Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Shedding of the IL-6R
Transgenic strategies to study...
Protein tools to study IL-6...
The sIL-6R/sgp130 buffer in the...
IL-6 Trans-signaling is...
Viral IL-6 as an example of...
Consequences for the therapeutic...
Acknowlegements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(9):1237-1247. doi:10.7150/ijbs.4989 This issue Cite
Review
IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6
Stefan Rose-John ![]()
Institute of Biochemistry, Christian-Albrechts-University of Kiel, Kiel, Germany.
Received 2012-8-6; Accepted 2012-8-15; Published 2012-10-24
Abstract
Interleukin-6 (IL-6) is a cytokine with many activities. It has functions in the regulation of the immune system and the nervous system. Furthermore, IL-6 is involved in liver regeneration and in the metabolic control of the body. On target cells, IL-6 binds to an 80 kDa IL-6 receptor (IL-6R). The complex of IL-6 and IL-6R associates with a second protein, gp130, which thereupon dimerizes and initiates intracellular signaling. Whereas gp130 is expressed on all cells, IL-6R is only present on few cells in the body including hepatocytes and some leukocytes. Cells, which do not express IL-6R cannot respond to the cytokine, since gp130 alone has no measurable affinity for IL-6. Interestingly, a soluble form of IL-6R (sIL-6R) comprising the extracellular portion of the receptor can bind IL-6 with a similar affinity as the membrane bound IL-6R. The complex of IL-6 and sIL-6R can bind to gp130 on cells, which do not express the IL-6R, and which are unresponsive to IL-6. This process has been called trans-signaling. Here I will review published evidence that IL-6 trans-signaling is pro-inflammatory whereas classic IL-6 signaling via the membrane bound IL-6R is needed for regenerative or anti-inflammatory activities of the cytokine. Furthermore, the detailed knowledge of IL-6 biology has important consequences for therapeutic strategies aimed at the blockade of the cytokine IL-6.
Keywords: IL-6, IL-6 receptor, shedding, soluble receptor, inflammation, inflammation associated cancer.
Introduction
Interleukin-6 (IL-6) is a four-helical protein of 184 amino acids. The cDNA of IL-6 was cloned in 1986 [1] and it was recognized that IL-6 belonged to a large family of cytokines, which all shared the four-helical protein topology [2]. The IL-6 receptor (IL-6R), a protein with an Ig-fold, binds IL-6 with nanomolar affinity [3]. Binding of IL-6 to the IL-6R does, however, not lead to signaling. The complex of IL-6 and IL-6R associates with the protein gp130 thereby inducing its dimerization and initiating intracellular signaling via the JAK/STAT pathway [4]. In addition to the activation of the canonical JAK/STAT pathway, the phosphatase SHP-2 is recruited to tyrosine phosphorylated gp130, becomes phosphorylated by JAK1 and thereupon mediates the activation of the Ras-Raf-MAPK signaling pathway [5, 6]. The contribution of the JAK/STAT pathway and the SHP-2 driven Ras-Raf-MAPK signaling pathway to pathophysiology has been addressed by using mice in which the portions of gp130, which trigger these respective pathways have been genetically modified [7]. Using these mice in models of rheumatoid arthritis led to the conclusion that the IL-6-gp130-STAT3 axis was fundamentally required for the orchestration of the inflammatory process in the animals [8]. From these results it can be expected that also in rheumatoid arthritis patients IL-6 mediated STAT3 activation will play a key role in the disease [9].
Interestingly, gp130 is also a receptor subunit of the receptor complexes for IL-11, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), oncostatin M (OSM), cardiotrophin-1 (CT-1), cardiotrophin like cytokine (CLC) and IL-27. These cytokines form the gp130 cytokine family [10].
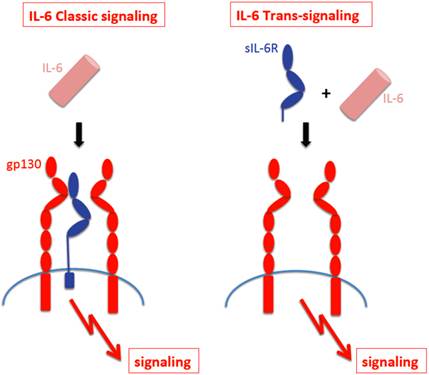
The receptor subunit gp130 has no measurable affinity for IL-6 nor the IL-6R [11]. As a consequence, IL-6 can only bind to and stimulate cells, which express the IL-6R. Cells, which only express gp130 are completely unresponsive to IL-6 [12]. Interestingly, a soluble form of the IL-6R (sIL-6R) has been found in body fluids such as urine and blood [13] and it was shown that in humans, the sIL-6R could be generated by two different mechanisms, limited proteolysis of the membrane bound receptor by the metalloprotease ADAM17 [14] and by translation of a differentially spliced mRNA [15]. It is thought that regulated generation of the sIL-6R occurs through shedding rather than through differential splicing [16]. Interestingly, in the mouse, only shedding but no differential splicing of the IL-6R mRNA has been detected [17]. The sIL-6R binds IL-6 with comparable affinity as the membrane bound IL-6R [18]. Gp130 expressing cells - even in the absence of IL-6R - can be stimulated by the complex of IL-6 and sIL-6R [19] and this process has been called trans-signaling [18]. In Fig. 1 it is depicted that cells which only express gp130 can only be stimulated by the complex of IL-6 and sIL-6R (right), whereas cells, which express IL-6R can respond to IL-6 alone (left).
IL-6 classic- signaling and IL-6 trans-signaling. IL-6 Classic-signaling requires membrane bound IL-6R and is restricted to hepatocytes, some epithelial cells and some leukocytes. IL-6 trans-signaling requires sIL-6R and is possible on all cells of the body since all cells express the gp130 protein.
