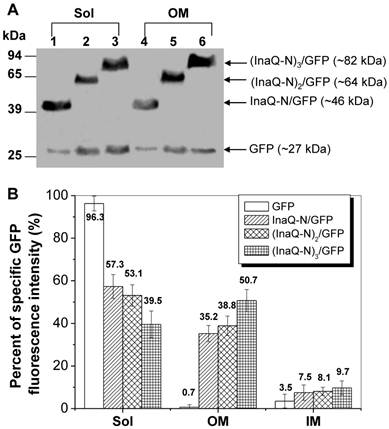
To investigate the effect of different repeats of the InaQ-N domain on the transmembrane transport and surface-display efficiencies, the 1-3 tandemly aligned InaQ-N repeats were used as anchoring motifs to target GFP onto the outer membrane from the cytoplasm of E. coli cells. Western blot analysis of the cell fractions show that each of the fusion proteins, InaQ-N/GFP, (InaQ-N)2/GFP, and (InaQ-N)3/GFP, which were expressed with the predicted molecular weights of approximately 46, 64, and 82 kDa, respectively (Fig. 6A), were found in both the total membrane fractions and the soluble cytoplasmic fractions of the corresponding cells. Interestingly, with the increase of InaQ-N repeats to 2 and 3, the amount of (InaQ-N)2/GFP and (InaQ-N)3/GFP in the outer membrane fractions (Fig. 6A, lane 2 and 3) appeared coordinately increased while compared to each residual protein in the ctoplasmic fractions [(InaQ-N)2/GFP in Fig. 6A, lane 5; (InaQ-N)3/GFP in Fig. 6A, lane 6], whereas the InaQ-N/GFP occurred in the cytoplasmic fraction (Fig. 6A, lane 1) rather than in the outer membrane fraction (Fig. 6A, lane 4). This is consistent with the quantitative determination of GFP intensities of the subcellular fractions, at which over 42.7%, 46.9%, and 60.5% of the total GFP intensity was recorded in total membrane fractions of the transformed cells expressing InaQ-N/GFP, (InaQ-N)2/GFP, and (InaQ-N)3/GFP, respectively (Fig. 6B), which exhibited an increased pattern of total membrane GFP-fluorescence percentage along with the increase of InaQ-N repeats, therefore indicating the increased InaQ-N repeats has resulted in the improved transmembrane transport efficiency of fusion proteins.
Expression profiles of E. coli JM109/pMB102, JM109/pMB109, and JM109/pMB110 cells expressing InaQ-N/GFP, (InaQ-N)2/GFP and (InaQ-N)3/GFP, respectively. (A) Western blot analysis of the cell fraction samples. Lanes 1 and 4, JM109/pMB102; lanes 2 and 5, JM109/pMB109; lanes 3 and 6, JM109/pMB110. (B) Percentage of specific GFP fluorescence intensities of subcellular fractions. E. coli JM109/pGFPuv expressing cellular GFP was used as the negative control of protein surface-immobilization. In (A) and (B), Sol, soluble cytoplasmic fraction; OM, outer membrane fraction; IM, inner membrane fraction.
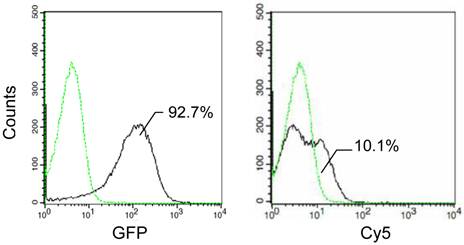
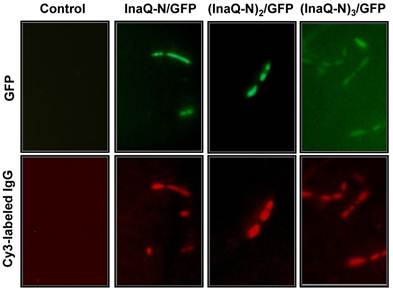
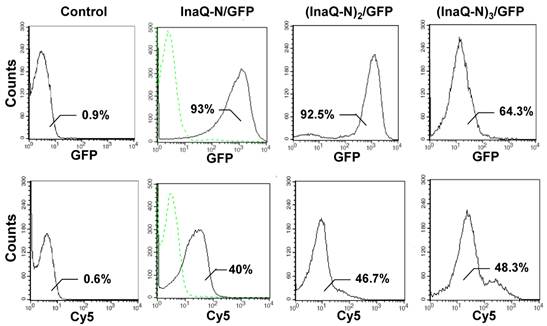
The surface localization of the fusion proteins with 1-3 tandemly aligned InaQ-N repeats and GFP in the transformed E. coli cells was examined via immunofluorescence microscopy (Fig. 7). Both GFP and Cy5 fluorescence were clearly visualized in the intact cells, thereby confirming that the anchors with 1-3 InaQ-N repeats were functional for the surface immobilization of GFP. Furthermore, FACS assays were performed to quantitatively detect the surface display efficiency of these fusion proteins in the transformed cells. As shown in Fig. 8, the increase in anchoring motifs with two or three repeats of InaQ-N resulted in a coordinated increase in the surface-immobilization efficiency. The three repeats of the InaQ-N-associated fusion protein showed the highest display efficiency despite the reduction in the total GFP fluorescence. These results are consistent with those noted in Western blot analysis, as was shown in Fig. 6A. Thus, the number of the InaQ-N repeats appeared to be crucial to the binding efficiency, and the use of tandemly aligned InaQ-N repeats could be considered a valuable approach to promote surface display capacity of foreign proteins.
