Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The ionotropic GluRs: Basic...
The metabotropic GluRs are...
Structure and function of the...
mGluR1 signaling in the nervous...
mGluR1 signaling in malignant...
Concluding Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(9):948-959. doi:10.7150/ijbs.6426 This issue Cite
Review
Glutamate, Glutamate Receptors, and Downstream Signaling Pathways
Stacey S. Willard, Shahriar Koochekpour ![]()
Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY, USA.
Received 2013-4-8; Accepted 2013-7-11; Published 2013-9-22
Abstract
Glutamate is a nonessential amino acid, a major bioenergetic substrate for proliferating normal and neoplastic cells, and an excitatory neurotransmitter that is actively involved in biosynthetic, bioenergetic, metabolic, and oncogenic signaling pathways. Glutamate signaling activates a family of receptors consisting of metabotropic glutamate receptors (mGluRs) and ionotropic glutamate receptors (iGluRs), both of which have been implicated in chronic disabling brain disorders such as Schizophrenia and neurodegenerative diseases like Alzheimer's, Parkinson's, and multiple sclerosis. In this review, we discuss the structural and functional relationship of mGluRs and iGluRs and their downstream signaling pathways. The three groups of mGluRs, the associated second messenger systems, and subsequent activation of PI3K/Akt, MAPK, NFkB, PLC, and Ca/CaM signaling systems will be discussed in detail. The current state of human mGluR1a as one of the most important isoforms of Group I-mGluRs will be highlighted. The lack of studies on the human orthologues of mGluRs family will be outlined. We conclude that upon further study, human glutamate-initiated signaling pathways may provide novel therapeutic opportunities for a variety of non-malignant and malignant human diseases.
Keywords: Glutamate, mGluR, iGluR, GRM1a, signaling.
Introduction
Research into the amino acid glutamate (Glu) and its role in neurotransmission and disease has a long history spanning over four decades. Recognized as the predominant excitatory neurotransmitter in the central nervous system (CNS), a byproduct of glutamine (Gln) deamination, a critical step in nitrogen metabolism and a source of energy for rapidly dividing cells, this non-essential amino acid has been well-studied for its role in cellular homeostasis [1].
In the nervous system, Glu is the primary fast excitatory neurotransmitter participating in a wide range of neural functions such as learning and memory, long-term potentiation and synaptic plasticity [2, 3]. The early observation that increasing amounts of Glu can cause excitotoxicity and neural cell death opened the door to a whole field of research implicating Glu toxicity in neurodegenerative diseases (NDDs). In depth study of Glu signaling mechanisms has facilitated the development of treatments for Glu-related NDDs such as Alzheimer's and Parkinson's disease and multiple schlerosis [4]. Most recently, Glu signaling has been implicated in cellular transformation and cancer progression in multiple organs such as brain, skin, breast and prostate [5].
Beginning with the many and varied Glu receptors (GluRs), Glu signaling has been studied extensively in normal and disease states. GluRs are divided into two families: ionotropic (iGluR) and metabotropic (mGluR) (reviewed in [6, 7]). Further, the ionotropic receptor family includes the N-methyl-D-aspartate (NMDA), a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor subfamilies - so named for the chemical agonist that selectively binds to the subfamily members. The iGluR subfamilies all share a common voltage-gated ion channel function. Metabotropic GluRs, on the contrary, are G-protein-coupled receptors (GPCRs) containing the classic seven transmembrane domain structure and initiate signaling cascades or cation influx upon Glu binding. Based on sequence homology, pharmacology and second messenger associations, mGluRs are further categorized into the group I, II and III subfamilies [8]. Although iGluR signaling plays a critical role in neural function and has been shown in a few cases to play a role in cancer progression, for the purposes of this review, we will provide a cursory discussion of iGluRs while focusing more on the detail of Glu-mGluR signaling mechanisms.