Shedding of the IL-6R
Using pulse-chase experiments it was demonstrated that it took 24 h to completely shed the IL-6R from cells. Stimulation of the cells with the phorbol ester PMA induced complete loss of the IL-6R from the cell surface within 1 h [14, 20]. The cleavage site was determined and it was shown that small deletion around the cleavage site completely abrogated PMA-induced shedding [21]. Subsequent studies established that treatment of cells with C-reactive protein (CRP) [22], bacterial toxins [23] and cholesterol depleting agents [24] as well as treatment of cells with bacterial metalloproteinases [25] induced shedding of the IL-6R. Since most of these treatments interfere with membrane integrity from the inside or outside it was hypothesized that also induction of apoptosis, which leads to reorientation of phosphatidylinositol within the membrane, might induce shedding of this cytokine receptor. Indeed, it turned out that early induction of apoptosis led to strong ADAM17 mediated shedding of the IL-6R [26]. It was established that in inflammatory processes, shedding of the IL-6R from neutrophils, which are the first cells to arrive at a site of damage, leads to the stimulation of endothelial cells, which do not express IL-6R on the cell surface and are therefore unresponsive to the cytokine. Stimulation of endothelial cells by the IL-6/sIL-6R complex leads to secretion of the mononuclear cell attracting cytokine MCP-1. Thereby, the shedding of the IL-6R acts as a gauge for the initial damage reflected by the numbers of neutrophils attracted [27].
On CD4-T-cells, IL-6 has been shown to strongly influence the balance between regulatory T-cells and TH17-cells [28]. The induction of TH17-cells by IL-6 was strongly increased by the presence of sIL-6R indicating a role for trans-signaling in this process [29]. Interestingly, sIL-6R is produced by naïve and memory CD4-T-cells when the T-cell receptor is activated [30]. It was shown that this local regulation of sIL-6R activity and IL-6 trans-signaling contributes to the status of the T-cell response [31].
A natural soluble form of gp130 (sgp130) is found in the circulation [32] at levels around 400 ng/ml. Furthermore, a shorter form of sgp130 was detected in normal human urine, plasma and the synovial fluid of patients with rheumatoid arthritis [33]. This form of sgp130 called gp130-RAPS only comprises the Ig-like N-terminal domain as well as the cytokine binding domain of gp130 and therefore has a molecular weight of only around 50 kDa [33]. It was shown that like the soluble, entire extracellular form of sgp130, the shorter form gp130-RAPS could bind the IL-6/sIL-6R complex [34]. The soluble forms of sgp130 were found not to be generated by proteolytic cleavage [35] but rather by translation from alternatively spliced mRNA [36].
Transgenic strategies to study IL-6 trans-signaling
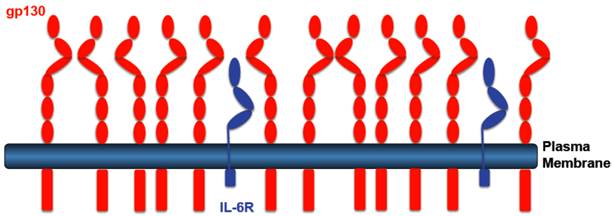
Transgenic mice were generated, which expressed a cDNA coding for the human sIL-6R from a liver promoter [37]. Since murine IL-6 does not bind to the human IL-6R [38], these mice showed no phenotype. However, when mice were injected with human IL-6 it turned out that the transgenically expressed sIL-6R protein led to sensitization towards IL-6 and to stabilization of IL-6 in the blood of the animals [37]. The sIL-6R transgenic mice were crossed with mice, which were transgenic for human IL-6 [39]. Double transgenic IL-6/sIL-6R mice were compared to IL-6 single transgenic mice. It turned out that only double transgenic IL-6/sIL-6R mice showed massive extramedullary hematopoiesis [40] together with hepatocellular proliferation [41, 42]. Since both phenotypes were not seen in single IL-6 transgenic mice, these results were the first indication that in vivo the response to the IL-6/sIL-6R complex can be substantially different from the response to IL-6 [43]. An explanation for the different response was that (i) hepatocytes express far more gp130 than IL-6R on their cell surface. Therefore, stimulation by IL-6 the presence of sIL-6R leads to far more gp130 dimers signaling into the cell (Fig. 2). (ii) In addition, we could show that while stimulation of cells with IL-6 leads to rapid internalization of IL-6 [44] and thereby to termination of signaling, addition of the IL-6/sIL-6R complex to cells resulted in very little internalization and prolonged signaling [45]. The combination of increased signal strength due to more gp130 signaling molecules with prolonged signaling due to reduced internalization resulted not only in a quantitative but also in a qualitative different cellular response.
Receptor distribution IL-6R expressing cells. Most IL-6R expressing cells express far more gp130 than IL-6R. IL-6 stimulation of such cells leads to the activation of only few gp130 molecules. IL-6 in the presence of the sIL-6R, however, will lead to stimulation of all gp130 molecules leading to a higher signal amplitude.
Protein tools to study IL-6 trans-signaling: Hyper-IL-6, sgp130Fc, L-gp130
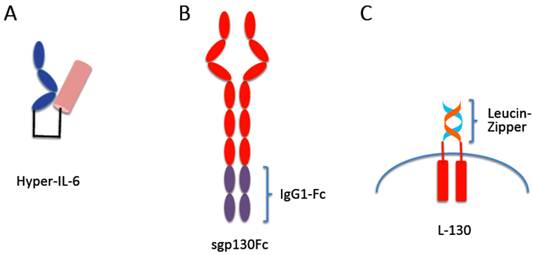
When together with Joachim Grötzinger we built a molecular model of the IL-6/sIL-6R protein complex [12] we realized that the C-terminus of the sIL-6R and the N-terminus of IL-6 were about 40Å apart. We therefore introduced at the cDNA level a flexible peptide linker spanning these 40Å between the C-terminus of the sIL-6R and the N-terminus of IL-6 (Fig. 3A) [12]. On gp130 expressing cells, the resulting protein called Hyper-IL-6 was shown to be 100-1,000 times more active than the separate proteins IL-6 and sIL-6R. This could be explained by the fact that the affinity of IL-6 to IL-6R is in the range of 1 nM whereas the complex of IL-6/IL-6R binds with 100 times higher affinity to gp130 [46]. Therefore, the preformed complex in Hyper-IL-6 showed higher efficacy than the two separate proteins.
