Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(2):149-155. doi:10.7150/ijbs.5617 This issue Cite
Research Paper
Pathogenic Mutation in VPS35 Impairs Its Protection against MPP+ Cytotoxicity
Fangfang Bi1, Fang Li1, Cao Huang2, Hongxia Zhou1, ![]()
1. Department of Neurology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
2. Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
Received 2012-11-28; Accepted 2013-1-18; Published 2013-1-26
Abstract
Parkinson's disease primarily results from progressive degeneration of dopaminergic neurons in the substantia nigra. Both neuronal toxicants and genetic factors are suggested to be involved in the disease pathogenesis. The mitochondrial toxicant 1-methyl-4-phenylpyridinium (MPP+) shows a highly selective toxicity to dopaminergic neurons. Recent studies indicate that mutation in the vacuolar protein sorting 35 (vps35) gene segregates with Parkinson's disease in some families, but how mutation in the vps35 gene causes dopaminergic cell death is not known. Here, we report that enhanced VPS35 expression protected dopaminergic cells against MPP+ toxicity and that this neuroprotection was compromised by pathogenic mutation in the gene. A loss of neuroprotective functions contributes to the pathogenesis of VPS35 mutation in Parkinson's disease.
Keywords: Parkinson's disease, vacuolar protein sorting 35, VPS35, 1-methyl-4-phenylpyridinium, MPP+.
Introduction
Parkinson disease primarily results from the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). While dopamine replenishment treatment temporally ameliorates the symptoms of Parkinson's disease including bradykinesia and muscle rigidity (1, 2), no treatment effectively prevents the disease from progression (3). Most Parkinson's disease cases are sporadic without a definitive cause, but about 10% of the cases have genetic mutations in the individual genes including alpha-synuclein, Leucine-Rich Repeat Kinase 2 (LRRK2), parkin, DJ-1, and PTEN-induced putative kinase 1 (PINK1) (4-11). Recent studies on human genetics suggest that mutations in the vacuolar protein sorting 35 (VPS35) gene segregate with Parkinson's disease in some families (12, 13). How VPS35 mutation leads to dopaminergic cell death remains to be determined.
VPS35 is a critical component of retromer complex that consists of VPS26, VPS29, and VPS35 in mammals (14-16). Retromer complex takes part in the recycling of lipids and proteins from plasmas membrane to Golgi complex and plays an important role in cell functions and survival. Deletion of VPS35 or VPS26a causes lethality in knockout mice (14, 15). VPS35 also regulates the transport of mitochondrial-anchored protein ligase from mitochondria to peroxisomes (17), implying that VPS35 regulates mitochondrial functions. Mitochondrial defect is implicated in PD pathogenesis. Deficiency in PINK1 or Parkin causes mitochondrial defects in animal models (18, 19). Rotenone and 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) induce dopaminergic neuron death by inhibiting mitochondrial complex functions (20, 21). MPTP itself is not toxic, but in glial cells MPTP is metabolized into the toxic cation 1-methyl-4-phenylpyridinium (MPP+) that can be taken up primarily by dopaminergic neurons (3, 22). MPP+ interferes with complex I of the electron transport chain in mitochondrion, causing cell death. It is not known whether VPS35 protects dopaminergic cells against the mitochondrial toxin MPP+ and how pathogenic mutation in VPS35 affects such a neuroprotective function.
Here we examined the effects of wild-type and mutant VPS35 on MPP+ cytotoxicity in dopaminergic N27 cells. VPS35 overexpression protected N27 cells against MPP+ toxicity and this protection was impaired by a pathogenic mutation.
