Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
IL-6 - an important regulator of...
Evidence for a role in human...
IL-6/STAT3 as critical mediators...
Therapeutic implications
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(9):1248-1253. doi:10.7150/ijbs.4614 This issue Cite
Review
Interleukin-6 - A Key Regulator of Colorectal Cancer Development
Maximilian J. Waldner, Sebastian Foersch, Markus F. Neurath ![]()
Department of Medicine 1, University of Erlangen-Nuremberg, Erlangen, Germany.
Received 2012-5-18; Accepted 2012-7-26; Published 2012-10-24
Abstract
Growing evidence proposes an important role for pro-inflammatory cytokines during tumor development. Several experimental and clinical studies have linked the pleiotropic cytokine interleukin-6 (IL-6) to the pathogenesis of sporadic and inflammation-associated colorectal cancer (CRC). Increased IL-6 expression has been related to advanced stage of disease and decreased survival in CRC patients. According to experimental studies, these effects are mediated through IL-6 trans-signaling promoting tumor cell proliferation and inhibiting apoptosis through gp130 activation on tumor cells with subsequent signaling through Janus kinases (JAKs) and signal transducer and activator of transcription 3 (STAT3).
During recent years, several therapeutics targeting the IL-6/STAT3 pathway have been developed and pose a promising strategy for the treatment of CRC. This review discusses the molecular mechanisms and possible therapeutic targets involved in IL-6 signaling in CRC.
Keywords: interleukin-6, IL-6, colorectal cancer
Introduction
In 1935, Rous and Beard were the first to describe the progression of virus-induced papillomas to carcinomas in rabbits [1]. During subsequent decades, it has been widely accepted that the development of human cancer follows a multi-stage process involving tumor initiation, promotion and progression [2]. These stages are paralleled by an accumulation of several mutations in genes regulating critical cellular pathways, which provide a growth advantage for individual tumor cells. In this regard, only a few genetic modifications enable the clonal expansion of normal cells during tumor initiation. Additional mutations further support tumor growth during promotion, and tumor cells finally develop a malignant phenotype including invasive growth and metastasis during progression [3, 4].
Concerning colorectal cancer (CRC), it has been known for several decades that carcinomas mostly develop from adenomas. In 1988, Vogelstein et al. described four specific mutations that accumulate during the progression of adenomas to carcinomas [5]. These mutations have subsequently been shown to involve so-called care- and gatekeeper genes, which enable genetic or epigenetic instability and support tumor growth respectively. Although several other mutations involved in CRC development have been added during recent years, most sporadic CRCs are believed to develop as a consequence of the mutations initially described by Vogelstein et al. leading to the so-called adenoma-carcinoma sequence.
Various factors have been shown to be responsible for the accumulation of mutations in CRC including inheritance and environmental factors (e.g. composition of diet, obesity, diabetes mellitus, smoking, alcohol consumption) [6]. Of note, also chronic inflammation is regarded as an important risk factor for the development of cancer. This is especially apparent in patients with inflammatory bowel diseases (IBD), which have an increased risk for the development of colitis-associated CRC depending on the duration and severity of intestinal inflammation [7]. Whereas the contribution of chronic inflammation to tumor development has been widely attributed to its ability to induce mutations (e.g. through reactive oxygen or nitrogen species) [8], recent data propose a direct effect of inflammation on tumor growth.
Several pro-inflammatory cytokines released by innate and adaptive immune cells have been shown to regulate cancer cell growth and thereby contribute to tumor promotion and progression. Among these, interleukin-6 (IL-6) seems to take a center stage in human cancer development. An increased expression of IL-6 has been detected and associated with an unfavourable prognosis in patients with various types of cancer including both sporadic and colitis-associated CRC. Experimental studies found an activation of important oncogenic pathways in cancer cells through IL-6.
In this article, we review the role of IL-6 during sporadic and inflammation-associated CRC development. Besides data from human CRC, molecular mechanisms of IL-6 signaling in experimental models of CRC will be discussed with an outlook on future therapeutic implications.