Designer proteins for the study of IL-6 signaling. (A) Hyper-IL-6 is a fusion protein between the sIL-6R (blue) and IL-6 (brown). In a molecular model, the C-terminus of sIL-6R was 40 Å apart from the N-terminus of IL-6. Therefore, we inserted a flexible peptide linker between sIL-6R and IL-6 (black) to connect both molecules. (B) The sgp130Fc protein consists of the extracellular portion of gp130 linked to the Fc domain of a human IgG1 antibody. The sgp130Fc protein blocks IL-6 trans-signaling without affecting IL-6 classic signaling. (C) In the L-gp130 protein, the entire extracellular portion of gp130 is replaced by the leucine zipper of the Jun protein, leading to constitutive dimerization and activation of the gp130 protein. This protein can be used to study the effects of permanent gp130 activation in transfected cells or in tissue specific transgenic animals.
Subsequently, the comparison between application of IL-6 and Hyper-IL-6 led to the characterization of many target cells of IL-6 trans-signaling. Cells, which would only respond to IL-6 in the presence of the sIL-6R include hematopoietic stem cells [47], many if not all neural cells [48, 49], smooth muscle cells [50] and embryonic stem cells [51, 52]. These results indicated that the IL-6 trans-signaling pathway was used in many cell types and tissues [53].
The receptor subunit gp130 does not show any affinity for IL-6 or IL-6R alone. Only the complex of IL-6 and IL-6R binds to cellular expressed gp130 [19] (Fig. 3B). We expressed the extracellular portion of gp130 as an IgG1-Fc fusion protein (sgp130Fc) and could show that also the soluble gp130 protein only interacts with the sIL-6R in the presence of IL-6 [11]. As a consequence, the soluble gp130 and the sgp130Fc protein inhibited IL-6 trans-signaling but had no inhibitory effect on classic IL-6 signaling, i.e. on the stimulation of cells by IL-6 via the membrane bound IL-6R [11]. Interestingly, the soluble gp130 protein did not show any species specificity: whereas murine IL-6 does not bind to the human IL-6R, the human and murine IL-6/sIL-6R complex bound to both, human and murine soluble gp130 [11]. Therefore, the sgp130Fc protein could be used as a molecular tool to discriminate between IL-6 classic signaling and IL-6 trans-signaling on human cells as well as in murine models of human diseases [54, 55]. Interestingly, it was shown that the sgp130Fc protein did not inhibit other gp130 cytokines such as CNTF, LIF, OSM and IL-27 [56]. The reason for this specificity is that these gp130 cytokines on the cell surface form a complex with a heterodimer of gp130 with LIF receptor, OSM receptor or WSX-1, respectively. Therefore the affinity of these cytokines to the dimeric sgp130 moiety in the sgp130Fc protein is much lower than to the membrane bound heterodimers of gp130 with LIF receptor, OSM receptor or WSX-1, respectively. Therefore, sgp130Fc fails to compete efficiently with these cell bound receptor complexes for binding of CNTF, LIF, OSM and IL-27 and consequently has little or no inhibitory activity [56]. Theoretically, an IL-11/sIL-11R complex could be inhibited by sgp130Fc. But to date, no naturally occurring sIL-11R has been detected neither in mouse nor in man.
So far it is unclear how binding of the IL-6/IL-6R or IL-6/sIL-6R complexes to gp130 leads to the transmission of the signal into the cell. It is, however, clear that dimerization of gp130 cannot be sufficient for signal initiation since gp130 even in the un-liganded state already forms a dimer [57, 58]. To address this question a gp130 cDNA was constructed, in which the entire region coding for the extracellular portion was replaced by DNA sequences coding for the leucine-zipper from the Jun protein [59, 60]. The presence of the jun-leucine-zipper led to IL-6 independent dimerization of gp130, activation and phosphorylation of JAK kinases and STAT3 and subsequently to transcription of STAT3 target genes (Fig. 3C). When transfected into IL-6 dependent cells, the presence of the L-gp130 protein led to longterm and stable cytokine-independent growth of the cells [59, 60]. Interestingly, when transfected into murine embryonic stem cells, L-gp130, even in the absence of leukemia inhibitory factor led to an upregulation of the transcription factor Oct4 accompanied by a stable and complete blockade of cellular differentiation [59]. It was concluded from these data that forced juxtaposition of the transmembrane domains of gp130 and consequently its cytoplasmic domains mediated by the jun-leucine-zipper was sufficient for gp130 signaling in the absence of extracellular stimulation. The dimerization of gp130 found in the absence of receptor stimulation [57, 58] apparently does not position the transmembrane domains and cytoplasmic domains of gp130 in a way that is sufficient to induce gp130 signaling. Interestingly, in a different study it had been observed that also the membrane bound IL-6R on cells was dimeric although the function of this dimerization so far remains unclear [61].
The sIL-6R/sgp130 buffer in the blood
Since all cells in the body express gp130 [9, 10], theoretically, all cells can be activated by the IL-6/sIL-6R complex. Since the cellular responses to the IL-6/sIL-6R complex can be dramatic, ranging from induction of hepatocyte proliferation to massive increase in hematopoiesis [40, 43] there must be a control mechanism to prevent IL-6 trans-signaling under steady-state conditions. Apparently, the concentrations of IL-6, sIL-6R and sgp130 have to be taken into account. In healthy subjects, IL-6 plasma levels are barely detectable and range between 2-6 pg/ml [62]. They are massively increased during inflammation and can reach levels of several µg/ml under septic conditions [63]. In patients with rheumatoid arthritis i.e. during chronic inflammatory conditions, IL-6 levels of up to 150 ng/ml have been described [64]. In contrast, levels of sIL-6R are in the range of 75 ng/ml [65] and sgp130 levels have been found at around 250-400 ng/ml [32, 66]. Thus, under steady state conditions, levels of sIL-6R and sgp130 are roughly 1000 times higher than IL-6 levels. Furthermore, as mentioned, IL-6 levels can increase up to 1 million fold in severe conditions [63], whereas sIL-6R and sgp130 levels have been reported to only increase not more than 2-5 fold during inflammation [67, 68]. These concentrations imply that IL-6, once secreted, will bind to sIL-6R in the plasma and this complex will associate with sgp130 and thereby be neutralized. Only when IL-6 levels exceed the levels of sIL-6R and sgp130, IL-6 can act systemically - as seen under septic conditions [63]. Under physiologic conditions IL-6 is thought to act in a paracrine fashion [69]. As recently reported, the sgp130Fc protein under conditions, when sIL-6R levels largely exceed levels of IL-6, can also block IL-6 classic signaling because then free IL-6 will bind to the excess of sIL-6R and this complex will bind to and will be neutralized by sgp130Fc [70]. It should be noted, however, that these calculations do not take into account the local cytokine and soluble cytokine receptor levels at the site of inflammation, which are largely unknown since they are mostly not experimentally accessible [9] .