Results and Discussion
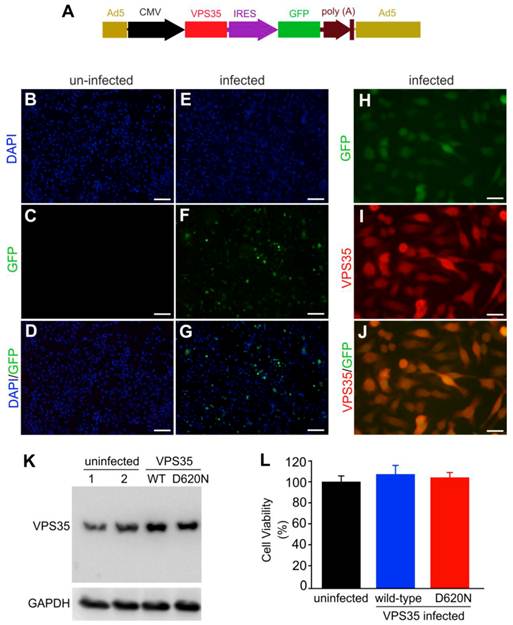
VPS35 mutation and mitochondrial defect are both implicated in Parkinson's disease. To examine how VPS35 modifies the cytotoxicity of the mitochondrial toxicant MPP+, we decided to overexpress human VPS35 with or without a pathogenic mutation in a dopaminergic cell line—N27 and thus to test the effects of wild-type and mutant VPS35 on MPP+ cytotoxicity (23). We chose an adenoviral system for robust and comparable gene expression (Figure 1A). To monitor transgene expression in living cells, we co-expressed green fluorescence protein (GFP) in the same transcript as human VPS35. As GFP was translated from the internal ribosome entry site (IRES), the robustness of GFP expression was relatively much low (Figure 1A-J). GFP was detected in the cells infected with adenoviral vector (Figure 1E-G) and not in the cells without adenoviral infection (Figure 1B-D). GFP fluorescence well colocalized with VPS35 immunostaining (Figure 1H-J). Immunoblotting confirmed that adenoviral vectors substantially expressed human VPS35 in infected cells (Figure 1K). Overexpression of wild-type or mutant VPS35 produced no effects on cell growth and survival as revealed by MTS viability assay (Figure 1L).
Mutant VPS35 overexpression produces no effect on cell growth in dopaminergic cell culture. (A) A diagram showing the construction of adenoviral vector that expresses wild-type or mutant (D620N substitution) human VPS35 gene. The open reading frame (ORF) of human VPS35 is driven by CMV promoter and the ORF of green fluorescence protein (GFP) gene is also integrated in the same transcript and is translated from the internal ribosome entry site (IRES). The expression cassette is recombined into adenoviral vector for producing viral particles in AD293 cells. (B-G) Fluorescence microscopy reveals that GFP was detectable in AD-VPS35 infected N27 cells (E-G) and not in uninfected cells (B-D). (H-J) Double-labeling fluorescence staining reveals that GFP colocalized with the immunostaining for VPS35 in AD-VPS35 infected cells. Scale bars: 100 µm (B-G) and 25 µm (H-J). (K) Western blotting reveals that VPS35 was overexpressed in the cells infected with adenovirus expressing wild-type (WT) or mutant (D620N) human VPS35. GAPDH immunoreactivity serves as an internal control for equal loading. (L) MTS assay reveals no effect of VPS35 overexpression on cell viability in N27 cell culture. N27 cells were infected with adenovirus expressing WT or D620N human VPS35 for 72 hours and cell viability was assayed with MTS kit. Data are means ± SEM (n = 5).
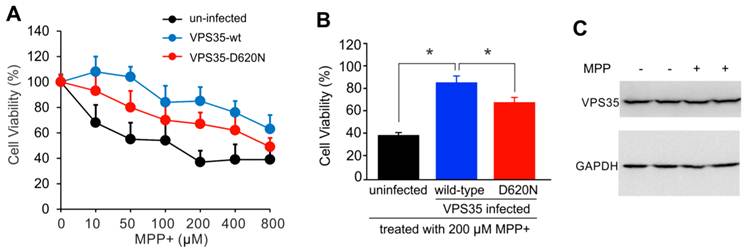
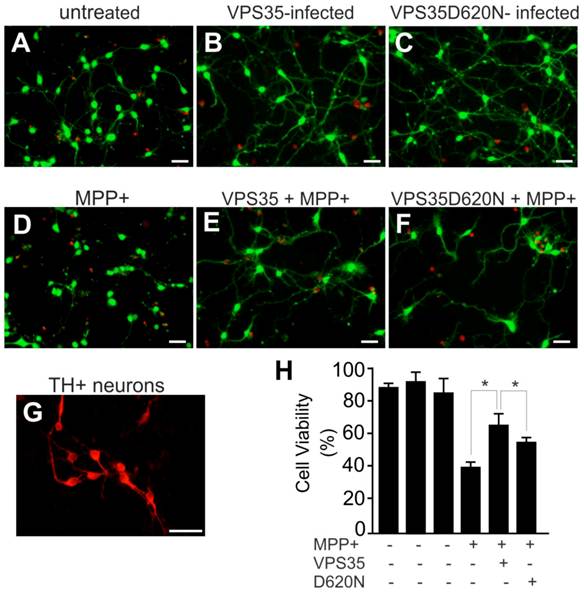
Adding mutant VPS35 to cultured dopaminergic cells was insufficient to induce severe phenotypes such as cell death (Figure 1), but pathogenic mutation may compromise the cell-protective functions of VPS35 particularly when the cells are challenged with exogenous stress. We decided to test whether VSP35 protects cells against the mitochondrial toxicant MPP+. Wild-type VPS35 protected N27 cells against MPP+ toxicity and this protection was decreased when VPS35 was pathogenically mutated (Figure 2). To extend our finding in cell line, we further tested VPS35 neuroprotection in primary culture. Rat mesencephalic culture was enriched with dopaminergic neurons and transduced with adenoviral vectors expressing human VPS35 with or without a pathogenic mutation. Viral expression of human VPS35 protected mesencephalic neurons against MPP+ toxicity and this neuroprotection was decreased when the gene contained a pathogenic mutation (Figure 3), confirming our finding N27 cell lines. As a member of retromer complex, VPS35 may facilitate the reuse of key transporters such as vesicular monoamine transporter type 2 (VMAT2). VMAT2 entraps toxicants such as MPP+ and protects against its neurotoxicity. Pathogenic mutation in VPS35 may decrease its normal functions and compromise its neuroprotective effects.