IL-6 - an important regulator of immune function
Following its initial description as a B cell differentiation factor in 1986 [9], a versatile role has been attributed to IL-6 for the regulation of innate and adaptive immunity. In fact, IL-6 is involved in the regulation of the acute phase response through the induction of acute phase proteins in hepatocytes, the differentiation of monocytes to macrophages, the proliferation and resistance against apoptosis of T cells and Th2 cytokine production [9-11]. Importantly, recent data suggest a critical role for IL-6 during chronic inflammation, since IL-6 is required for the induction of effector Th17 cells and inhibits the differentiation of regulatory T cells.
IL-6 is produced by various cell types including monocytes, macrophages, fibroblasts, keratinocytes, endothelial cells, B cells, T cells, and also several tumor cells [12]. However, monocytes and macrophages seem to be the predominant producers of IL-6 during acute and T cells during chronic inflammation [13]. In these cells, IL-6 expression is regulated through the activation of several transcription factors such as NF-kB, C/EBPbeta (CAAT/enhancer-binding protein beta) or AP-1 (activator protein 1). The regulation of IL-6 expression through these transcription factors enables a rather unspecific upregulation of this cytokine during nearly every type of inflammation.
IL-6 binds to the membrane-bound IL-6 receptor alpha (mIL-6R, CD126) subunit of the IL-6 receptor on target cells. This complex then associates with a homodimer of the second receptor subunit, glycoprotein 130 (gp130, CD130), and thereby enables the activation of subsequent downstream signaling. This so-called “classic signaling” is restricted to cells expressing both the mIL-6R subunit and gp130. Although gp130 is widely expressed, mIL-6R expression is limited to hepatocytes and some leukocytes [14].
Importantly, a soluble form of IL-6R (sIL-6R) enables IL-6 signaling in cells that do not express mIL-6R through “trans-signaling”. sIL-6R is either produced by limited proteolysis of mIL-6R through the metalloproteinase ADAM17 or translation from a splice variant of IL-6R mRNA [14]. sIL-6R binds IL-6 with a similar affinity as mIL-6R and the complex of sIL-6R and IL-6 can interact with gp130 on target cells that don't express mIL-6R.
As signal transduction during both classic and trans-signaling is mediated through gp130, both induce the activation of identical intracellular pathways [11]. These include the activation of Janus kinases (JAKs) with a subsequent activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) through phosphorylation. STAT3 activation has been shown to be an important step for promotion and progression through the induction of various target genes. These target genes are involved in tumor cell survival (e.g. Bcl-2, Survivin, Mcl-1), proliferation (e.g. c-Myc, Cyclin D1, Cyclin B), angiogenesis (e.g. HIF1alpha, VEGF), metastasis (e.g. MMP2, MMP9), cell adhesion (e.g. ICAM-1, TWIST1), inflammation (e.g. IL-6, IL-17, IL-23, Cox2) and others (for review see [15, 16]). Among these, suppressor of cytokine signaling 3 (SOCS3) is a direct inhibitor of STAT3 signaling. Although the influence of these pathways on immune cells has been known for several years, only recent data provide a molecular insight on the importance of IL-6 signaling during tumor development.
Evidence for a role in human colorectal cancer
Concerning the role of IL-6 in B cell differentiation, it's not surprising that multiple myeloma was among the first types of cancer that have been shown to be influenced by IL-6. In fact, IL-6 acts as an auto- and paracrine growth factor for myeloma cells and antibodies against IL-6 inhibit myeloma cell growth in vitro and in vivo [17]. Today, IL-6 is regarded as an important tumor promoting factor in various types of human cancer including glioma, lymphoma, melanoma as well as breast, ovarian, pancreatic, prostate, renal and, of course, colorectal cancer.
Various studies have found an increased expression of IL-6 in patients with CRC, where IL-6 levels are elevated in the serum of patients and in tumor tissue itself [18-20].
According to a review article by Knüpfer and Preiss, IL-6 expression can be associated with tumor stage, size, metastasis and survival of patients with CRC [21].