In two recent publications, it was shown that a single nucleotide polymorphism in the IL-6R gene changing glutamine 258 into alanine resulted in higher serum concentrations of sIL-6R was connected to a lower risk of coronary heart disease [71, 72]. Interestingly, this amino acid is positioned exactly at the cleavage site of the human IL-6R [21]. This effect of this single nucleotide polymorphism was explained with the loss of membrane bound IL-6R from hepatocytes, monocytes, and macrophages and a concomitant loss of IL-6 signaling in these cells [73]. An alternative explanation would be that an increase in sIL-6R levels would increase the capacity of the sIL-6R/sgp130 buffer in the blood and therefore lead to reduced overall IL-6 activity [74]. Recently, it was shown that by specifically blocking IL-6 trans-signaling with the sgp130Fc protein, we could significantly reduce the extent of atherosclerosis in hypercholesterolemic LDL receptor negative mice [68].
IL-6 Trans-signaling is pro-inflammatory
With the sgp130Fc protein as a molecular tool it was possible to discriminate between IL-6 classic signaling and IL-6 trans-signaling not only in vitro but also in vivo. In gp130 dependent mouse models of human diseases, global IL-6 signaling could be blocked by the use of anti-IL-6 or anti-IL-6R neutralizing antibodies. In a parallel experiment, IL-6 trans-signaling could be blocked with the help of the sgp130Fc protein. These experiments allowed to decide whether the gp130 driven disease model was based on IL-6 classic signaling or IL-6 trans-signaling [54, 55]. The sgp130Fc could be applied by injection of the recombinant protein or was synthesized in vivo in transgenic mice expressing the sgp130Fc from liver cells. Using this approach, we could show that in models of inflammation, of autoimmune diseases and of inflammation associated cancer, blockade of IL-6 trans-signaling by sgp130Fc was sufficient to block the inflammatory progress (Table 1). In the case of mycobacterium tuberculosis infection, treatment of the animals with sgp130Fc protein did not lead to an increase of bacterial burden in lung, liver and spleen, whereas treatment with neutralizing TNFα-antibodies resulted in a 10-100 fold increase in mycobacterial colony forming units in the respective organs [75].
Efficacy of sgp130Fc in animal models of human diseases.
| Disease | Mouse model | Reference |
|---|---|---|
| Intestinal Inflammation | DSS-colitis, SAMP/YIT mouse model | [98, 99] |
| Rheumatoid Arthritis | Antigen induced arthritis, collagen induced arthritis | [8, 34, 64] |
| Acute Inflammation | Air pouch model | [26, 100] |
| Sepsis | Cecal ligation and puncture sepsis, LPS-mediated septic shock | [79, 101] |
| Arterosclerosis | Hypercholesterolemic LDL-receptor negative mice | [68] |
| Asthma | Late-phase murine asthma model after OVA sensitization | [67, 102] |
| Colon Cancer | AOM/DSS model, DSS only model | [78, 103] |
| Ovarian Cancer | Xenograft model of human ovarian cancer cells in NOD/SCID mice | [104] |
| Pancreatic Cancer | Pancreatic cancer caused by expression of oncogenic G12D K-ras | [105] |
In a recent study comparing the susceptibility of IL-6-/- mice and wt mice in an inflammatory colon cancer model (AOM/DSS), it was shown that IL-6-/- mice exhibited less tumors but more inflammation than wt mice [76]. It turned out that IL-6-/- mice were impaired in regenerating the irritated intestinal epithelium. A similar protective effect of IL-6 had already been noted in mice infected with Citrobacter rodentium [77]. From these experiments it was concluded that in the intestine upon wounding, the regenerative activities of IL-6 are needed for wound healing [76] and that this activity was most likely mediated via the membrane bound IL-6R [69, 78]. In a recent study, we used a standardized cecal ligation and puncture (CLP) sepsis model, leading to the death of about 60% of the animals [79]. In this model, we could demonstrate that global blockade with a neutralizing IL-6 antibody did not lead to a significant increase in survival of the animals. In contrast, when only IL-6 trans-signaling was blocked with 0.5 mg/kg sgp130Fc, 100% of the animals survived. Interestingly, CLP led an upregulation of the acute phase protein serum amyloid A (SAA). This SAA induction was inhibited by global IL-6 blockade but was unaffected by treatment of the mice with sgp130Fc [79]. Likewise, apoptosis of intestinal epithelial cells was detected in mice after CLP, which was only slightly reduced after global IL-6 blockade but was completely blocked after treatment with sgp130Fc [79]. These results clearly showed a benefit of the more specific blockade of IL-6 trans-signaling as compared to global IL-6 inhibition by neutralizing antibodies.