Pathogenic mutation in VPS35 impairs its protection against MPP+ toxicity to N27 cells. (A) MTS assay reveals that MPP+ dose-dependently induced cell death in N27 cell culture and that VPS35 overexpression protected the cells against MPP+ cytotoxicity. (B) Pathogenic mutation in VPS35 impaired its protection against MPP+ toxicity. Cells were infected with adenovirus expressing wild-type or mutant (D620N substitution) human VPS35 for 24 hours and then were treated with MPP+ at indicated concentrations for 48 hours. Cell viability was assayed with MTS kit afterwards. Data are means ± SEM (n = 5). * p < 0.05. (C) Immunoblotting reveals no effects of MPP+ (MPP) on endogenous VPS35 expression in N27 cells. GAPDH immunoreactivity serves as an internal control for equal loading. N27 cells were treated with MPP+ (200µM) for 48 hours and were then harvested for analysis of VPS35 induction.
Pathogenic mutation in VPS35 impairs its protection against MPP+ toxicity to primary ventral mesencephalic neurons. (A-F) Representative photos show live (green) and dead (red) cells in rat mesencephalic culture. Ventral mesencephalon was dissected from rat embryos (E-15) and mesencephalic neurons were cultured for 4 days before viral transduction. Primary neurons were transduced with adenoviral vectors and were further treated with MPP+ at 24 hours after viral transduction. Cell viability was assessed at 48 hours after MPP+ treatment. (G) Rat ventral mesencephalic culture was immunostained with an antibody against rat tyrosine hydroxylase (TH) to label dopaminergic neurons. Scale bars: 40 µm (A-G). (H) Cell viability in rat mesencephalic culture was quantitated. Data are means ± SEM (n = 7). * p < 0.05.
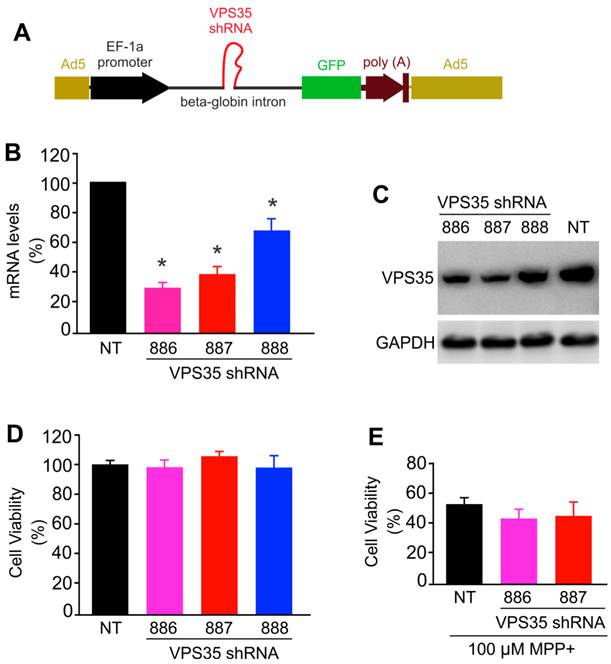
To further test neuroprotective functions for VPS35, we attempted to knock down VPS35 expression in N27 cells. We used adenoviral vectors to achieve desirable expression and expressed VPS35 shRNA within an artificial intron upstream of GFP gene. Therefore, shRNA expression can be monitored indirectly by observing GFP fluorescence (Figure 4A). We selected three shRNA targeting sites within the opening reading frame (ORF) of rat VPS35 and found that two of the three shRNA knocked down VPS35 expression efficiently (Figure 4B-C). Reducing VPS35 expression by about 70% did not affect the viability of N27 cell culture and the protection of VPS35 against MPP+ toxicity (Figure 4D-E). A significant residue of VPS35 protein was observed in knockdown cells (Figure 4C); thus, partial VPS35 reduction might be insufficient to produce a marked effect on its functions. Indeed, heterozygous VPS35 knockout mice do not develop severe phenotypes though the homozygous knockout embryos die at early embryonic stages (14). A minimal level of VPS35 expression appears to be sufficient for maintaining its functions.