Micrographs of E. coli JM109/pMB102, JM109/pMB109, and JM109/pMB110 expressing InaQ-N/GFP, (InaQ-N)2/GFP and (InaQ-N)3/GFP, respectively. The panels show the phase-contrast microscopy and fluorescence microscopy images using green and red emission filters. For immunofluorescence microscopy, the cells were treated with anti-GFP monoclonal antibodies, followed with Cy3-conjugated secondary antibodies. E. coli JM109 cells were used as the negative control.
Flow cytometric analysis of E. coli JM109/pMB102, JM109/pMB109, and JM109/pMB110 cells. The cells were labeled with primary monoclonal anti-GFP antibodies, followed with secondary Cy5-conjugated antibodies. The value in each histogram indicates the percentage of total GFP or Cy5-labeled fluorescent cells. E. coli JM109 cells were used as the negative control.
As a commonly used carrier protein for various developed cell-surface display systems for Gram-negative bacteria in the past decade, the further promotion of the transmembrane transport activity of this protein as a means of improving the surface display efficiency of target proteins has been a subject of significant interest. Unfortunately, although previous studies have been validated the functionality of various INP-based anchoring motifs using the full-length INP, the truncated INP with only N- and C-terminal domains, the alone N-terminal domain, or even N- and C-terminal domains with partially prolonged sequences from CRD region [9, 15, 22, 23], inefficient translocation activity was noted as a result of usually over 50% expressed proteins retained in the cell cytoplasm [15, 16, 23]. Considering that the N-terminal domain is the only relatively hydrophobic region, we performed a strategy to improve the transmembrane transport activity of living cells by increasing the number of InaQ-N anchors. The significant increase of the surface display efficiency with the increase of tandemly aligned InaQ-N repeats reflects that this strategy is applicable. It is of interest to further investigate the critical InaQ-N repeat numbers that allows for more efficient transport of InaQ-N based fusion proteins in this system.
In summary, the present study demonstrates that the N-terminal domain is responsible for the transmembrane transport and membrane-binding activity of a newly characterized INP-gene variant (InaQ). Although no clear signal sequences were identified, the first 18 aa of InaQ-N was found essential to protein secretion. The C-terminal domain was non-functional in extracellular secretion. Increased membrane transport and surface immobilization efficiencies were observed at a low IPTG concentration (0.1 mmol/l). Furthermore, the increase in the number of anchoring motifs (with two or three InaQ-N repeats) resulted in a coordinated increase in the surface-immobilization efficiency. The results of this study can serve as a molecular basis for improving the performance of INP-based cell surface-display systems.
Supplementary Material
Fig.S1 - Hydrophobicity/hydrophilicity and signal peptide prediction of the InaQ domains using bioinformatics tools.