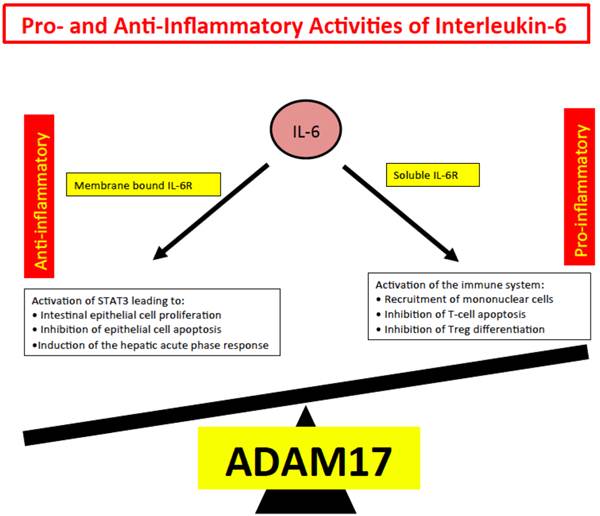
We inferred from all these experiments, that IL-6 trans-signaling represents the pro-inflammatory part of IL-6 biologic activities whereas IL-6 classic signaling comprises the anti-inflammatory or regenerative activities of IL-6 such as regeneration of intestinal epithelial cells [76], inhibition of epithelial apoptosis and the activation of the hepatic acute phase response [79]. Since shedding of the IL-6R is mainly governed by the protease ADAM17 [14, 80], we speculate that ADAM17 has a decisive role in inflammation and cancer [17] (Fig. 4).
Pro- and anti-inflammatory activities of IL-6. Anti-inflammatory activities of IL-6 include STAT3 dependent regeneration of epithelial cells and the induction of the hepatic acute phase response. These activities are dependent on the membrane bound IL-6R. Pro-inflammatory activities of IL-6 include recruitment of inflammatory cells, inhibition of apoptosis of inflammatory cells and inhibition of regulatory T-cell differentiation.
Viral IL-6 as an example of pathophysiologic trans-signaling
The genome of Human Herpesvirus 8 (HHV8) also called Kaposi's sarcoma-associated herpesvirus codes for a viral form of IL-6, designated vIL-6 [81]. Although this viral homolog shares only 25% sequence identity with human IL-6 it could be demonstrated that it stimulates cells in a gp130 dependent manner [82]. Interestingly, it could be shown that vIL-6 directly bound to gp130 without the need of soluble of membrane bound IL-6R and that association with gp130 was sufficient to induce STAT3-dependent proliferation of cells [83, 84]. This was confirmed by the structure of vIL-6 bound to the extracellular portion of gp130 [85] and by the definition of the amino acid sequences responsible for the ability of vIL-6 to directly bind to and activate gp130 [86]. In addition, it could be shown that vIL-6 not only stimulated cells from the outside but could also act from within the cell [87, 88], which might be important for the pathophysiologic role of vIL-6 and needs to be considered when strategies of vIL-6 neutralization are assessed [87, 88].
Since vIL-6 is the only known cytokine, which can stimulate gp130 without the help of an additional receptor protein, we speculated that vIL-6 was a natural model for IL-6 trans-signaling [89-91]. Indeed, it could be shown that vIL-6 affects neutrophil infiltration during acute inflammation in a similar way as the IL-6/sIL-6R complex [92]. Furthermore, in mice transgenic for vIL-6 we observed development of a phenotype, which closely resembled multicentric Castleman disease [93]. Interestingly, when we crossed the vIL-6 transgenic mice with IL-6-/- mice, the phenotype disappeared, indicating that vIL-6 collaborated with murine IL-6 in the development of the pathophysiology [93]. This might be very relevant for the situation in patients because HHV8 has been associated with multicentric Castleman disease and 100% of HIV positive patients and 50% of HIV negative patients harbor the virus [94]. Since IL-6R neutralizing antibodies are already in the clinic for the treatment of autoimmune diseases, neutralization of human IL-6 should be considered for the treatment of multicentric Castleman disease. Indeed, it was already noted in a clinical trial with IL-6R neutralizing antibodies that patients with multicentric Castleman disease, who were HHV8 positive, improved upon neutralization of human IL-6 activity [95].
Consequences for the therapeutic targeting of IL-6
Activities of IL-6 can be regenerative and protective during infection and inflammation [76, 77, 96]. On the other hand, IL-6 is needed for the activation of the immune system (Fig. 4). Indeed it has been shown that neutralization of IL-6 by an anti-IL-6R antibody has beneficial effects in several autoimmune diseases such as Rheumatoid Arthritis, Castleman's disease and juvenile idiopathic arthritis and has been approved for treatment of these diseases [97]. In clinical trials, the benefit from blocking IL-6R activity for the patients was at least as high as the benefit from blocking TNFα activity. Autoimmune diseases are not cured by anti-cytokine therapy but only suppressed. Therefore, patients will likely have to be treated for the rest of their lives. Therefore, side effects will be an important issue of consideration for such therapies in the future.
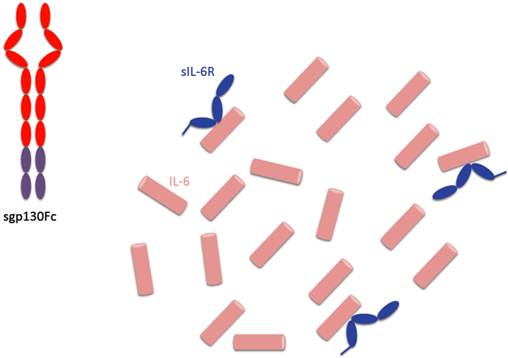
The principle of cytokine blockade differs between neutralizing antibodies and soluble receptor binding proteins. The anti-human IL-6R antibody tocilizumab blocks both, IL-6 classic signaling and IL-6 trans-signaling (Fig. 1) and therefore blocks both, pro- and anti-inflammatory activities of IL-6 (Fig. 4). The IL-6 trans-signaling inhibitor sgp130Fc, however, binds only IL-6 molecules, which are bound by the sIL-6R (Fig. 5). Since during inflammatory states IL-6 concentrations at the site of inflammation are likely to largely exceed concentrations of sIL-6R, many IL-6 molecules are not blocked and can induce regeneration of intestine epithelial cells or induce the hepatic acute phase reaction and thereby support the innate immune response of the body in defense of bacterial infection. Therefore we are looking forward to the first clinical trials with sgp130Fc, which are planned in early 2013.
Consequences of specific IL-6 trans-signaling blockade by sgp130Fc. The sgp130Fc protein only binds to IL-6 molecules, which are complexed with sIL-6R. Therefore, in the presence of an excess of IL-6, not all IL-6 molecules are neutralized and can induce regeneration of epithelial cells or trigger the hepatic acute phase response.