VPS35 expression knockdown produces no effect on VPS35-mediated protection against MPP+ toxicity. (A) A diagram showing the construction of adenoviral vector for expressing short-hairpin RNA (shRNA) against VPS35. VPS35 shRNA was inserted in the intron for co-expression with GFP. (B) Quantitative PCR reveals that the VPS35 mRNA level was reduced by expressing VPS35 shRNA. Data are means + SEM (n = 3). (C) Immunoblotting reveals that VPS35 expression was knocked down at protein levels by VPS35-shRNA. (D) Knocking down VPS35 expression produced no effect on cell viability in N27 cell culture. Data are means ± SEM (n =5). Cells were infected with adenovirus expressing VSP35 shRNA (shRNA-886, -887, or -888) for 72 hours and VPS35 knockdown and cell viability was assayed afterwards (B-D). (E) N27 cells were infected with shRNA viral vector for 24 hours and then were treated with MPP+ for 48 hours. After MPP+ treatment, cell viability was determined with MTS kit. Data are means ± SEM (n = 5). * p < 0.05.
In summary, our studies showed that VPS35 provides protection for dopaminergic cells against the mitochondrial toxicant MPP+. The protective function of VPS35 is compromised by a pathogenic mutation. A limited expression of VPS35 appears sufficient to maintain its functions, at least in cell culture.
Materials and Methods
Production of adenoviral vectors for expressing human VPS35. Human VPS35 open reading frame (ORF) was PCR-cloned from human brain cDNA pools (Invitrogen) and the pathogenic mutation D620N was introduced by PCR-based mutagenesis. Resulting VPS35 ORF was verified by sequencing and was inserted between Pme1 and Xho1 sites of pacAd5CMV-IRES-GFP plasmid (Cell Biolabs, San Diego, CA, USA). The following three regions in rat VPS35 ORF were selected for constructing shRNA vectors: 5'-GGCTGTGAAAGTTCAATCATTCC-3' (shRNA-886), 5'-CAAGCTGATGGACGCTCTGAAACA-3' (shRNA-887), and 5'-GGACTTCTATGTTATCACCAAAGA-3' (shRNA-888). Individual shRNA was built to target one of the three selected regions using an established protocol (24). Each shRNA was inserted between BamH1 and Nhe1 sites of pacAd5miR-GFP vector (Cell Biolabs, San Diego, CA, USA). Individual transgene expression adenoviral vector was co-transfected with pacAd5-9.2-100 vector into Ad293 cells for homologous recombination and for viral particle formation. Adenoviral particles were harvested and amplified in Ad293 cells. Raw viral particles were used for infecting N27 cells.
Cell culture and viability assay. Ad293 cells were cultured in DMEM medium supplemented with 10% FBS and antibiotics as instructed by the provider (Cell Biolabs, San Diego, CA, USA). N27 cells were cultured in DMEM medium supplemented with 10% FBS and antibiotics (ampicillin/streptomycin). In the morning of adenovirus infection, cells were split into 24-well culture plate at a confluence of 40% and were infected with individual adenoviral vector 6 hours after plating. Each well of 24-well culture plate was added with 10 µl of original adenoviral solution. GFP expression was monitored under a fluorescence microscope. Infected cells were further challenged with MPP+ (Sigma) at indicated concentrations for defined times. Cell viability was determined using a 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay according to manufacturer's instruction (Promega) as described previously (25). Statistical significance between experimental groups was analyzed by unpaired t test and p value less than 0.05 was considered of significance. Dopaminergic neurons were obtained from rat embryos at embryonic day 15 using a published protocol (26).