Although data on IL-6 expression in sporadic CRC are well proven, there is an on-going debate about the source of IL-6 expression in non-inflammation-associated cancer. For instance, Belluco et al. described an association of a polymorphism of the IL-6 promoter with serum levels of this cytokine in patients with CRC [22]. An additional mechanism could be an amplification of the IL-6 gene, as reported in patients with glioblastoma, although this mechanism has not been shown for CRC so far [23]. Another explanation for increased IL-6 levels could be an infiltration of tumors with IL-6 secreting inflammatory cells as seen in colitis-associated cancer (CAC).
As mentioned above, patients with inflammatory bowel diseases such as Crohn's disease (CD) and ulcerative colitis (UC) have an increased risk for the development of CAC. For instance, the cumulative risk for the development of CRC in patients with UC is about 17.8% after 30 years of disease [24]. In patients with large bowel involvement of CD, there is a 8.3% risk for CRC over a period of 30 years [25].
Similar to CRC, IL-6 expression is increased in patients with IBD [26]. In fact, various studies have shown that IL-6 is an important regulator of IBD pathogenesis, mainly through its effect on immune cell function [11]. For instance, IL-6 trans-signaling has been shown to activate T cells in the lamina propria of patients with IBD and induces resistance of these cells against apoptosis through upregulation of anti-apoptotic factors such as Bcl-2 and Bcl-xl [27].
Due to the correlation of IL-6 expression with CRC prognosis and the increased expression of IL-6 in patients with IBD, IL-6 is thought to act as a link between chronic inflammation and tumor development. Importantly, Corvinus et al. could show an increased phosphorylation of STAT3 in CRC cells, but not in normal intestinal epithelial cells [28]. These data propose a functional relevance for IL-6 directly acting on tumor cells in sporadic CRC. This direct effect of IL-6 on colorectal cancer cells is likely mediated through trans-signaling, as intestinal epithelial cells usually do not express mIL-6R [14].
In fact, evidence for this hypothesis comes from various experimental studies in mouse models of colitis-associated cancer.
IL-6/STAT3 as critical mediators of cancer cell proliferation
In 1992, Lahm et al. were among the first to describe a growth-promoting effect of IL-6 on colorectal cancer cell lines in vitro [29]. However, it was not until 2004 that Becker et al. were able to show that IL-6, secreted by lamina propria T cells and macrophages, is also important for the development of CAC in vivo [30]. In the widely used AOM+DSS mouse model of CAC, the authors found an IL-6 dependent growth of intestinal tumors that was dependent on IL-6 trans-signaling in intestinal epithelial cells, possibly with downstream activation of STAT3. The tumor promoting effect of IL-6 could be inhibited through treatment with anti-IL-6R antibodies or sgp130Fc, a designer variant of soluble gp130 that specifically blocks trans-signaling. Furthermore, TGFβ signaling repressed IL-6 trans-signaling in tumor cells.
Similarly, Grivennikov et al. found reduced tumor development in IL-6 -/- mice exposed to the AOM+DSS model [31]. In this work, the authors underlined the importance of an IL-6 dependent STAT3 in tumor cells as critical for proliferation and the inhibition of apoptosis. These data were complemented by Bollrath et al. showing increased CAC development following AOM+DSS treatment in gp130Y757F mice, which have STAT3 hyperactivation, and attenuated tumor development in conditional knockout mice with a specific deletion of STAT3 in intestinal epithelial cells [32]. The effect of STAT3 on tumor cells was mediated through the expression of various regulators of G1/S and G2/M cell cycle progression. Interestingly, in both studies defective IL-6/STAT3 signaling was impairing intestinal inflammation in AOM+DSS treated animals. In this regards, IL-6 and STAT3 were proposed as important regulators of intestinal homeostasis.
In addition to IL-6 and STAT3, in a study by Rigby et al. the specific deletion of SOCS3 in intestinal epithelial cells of mice treated with AOM+DSS was associated with increased tumor development [33]. In contrast, overexpression of SOCS3 in vitro was able to reduce proliferation of CRC cell lines.