Acknowledgements
This work was supported by grants from 31070111 and 30370026 from the National Natural Science Foundation of China, and in part by 2012MBDX011 from the Fundamental Research Funds for the Central Universities and by 30670054 from the National Natural Science Foundation of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bender C, Alarcon-Chaidez F, Gross D. Pseudomonas syringae phytotoxins: mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol Mol Biol Rev. 1999;63:266-292
2. Collmer A, Badel JL, Charkowski AO. et al. Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc Natl Acad Sci USA. 2000;97:8770-8777
3. Hirano SS, Upper CD. Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev. 2000;64:624-653
4. Cochet N, Widehem P. Ice crystallization by Pseudomonas syringae. Appl Microbiol Biotechnol. 2000;54:153-161
5. Gurian-Sherman D, Lindow SE. Bacterial ice nucleation: significance and molecular basis. FASEB J. 1993;7:1338-1343
6. Lee SY, Choi JH, Xu Z. Microbial cell-surface display. Trends Biotechnol. 2003;21:45-52
7. Daugherty PS. Protein engineering with bacterial display. Curr Opin Struct Biol. 2007;17:474-480
8. Kawahara H. The structures and functions of ice crystal-controlling proteins from bacteria. J Biosci Bioeng. 2002;94:492-496
9. Jung HC, Lebeault JM, Pan JG. Surface display of Zymomonas mobilis levansucrase by using the ice-nucleation protein of Pseudomonas syringae. Nat Biotechnol. 1998;16:576-580
10. Schmid D, Pridmore D, Capitani G. et al. Molecular organisation of the ice nucleation protein InaV from Pseudomonas syringae. FEBS Lett. 1997;414:590-594
11. Green RL, Warren GJ. Physical and functional repetition in a bacterial ice nucleation gene. Nature. 1985;317:645-648
12. Turner MA, Arellano F, Kozloff LM. Components of ice nucleation structures of bacteria. J Bacteriol. 1991;173:6515-6527
13. Kozloff LM, Turner MA, Arellano F. et al. Phosphatidylinositol, a phospholipid of ice-nucleating bacteria. J Bacteriol. 1991;173:2053-2060
14. Ferguson MA, Williams AF. Cell-surface anchoring of proteins via glycosyl-phosphatidylinositol structures. Annu Rev Biochem. 1988;57:285-320
15. Li L, Kang DG, Cha HJ. Functional display of foreign protein on surface of Escherichia coli using N-terminal domain of ice nucleation protein. Biotechnol Bioeng. 2004;85:214-221
16. Li Q, Yu Z, Shao X. et al. Improved phosphate biosorption by bacterial surface display of phosphate-binding protein utilizing ice nucleation protein. FEMS Microbiol Lett. 2009;299:44-52
17. Wu ML, Tsai CY, Chen TH. Cell surface display of Chi92 on Escherichia coli using ice nucleation protein for improved catalytic and antifungal activity. FEMS Microbiol Lett. 2006;256:119-125
18. Lee JS, Shin KS, Pan JG. et al. Surface-displayed viral antigens on Salmonella carrier vaccine. Nat Biotechnol. 2000;18:645-648
19. Xu Y, Liu Q, Zhou L. et al. Surface display of GFP by Pseudomonas syringae truncated ice nucleation protein in attenuated Vibrio anguillarum strain. Mar Biotechnol (NY). 2008;10:701-708
20. Shimazu M, Nguyen A, Mulchandani A. et al. Cell surface display of organophosphorus hydrolase in Pseudomonas putida using an ice-nucleation protein anchor. Biotechnol Prog. 2003;19:1612-1614
21. Chungjatupornchai W, Fa-aroonsawat S. Translocation of green fluorescent protein to cyanobacterial periplasm using ice nucleation protein. J Microbiol. 2009;47:187-192
22. Jung HC, Park JH, Park SH. et al. Expression of carboxymethylcellulase on the surface of Escherichia coli using Pseudomonas syringae ice nucleation protein. Enzyme Microb Technol. 1998;22:348-354
23. Li Q, Ni H, Meng S. et al. Suppressing Erwinia carotovora pathogenicity by projecting N-acyl homoserine lactonase onto the surface of Pseudomonas putida cells. J Microbiol Biotechnol. 2011;21:1330-1335
24. Sambrook J, Russell DW. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. 2001
25. Nielsen H, Engelbrecht J, Brunak S. et al. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1-6
26. Shi H, Wen SW. Display of green fluorescent protein on Escherichia coli cell surface. Enzyme Microb Technol. 2001;28:25-34
Author contact
![]() Corresponding author: Tel.: +86-27-87286952; Fax: +86-27-87280670; E-mail: lilinhzau.edu.cn (L. Li).
Corresponding author: Tel.: +86-27-87286952; Fax: +86-27-87280670; E-mail: lilinhzau.edu.cn (L. Li).