Acknowlegements
The work of SRJ was funded by the Deutsche Forschungsgemeinschaft, Bonn (SFB654, Project C5; SFB841, project C1; SFB877, project A1) and by the Cluster of Excellence 'Inflammation at Interfaces'.
Competing Interests
Dr. Rose-John is an inventor on patents describing the function of sgp130Fc. He is also a shareholder of the CONARIS Research Institute (Kiel, Germany), which is commercially developing sgp130Fc proteins as therapeutics for inflammatory diseases.
References
1. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T. et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73-6
2. Bazan JF. Haemopoietic receptors and helical cytokines. Immunol Today. 1990;11:350-4
3. Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B. et al. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825-8
4. Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149-57
5. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1-20
6. Scheller J, Grötzinger J, Rose-John S. Updating IL-6 classic- and trans-signaling. Signal Transduction. 2006;6:240-59
7. Ernst M, Jenkins BJ. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23-32
8. Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW. et al. Therapeutic targeting of IL-6 trans-signaling counteracts STAT3 control of the inflammatory infiltrate in experimental arthritis. J Immunol. 2009;182:613-22
9. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375-83 doi:10.1172/JCI57158
10. Kishimoto T. Interleukin-6: from basic science to medicine--40 years in immunology. Annu Rev Immunol. 2005;23:1-21
11. Jostock T, Müllberg J, Özbek S, Atreya R, Blinn G, Voltz N. et al. Soluble gp130 is the natural inhibitor of soluble IL-6R transsignaling responses. Eur J Biochem. 2001;268:160-7
12. Fischer M, Goldschmitt J, Peschel C, Kallen KJ, Brakenhoff JPJ, Wollmer A. et al. A designer cytokine with high activity on human hematopoietic progenitor cells. Nat Biotech. 1997;15:142-5
13. Novick D, Engelmann H, Wallach D, Rubinstein M. Soluble cytokine receptors are present in normal human urine. J Exp Med. 1989;170:1409-14
14. Müllberg J, Schooltink H, Stoyan T, Gunther M, Graeve L, Buse G. et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23:473-80
15. Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96-100
16. Dimitrov S, Lange T, Benedict C, Scheller J, Rose-John S, Jones S. et al. Sleep Enhances IL-6 Trans-Signaling in Humans. Faseb J. 2006;20:2174-2176
17. Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch of inflammatory and regenerative responses? Trends Immunol. 2011;32:380-387
18. Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300:281-90
19. Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T. et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573-81
20. Müllberg J, Schooltink H, Stoyan T, Heinrich PC, Rose-John S. Protein kinase C activity is rate limiting for shedding of the interleukin-6 receptor. Biochem Biophys Res Commun. 1992;189:794-800
21. Müllberg J, Oberthur W, Lottspeich F, Mehl E, Dittrich E, Graeve L. et al. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J Immunol. 1994;152:4958-68
22. Jones SA, Novick D, Horiuchi S, Yamamoto N, Szalai AJ, Fuller GM. C-reactive protein: a physiological activator of interleukin 6 receptor shedding. J Exp Med. 1999;189:599-604
23. Walev I, Vollmer P, Palmer M, Bhakdi S, Rose-John S. Pore-forming toxins trigger shedding of receptors for interleukin 6 and lipopolysaccharide. Proc Natl Acad Sci USA. 1996;93:7882-7
24. Matthews V, Schuster B, Schütze S, Kallen K-J, Rose-John S. Cholesterol depletion of the plasma membrane triggers shedding of the human interleukin-6 receptor by TACE and independently of PKC. J Biol Chem. 2003;278:38829-39
25. Vollmer P, Walev I, Rose-John S, Bhakdi S. Novel pathogenic mechanism of microbial metalloproteinases: liberation of membrane-anchored molecules in biologically active form exemplified by studies with the human interleukin-6 receptor. Infect Immun. 1996;64:3646-51
26. Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA. et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the pro-inflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748-55
27. DeLeo FR. Attractive shedding. Blood. 2007;110:1711-2
28. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235-8
29. Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J. et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J Immunol. 2007;179:2041-5
30. Briso EM, Dienz O, Rincon M. Cutting Edge: Soluble IL-6R Is Produced by IL-6R Ectodomain Shedding in Activated CD4 T Cells. J Immunol. 2008;180:7102-6
31. Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R. et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol. 2010;184:2130-9
32. Narazaki M, Yasukawa K, Saito T, Ohsugi Y, Fukui H, Koishihara Y. et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood. 1993;82:1120-6
33. Zhang JG, Zhang Y, Owczarek CM, Ward LD, Moritz RL, Simpson RJ. et al. Identification and characterization of two distinct truncated forms of gp130 and a soluble form of leukemia inhibitory factor receptor alpha-chain in normal human urine and plasma. J Biol Chem. 1998;273:10798-805
34. Richards PJ, Nowell MA, Horiuchi S, McLoughlin RM, Fielding CA, Grau S. et al. Functional characterization of a soluble gp130 isoform and its therapeutic capacity in an experimental model of inflammatory arthritis. Arthr Rheumat. 2006;54:1662-72
35. Müllberg J, Dittrich E, Graeve L, Gerhartz C, Yasukawa K, Taga T. et al. Differential shedding of the two subunits of the interleukin-6 receptor. FEBS Lett. 1993;332:174-8
36. Diamant M, Hansen MB, Rieneck K, Svenson M, Yasukawa K, Bendtzen K. Differential interleukin-6 (IL-6) responses of three established myeloma cell lines in the presence of soluble human IL-6 receptors. Leuk Res. 1996;20:291-301
37. Peters M, Jacobs S, Ehlers M, Vollmer P, Müllberg J, Wolf E. et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med. 1996;183:1399-406