Whereas most effects of STAT3 have been attributed to a direct regulation of cell cycle progression through this transcription factor, other mechanisms supporting tumor growth have been described. In a recent study, we were able to show that IL-6 induces the expression of VEGFR2 (vascular endothelial growth factor receptor 2) in intestinal epithelial cells and enables an auto-/paracrine feedback loop, which promotes proliferation of tumor cells in the AOM+DSS model of CAC through VEGFR2-dependent STAT3 activation [34].
Therapeutic implications
Despite substantial progress of CRC treatment during recent years, CRC still belongs to the leading causes of cancer related death in industrialized countries. Therefore, new therapeutics, especially for patients with advanced disease, are desperately required. According to the growing evidence supporting a critical role for IL-6/STAT3 signaling during various aspects of both sporadic and inflammation-associated CRC development, therapeutics targeting this pathway could be promising options for affected patients.
In fact, several therapeutics inhibiting the IL-6/STAT3 pathway have been developed for the treatment of human disease. These include anti-IL-6 or anti-IL-6R antibodies, soluble gp130Fc (sgp130Fc) and selective small molecule JAK inhibitors [35] (Table 1).
Therapeutic strategies targeting IL-6 signaling in CRC.
| Strategy | Compound | Disease | Phase |
|---|---|---|---|
| IL-6 inhibition | Siltuximab (CNTO328, Centocor, Inc.) | Solid cancer including CRC | Phase I + II (NCT00841191) |
| sIL-6R and mIL-6R inhibition | Tocilizumab (Chugal, Roche) | Experimental models of cancer | Preclinical |
| IL-6 trans-signaling inhibition | sgp130Fc | Experimental CRC | Preclinical |
| JAK2 Inhibition | CEP-33779 (Cephalon, Inc.) | Experimental CRC | Preclinical |
Anti-IL-6 antibodies were among the first therapeutics targeting the IL-6/STAT3 pathway to be used in clinical studies on human cancer during the 1990s. These include the treatment of multiple myeloma or AIDS-associated Kaposi's sarcoma [36, 37]. However, these treatments only produced a limited response, and, as the antibody used (BE-8) was from murine origin, an immune response against this therapeutic was induced in treated patients [38].
As a consequence, the chimeric, murine-human monoclonal anti-IL-6 antibody siltuximab was generated during subsequent years. Although there are currently no data on the use of siltuximab in patients with CRC, a study analyzing the effect of siltuximab in various types of solid cancer including CRC has just been finished and the results are awaited eagerly (ClinicalTrials.gov Identifier: NCT00841191). However, initial clinical studies using siltuximab in patients with metastatic renal cancer, ovarian cancer or prostate cancer provided mixed results [39-41]. A possible explanation might come from the fact that antibody-associated IL-6 is not cleared from the circulation and thus increases systemic IL-6 concentrations [35]. Antibodies targeting IL-6R could overcome these problems.
In this regard, the anti-IL-6R antibody tocilizumab was developed [42]. So far, several clinical studies have shown a promising effect of tocilizumab in chronic inflammatory diseases and led to the approval of this antibody for the treatment of rheumatoid arthritis and juvenile idiopathic arthritis by the FDA [43]. Although there are preclinical data on a therapeutic effect of tocilizumab in cancer, clinical trials in human cancer are missing so far.
Another strategy to inhibit IL-6/STAT3 signaling is the specific inhibition of JAK activation. For instance, the small molecule JAK1 and 2 inhibitor ruxolitinib has shown promising results in a clinical trial in patients with post myeloproliferative neoplasms and acute myeloid leukemia [44]. Currently, there are no clinical data on the use of ruxolitinib in CRC. However, CEP-33779, another JAK inhibitor, could successfully reduce tumor growth in experimental CAC [45]
All of the treatments mentioned above inhibit both classical and trans-signaling, and therefore also block physiological functions of IL-6. In contrast, a specific inhibition of trans-signaling could be achieved by sgp130Fc. sgp130Fc is a designer cytokine that specifically binds IL-6/sIL-6R complexes and therefore only blocks trans-signaling. In fact, sgp130Fc has been shown to be effective for the treatment of experimental CAC in the study by Becker et al. [30]. The substance will soon enter clinical development and it will be interesting to see its effect on human cancer [46].