Immunoblotting and immunofluorescence staining. Cells were washed with cooled PBS twice and then were mechanically scraped off culture plates. Cells were lyzed in 1X RIPA buffer and cleared cell lysates were determined for protein concentration. Protein composition in cell lysates was resolved on SDS-PAGE gel and transferred onto GeneScreen Plus membrane (Perkin Elmer, Wellesley, MA, USA). Immunoreactivity for VPS35 and GAPDH was determined by incubating the membrane with primary antibody to VPS35 (Novus, Littleton, Colorado, USA) or GAPDH (Sigma). Primary antibodies were diluted at 1:2000 and the secondary antibodies were diluted at 1:1000. Immunoreactivity was visualized with Super-Signal kit (27). For double-labeling fluorescence staining, cells were fixed in 4% paraformaldehyde for 30 minutes and washed in 1X PBS twice. Fixed cells were incubated with primary and secondary antibodies sequentially and immunoreactivity was visualized with a Nikon fluorescence microscope as described previously (28).
Acknowledgements
This work is supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NS073829 to H. Z.). The content is the author's responsibility and does not necessarily represent the official view of the NIH institutes.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schulz JB. Mechanisms of neurodegeneration in idiopathic Parkinson's disease. Parkinsonism Relat Disord 13 Suppl. 2007;3:S306-308
2. Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB. Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem. 2001;79:63-70
3. Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:10433-10438
4. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R. et al. Mu-tation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045-2047
5. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-608
6. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-259
7. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG. et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158-1160
8. Zimprich A, Biskup S, Leitner P, Licht-ner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-607
9. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de, Munain A, Aparicio S, Gil AM, Khan N. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595-600
10. Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease al-pha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41-50
11. Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG. TDP-43 Potentiates Alpha-synuclein Toxicity to Dopaminergic Neurons in Transgenic Mice. Int J Biol Sci. 2011;7:234-243
12. Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Hau-benberger D, Spielberger S, Schulte EC, Lichtner P. et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168-175
13. Vilarino-Guell C, Wider C, Ross O.A, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA. et al. VPS35 mutations in Par-kinson disease. Am J Hum Genet. 2011;89:162-167
14. Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH. et al. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol. 2011;195:765-779
15. Lee JJ, Radice G, Perkins C.P, Costantini F. Identification and characteri-zation of a novel, evolutionarily conserved gene disrupted by the murine H beta 58 embryonic lethal transgene insertion. De-velopment. 1992;115:277-288
16. Collins BM, Skinner CF, Watson PJ, Seaman MN, Owen DJ. Vps29 has a phosphoesterase fold that acts as a protein interaction scaffold for retromer assembly. Nat Struct Mol Biol. 2005;12:594-602
17. Braschi E, Goyon V, Zunino R, Mo-hanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol. 2010;20:1310-1315
18. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162-1166
19. Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3:242-250
20. Jenner P. The contribution of the MPTP-treated primate model to the devel-opment of new treatment strategies for Parkinson's disease. Parkinsonism Relat Disord. 2003;9:131-137
21. Dawson T, Mandir A, Lee M. Animal models of PD: pieces of the same puzzle? Neuron. 2002;35:219-222
22. Szego EM, Outeiro TF, Kermer P, Schulz JB. Impairment of the septal cho-linergic neurons in MPTP-treated A30P al-pha-synuclein mice. Neurobiol Aging. 2013;34:589-601
23. Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285:11740-11752
24. Xia XG, Zhou H, Samper E, Melov S, Xu Z. Pol II-expressed shRNA knocks down Sod2 gene expression and causes phenotypes of the gene knockout in mice. PLoS Genet. 2006;2:27
25. Xia XG, Zhou H, Zhou S, Yu Y, Wu R, Xu Z. An RNAi strategy for treatment of amyotrophic lateral sclerosis caused by mutant Cu,Zn superoxide dismutase. J Neurochem. 2005;92:362-367
26. Schneider JS, Huang FN, Vemuri MC. Effects of low-level lead exposure on cell survival and neurite length in primary mesencephalic cultures. Neurotoxicol Teratol. 2003;25:555-559
27. Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909-919
28. Huang C, Tong J, Bi F, Wu Q, Huang B, Zhou H, Xia XG. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum Mol Genet. 2012;21:4602-4614
Author contact
![]() Corresponding author: H.Z. (Hongxia.zhouedu), phone: 215-503-9156; fax: 215-923-3808; address: JAH506, 1020 Locust Street, Philadelphia, PA 19107.
Corresponding author: H.Z. (Hongxia.zhouedu), phone: 215-503-9156; fax: 215-923-3808; address: JAH506, 1020 Locust Street, Philadelphia, PA 19107.