38. van Dam M, Müllberg J, Schooltink H, Stoyan T, Brakenhoff JP, Graeve L. et al. Structure-function analysis of interleukin-6 utilizing human/murine chimeric molecules. Involvement of two separate domains in receptor binding. J Biol Chem. 1993;268:15285-90
39. Fattori E, Della Rocca C, Costa P, Giorgio M, Dente B, Pozzi L. et al. Development of progressive kidney damage and myeloma kidney in interleukin-6 transgenic mice. Blood. 1994;83:2570-9
40. Peters M, Schirmacher P, Goldschmitt J, Odenthal M, Peschel C, Dienes HP. et al. Extramedullary expansion of hematopoietic progenitor cells in IL-6/sIL-6R double transgenic mice. J Exp Med. 1997;185:755-66
41. Schirmacher P, Peters M, Ciliberto G, Fattori E, Lotz J, Meyer zum Büschenfelde KH. et al. Hepatocellular Hyperplasia, Plasmacytoma Formation, and Extracellular Hematopoiesis in Interleukin (IL)-6/Soluble IL-6 Receptor Double-Transgenic Mice. Am J Pathol. 1998;153:639-48
42. Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M. et al. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. Embo J. 1998;17:5588-97
43. Peters M, Müller A, Rose-John S. Interleukin-6 and soluble Interleukin-6 Receptor: Direct Stimulation of gp130 and Hematopoiesis. Blood. 1998;92:3495-504
44. Dittrich E, Rose-John S, Gerhartz C, Mullberg J, Stoyan T, Yasukawa K. et al. Identification of a region within the cytoplasmic domain of the interleukin-6 (IL-6) signal transducer gp130 important for ligand-induced endocytosis of the IL-6 receptor. J Biol Chem. 1994;269:19014-20
45. Peters M, Blinn G, Solem F, Fischer M, Meyer zum Büschenfelde K-H, Rose-John S. In Vivo and in vitro Activity of the gp130 Stimulating Designer Cytokine Hyper-IL-6. J Immunol. 1998;161:3575-81
46. Rose-John S, Schooltink H, Lenz D, Hipp E, Dufhues G, Schmitz H. et al. Studies on the structure and regulation of the human hepatic interleukin-6 receptor. Eur J Biochem. 1990;190:79-83
47. Audet J, Miller CL, Rose-John S, Piret JM, Eaves CJ. Distinct role of gp130 activation in promoting self-renewal divisions by mitogenically stimulated murine hematopoietic cells. Proc Natl Acad Sci USA. 2001;98:1757-62
48. März P, Cheng J-C, Gadient RA, Patterson P, Stoyan T, Otten U. et al. Sympathetic Neurons can produce and respond to Interleukin-6. Proc Natl Acad Sci USA. 1998;95:3251-6
49. März P, Heese K, Dimitriades-Schmutz B, Rose-John S, Otten U. Role of Interleukin-6 and soluble IL-6 Receptor in Region Specific Induction of Astrocytic Differentiation and Neurotrophin Expression. Glia. 1999;26:191-200
50. Klouche M, Bhakdi S, Hemmes M, Rose-John S. Novel Path of activation of primary human smooth muscle cells: upregulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J Immunol. 1999;163:4583-9
51. Viswanathan S, Benatar T, Rose-John S, Lauffenburger DA, Zandstra PW. Ligand/receptor signaling threshold (LIST) model accounts for gp130-mediated embryonic stem cell self-renewal responses to LIF and HIL-6. Stem cells (Dayton, Ohio). 2002;20:119-38
52. Humphrey RK, Beattie GM, Lopez AD, Bucay N, King CC, Firpo M. et al. Maintenance of pluripotency in human embryonic stem cells is Stat3 independent. Stem cells (Dayton, Ohio). 2004;22:522-30
53. Rose-John S, Scheller J, Elson G, Jones S. Interleukin-6 Biology is Coordinated by Membrane-Bound and Soluble Receptors: Role in Inflammation and Cancer. J Leuk Biol. 2006;80:227-36
54. Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11:613-24
55. Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin Ther Targets. 2012;16:225-36
56. Scheller J, Schuster B, Holscher C, Yoshimoto T, Rose-John S. No inhibition of IL-27 signaling by soluble gp130. Biochem Biophys Res Commun. 2005;326:724-8
57. Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC. et al. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. Journal of cell science. 2005;118:5129-40
58. Tenhumberg S, Schuster B, Zhu L, Kovaleva M, Scheller J, Kallen KJ. et al. gp130 Dimerization in the Absence of Ligand: preformed Cytokine Receptor Complexes. Biochem Biophys Res Commun. 2006;346:649-57
59. Stuhlmann-Laeisz C, Lang S, Chalaris A, Paliga K, Sudarman E, Eichler J. et al. Forced dimerization of gp130 leads to constitutive STAT3 activation, cytokine independent growth and blockade of differentiation of embryonic stem cells. Mol Biol Cell. 2006;17:2986-95
60. Suthaus J, Tillmann A, Lorenzen I, Bulanova E, Rose-John S, Scheller J. Forced homo- and heterodimerization of all gp130-type receptor complexes leads to constitutive ligand-independent signaling and cytokine-independent growth. Mol Biol Cell. 2010;21:2797-807
61. Schuster B, Meinert W, Rose-John S, Kallen K-J. The human interleukin-6 (IL-6) receptor exists as a preformed dimer in the plasma membrane. FEBS Lett. 2003;538:113-6
62. Scambia G, Testa U, Panici PB, Martucci R, Foti E, Petrini M. et al. Interleukin-6 serum levels in patients with gynecological tumors. Int J Cancer. 1994;57:318-23
63. Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. J Exp Med. 1989;169:333-8
64. Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N. et al. Soluble IL-6 Receptor Governs IL-6 Activity in Experimental Arthritis: Blockade of Arthritis Severity by Soluble Glycoprotein 130. J Immunol. 2003;171:3202-9
65. Honda M, Yamamoto S, Cheng M, Yasukawa K, Suzuki H, Saito T. et al. Human soluble IL-6 receptor: its detection and enhanced release by HIV infection. J Immunol. 1992;148:2175-80
66. Padberg F, Feneberg W, Schmidt S, Schwarz MJ, Korschenhausen D, Greenberg BD. et al. CSF and serum levels of soluble interleukin-6 receptors (sIL-6R and sgp130), but not of interleukin-6 are altered in multiple sclerosis. J Neuroimmunol. 1999;99:218-23
67. Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ. et al. The IL-6R alpha chain controls lung CD4+CD4+CD25+T regulatory cell development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313-25
68. Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A. et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:281-90
69. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878-88
70. Garbers C, Thaiss W, Jones GW, Waetzig GH, Lorenzen I, Guilhot F. et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J Biol Chem. 2011;286:42959-70
71. Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN. et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379:1205-13
72. Hingorani AD, Casas JP. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214-24
73. Boekholdt SM, Stroes ES. The interleukin-6 pathway and atherosclerosis. Lancet. 2012;379:1176-8
74. Scheller J, Rose-John S. The interleukin 6 pathway and atherosclerosis. Lancet. 2012;380:338
75. Sodenkamp J, Waetzig GH, Scheller J, Seegert D, Grotzinger J, Rose-John S. et al. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology. 2012 in press
76. Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S. et al. IL-6 and STAT3 signaling is required for survival of intestinal epithelial cells and colitis associated cancer. Cancer Cell. 2009;16:103-13
77. Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ. et al. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J Immunol. 2008;180:6816-26
78. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491-501
79. Barkhausen T, Tschernig T, Rosenstiel P, van Griensven M, Vonberg RP, Dorsch M. et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit Care Med. 2011;39:1407-13
80. Chalaris A, Gewiese J, Paliga K, Fleig L, Schneede A, Krieger K. et al. ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by gamma-secretase. Biochim Biophys Acta. 2010;1803:234-45
81. Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, Friedman Kien AE. et al. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol. 1997;71:839-42
82. Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. A Kaposi`s Sarcoma-associated Herpesvirus-encoded Cytokine Homolog (vIL-6) Activates Signaling through the Shared gp130 Receptor Subunit. J Biol Chem. 1997;272:19625-31
83. Müllberg J, Geib T, Jostock T, Hoischen SH, Vollmer P, Voltz N. et al. IL-6-Receptor Independent Stimulation of Human gp130 by Viral IL-6. J Immunol. 2000;164:4672-7
84. Hoischen SH, Vollmer P, Marz P, Ozbek S, Gotze KS, Peschel C. et al. Human herpes virus 8 interleukin-6 homologue triggers gp130 on neuronal and hematopoietic cells. Eur J Biochem / FEBS J. 2000;267:3604-12
85. Chow D, He X, Snow AL, Rose-John S, Garcia KC. Structure of an extracellular gp130 cytokine receptor signaling complex. Science. 2001;291:2150-5
86. Adam N, Rabe B, Suthaus J, Grotzinger J, Rose-John S, Scheller J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J Virol. 2009;83:5117-26
87. Kovaleva M, Bussmeyer I, Rabe B, Grotzinger J, Sudarman E, Eichler J. et al. Abrogation of viral interleukin-6 (vIL-6)-induced signaling by intracellular retention and neutralization of vIL-6 with an anti-vIL-6 single-chain antibody selected by phage display. J Virol. 2006;80:8510-20
88. Meads MB, Medveczky PG. Kaposi's sarcoma-associated herpesvirus-encoded viral interleukin-6 is secreted and modified differently than human interleukin-6: evidence for a unique autocrine signaling mechanism. J Biol Chem. 2004;279:51793-803
89. Klouche M, Brockmeyer N, Knabbe C, Rose-John S. Human herpesvirus 8-derived viral IL-6 induces PTX3 expression in Kaposi's sarcoma cells. Aids. 2002;16:F9-18
90. Klouche M, Carruba G, Castagnetta L, Rose-John S. Virokines in the pathogenesis of cancer: focus on human herpesvirus 8. Ann N Y Acad Sci. 2004;1028:329-39
91. Suthaus J, Adam N, Grotzinger J, Scheller J, Rose-John S. Viral Interleukin-6: Structure, pathophysiology and strategies of neutralization. Eur J Cell Biol. 2011;90:495-504
92. Fielding CA, McLoughlin RM, Colmont CS, Kovaleva M, Harris DA, Rose-John S. et al. Viral IL-6 blocks neutrophil infiltration during acute inflammation. J Immunol. 2005;175:4024-9
93. Suthaus J, Stuhlmann-Laeisz C, Tompkins VS, Rosean TR, Klapper W, Tosato G. et al. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric Castleman disease in mice. Blood. 2012;119:5173-81
94. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals Hatem D, Babinet P. et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276-80
95. Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S. et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106:2627-3
96. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91-102
97. Tanaka T, Narazaki M, Kishimoto T. Therapeutic Targeting of the Interleukin-6 Receptor. Annu Rev Pharmacol Toxicol. 2012;52:199-219
98. Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S. et al. Blockade of IL-6 transsignaling abrogates established experimental colitis in mice by suppression of the antiapoptotic resistance of lamina propria T cells. Nat Med. 2000;6:583-8
99. Mitsuyama K, Matsumoto S, Rose-John S, Suzuki A, Hara T, Tomiyasu N. et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263-9
100. Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS. et al. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111:1021-8
101. Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ. et al. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. Journal of immunology. 2011;186:1199-208
102. Finotto S, Eigenbrod T, Karwot R, Boross I, Doganci A, Ito H. et al. Local blockade of IL-6R signaling induces lung CD4+ T cell apoptosis in a murine model of asthma via regulatory T cells. Int Immunol. 2007;19:685-93
103. Matsumoto S, Hara T, Mitsuyama K, Yamamoto M, Tsuruta O, Sata M. et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543-51
104. Lo CW, Chen MW, Hsiao M, Wang S, Chen CA, Hsiao SM. et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2011;71:424-34
105. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G. et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456-69
Author contact
![]() Corresponding author: Stefan Rose-John, Department of Biochemistry, Christian-Albrechts-Universität zu Kiel, Olshausenstrasse 40, D-24098 Kiel, Germany, +49-431-880 3336; Fax: +49-431-880 5007; e-mail: rosejohnuni-kiel.de.
Corresponding author: Stefan Rose-John, Department of Biochemistry, Christian-Albrechts-Universität zu Kiel, Olshausenstrasse 40, D-24098 Kiel, Germany, +49-431-880 3336; Fax: +49-431-880 5007; e-mail: rosejohnuni-kiel.de.