IL-6 signaling in colorectal cancer. IL-6 secreted by cells of the innate or adaptive immune system binds to soluble IL‑6R (sIL‑6R). The complex of IL‑6 and sIL‑6R interacts with gp130 on tumor cells through trans‑signaling and induces subsequent activation of Janus kinases (JAKs) and phosphorylation of signal transducer and activator transcription 3 (STAT3). A homodimer of phosphorylated STAT3 then translocates to the nucleus and induces the transcription of several target genes promoting proliferation, cell growth and the inhibition of apoptosis.
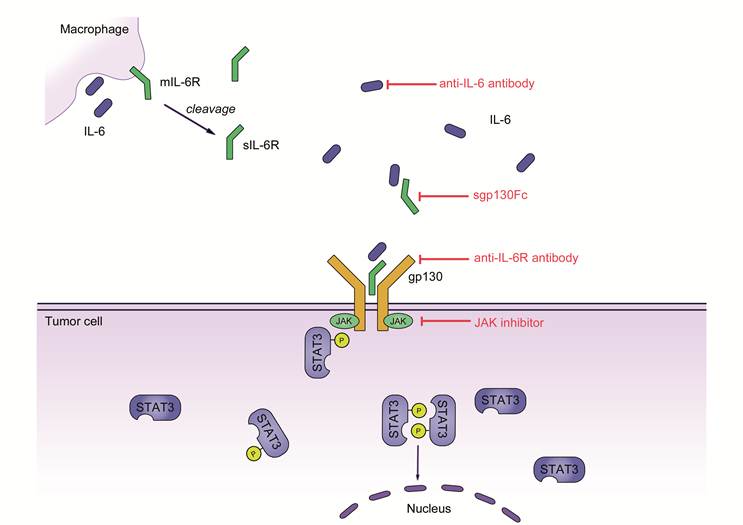
Conclusion
IL-6 has been shown to be an important tumor promoting cytokine that enforces proliferation and anti-apoptotic effects in tumor cells. Clinical and experimental data strongly propose a contribution of IL-6 signaling to the development of both sporadic and colitis-associated colorectal cancer development. In this regard, several components of the IL-6 signaling pathway such as IL-6, IL-6R, JAK have been proposed a promising targets for CRC therapy. As initial clinical studies using anti-IL-6 therapy are on their way, it will be interesting to see, if preclinical data will live up to their promise.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rous P, Beard JW. The Progression to Carcinoma of Virus-Induced Rabbit Papillomas (Shope). J Exp Med. 1935;62:523-48
2. Pitot HC. The molecular biology of carcinogenesis. Cancer. 1993;72:962-70
3. Foulds L. The natural history of cancer. J Chronic Dis. 1958;8:2-37
4. Foulds L. The experimental study of tumor progression: a review. Cancer Res. 1954;14:327-39
5. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M. et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525-32
6. Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B. et al. Colorectal cancer. Lancet. 2010;375:1030-47
7. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807-16
8. Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14:433-9
9. Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16:249-84
10. Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39:531-6
11. Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22:83-9
12. Kishimoto T. The biology of interleukin-6. Blood. 1989;74:1-10
13. Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109-19
14. Yamamoto K, Rose-John S. Therapeutic blockade of interleukin-6 in chronic inflammatory disease. Clin Pharmacol Ther. 2012;91:574-6
15. Jarnicki A, Putoczki T, Ernst M. Stat3: linking inflammation to epithelial cancer - more than a "gut" feeling? Cell Div. 2010;5:14
16. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798-809
17. Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science. 1992;258:593-7
18. Komoda H, Tanaka Y, Honda M, Matsuo Y, Hazama K, Takao T. Interleukin-6 levels in colorectal cancer tissues. World J Surg. 1998;22:895-8
19. Chung YC, Chang YF. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J Surg Oncol. 2003;83:222-6
20. Galizia G, Orditura M, Romano C, Lieto E, Castellano P, Pelosio L. et al. Prognostic significance of circulating IL-10 and IL-6 serum levels in colon cancer patients undergoing surgery. Clin Immunol. 2002;102:169-78
21. Knupfer H, Preiss R. Serum interleukin-6 levels in colorectal cancer patients--a summary of published results. Int J Colorectal Dis. 2010;25:135-40
22. Belluco C, Nitti D, Frantz M, Toppan P, Basso D, Plebani M. et al. Interleukin-6 blood level is associated with circulating carcinoembryonic antigen and prognosis in patients with colorectal cancer. Ann Surg Oncol. 2000;7:133-8
23. Tchirkov A, Khalil T, Chautard E, Mokhtari K, Veronese L, Irthum B. et al. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer. 2007;96:474-6
24. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526-35
25. Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment Pharmacol Ther. 2006;23:1097-104
26. Atreya R, Neurath MF. New therapeutic strategies for treatment of inflammatory bowel disease. Mucosal Immunol. 2008;1:175-82
27. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S. et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583-8
28. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB. et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545-55
29. Lahm H, Petral-Malec D, Yilmaz-Ceyhan A, Fischer JR, Lorenzoni M, Givel JC. et al. Growth stimulation of a human colorectal carcinoma cell line by interleukin-1 and -6 and antagonistic effects of transforming growth factor beta 1. Eur J Cancer. 1992;28A:1894-9
30. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491-501
31. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103-13
32. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91-102
33. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833-41
34. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R. et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med. 2010;207:2855-68
35. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375-83
36. Bataille R, Barlogie B, Lu ZY, Rossi JF, Lavabre-Bertrand T, Beck T. et al. Biologic effects of anti-interleukin-6 murine monoclonal antibody in advanced multiple myeloma. Blood. 1995;86:685-91
37. Racadot E, Audhuy B, Duvernoy H, Thyss A, Lang JM, Wijdenes J. et al. Clinical and immunological follow-up of patients with AIDS-associated Kaposi's sarcoma treated with an anti-IL-6 monoclonal antibody. Cytokines Mol Ther. 1995;1:133-8
38. Legouffe E, Liautard J, Gaillard JP, Rossi JF, Wijdenes J, Bataille R. et al. Human anti-mouse antibody response to the injection of murine monoclonal antibodies against IL-6. Clin Exp Immunol. 1994;98:323-9
39. Rossi JF, Negrier S, James ND, Kocak I, Hawkins R, Davis H. et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br J Cancer. 2010;103:1154-62
40. Guo Y, Nemeth J, O'Brien C, Susa M, Liu X, Zhang Z. et al. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin Cancer Res. 2010;16:5759-69
41. Karkera J, Steiner H, Li W, Skradski V, Moser PL, Riethdorf S. et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate. 2011;71:1455-65
42. Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T. et al. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993;53:851-6
43. Tanaka T, Narazaki M, Kishimoto T. Therapeutic targeting of the interleukin-6 receptor. Annu Rev Pharmacol Toxicol. 2012;52:199-219
44. Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C. et al. Phase II study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including post myeloproliferative neoplasms (MPN) acute myeloid leukemia (AML). Blood. 2012;119(20):4614-8
45. Seavey MM, Lu LD, Stump KL, Wallace NH, Hockeimer W, O'Kane TM. et al. Therapeutic efficacy of CEP-33779, a novel selective JAK2 inhibitor, in a mouse model of colitis-induced colorectal cancer. Mol Cancer Ther. 2012;11:984-93
46. Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert Opin Ther Targets. 2012;16:225-36
Author contact
![]() Corresponding author: Prof. Dr. med. Markus F. Neurath, Department of Medicine 1, University of Erlangen-Nuremberg, Ulmenweg 18, 91054 Erlangen, Germany. Tel.: +49 9131 85 35200 Fax: +49 9131 85 35209 markus.neurathde.
Corresponding author: Prof. Dr. med. Markus F. Neurath, Department of Medicine 1, University of Erlangen-Nuremberg, Ulmenweg 18, 91054 Erlangen, Germany. Tel.: +49 9131 85 35200 Fax: +49 9131 85 35209 markus.neurathde.