The ionotropic GluRs: Basic functions and implications in disease states
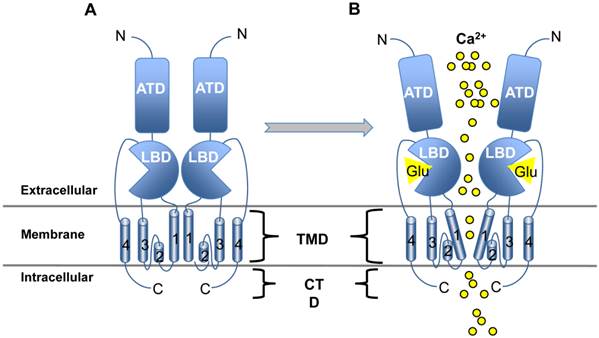
In general, all iGluRs are quaternary ligand-gated ion channels that allow cation influx upon Glu binding [7]. Each of the four subunits that comprise the channel share a similar structure of four well-conserved domains including the extracellular amino-terminal (ATD), ligand-binding (LBD), transmembrane (TMD) and intracellular carboxy-terminal (CTD) domains (Figure 1A). Glu or agonist interaction with the LBD results in a conformation shift which changes the angle of the TMD regions, opening a pore in the membrane where influx of sodium, potassium and/or calcium occurs (Figure 1B). Interestingly, the NMDA family of iGluRs can also be activated by voltage gating and are often found paired with mGluRs. If enough iGluRs are stimulated simultaneously, high concentrations of cation influx will result in an action potential - the fastest type of excitatory synaptic transmission throughout the central and peripheral nervous systems (CNS, PNS) and the retina [7]. After the signal is received by the target cell, excitatory amino acid transporters (EAATs; also known as glutamate transporters) expressed on postsynaptic and supporting glial cells are tasked with emptying the synaptic cleft of Glu to effectively turn off the signal and reset the system for generation and propagation of new action potentials [7].
iGluR structure and activation. iGluRs share the same basic structure: Beginning at the N terminus (N), the amino terminal domain (ATD) is followed by the ligand binding domain (LBD), the four transmembrane domains (TMD; numbered 1-4) and the C-terminal domain (CTD). Panel A illustrates the native resting conformation of iGluR. Only two subunits are shown, but the entire molecule consists of four subunits which are arranged in such a way as to create a channel through the plasma membrane. Upon Glu binding (Panel B), a conformational change in TMD domain 1 causes opening of the channel where Ca2+ passes into the cell by diffusion.
Prolonged stimulation of the iGluRs and other post-synaptic signaling components results in a phenomenon called excitotoxicity - a major contributor to NDDs and nervous system injuries [9]. Cells associated with trauma or disease release Glu, stimulating further Glu release by normal signaling mechanisms, and the resulting excessive Ca2+ influx leads to aberrant over-activation of proteases and caspases [4, 10]. Since EAATs require normal membrane voltage to function, they are not able to overcome the bombardment of cations caused by the overstimulation of iGluRs and apoptosis can occur, further exacerbating the injury and/or contributing to disease onset and progression [11, 12]. In fact, ischemia and hypoxia after CNS injury results in such a buildup of Glu in the synaptic cleft that depolarization and EAAT activity is inhibited leading to paralysis. Both NMDA and AMPA receptor subfamily members have been implicated in excitotoxicity after nervous system trauma [13].
Overstimulation of Glu signaling via iGluRs has been shown to contribute to NDDs [13]. It is clear that reactive oxygen species (ROS) and overstimulation of calcium-dependent signaling molecules is responsible, at least in part, for neuronal cell death by a similar mechanism as outlined for nervous system trauma [9]. Research into exactly which iGluRs are involved in disease onset and progression has led to treatments with varying success rates (reviewed in [4, 14]).
The metabotropic GluRs are atypical G-protein coupled receptors
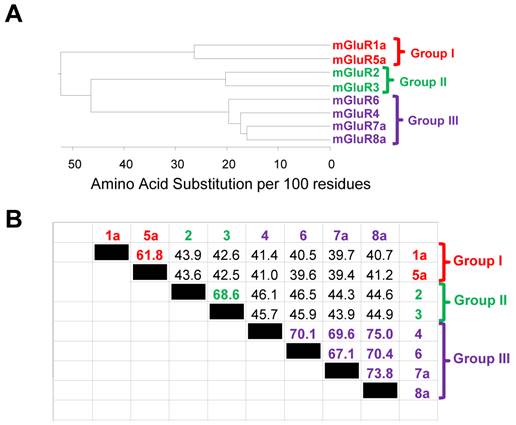
The mGluR superfamily consists of 12 members encoded by eight genes (reviewed in [15]). This is accomplished by multiple alternative splicing products produced by some mGluR genes, while others encode only one (Figure 2). For example, mGluR1 encodes five isoforms while there is only one isoform of mGluR2. Within the superfamily, the three aforementioned subgroups (I, II and III) emerge based on amino acid sequence homology, agonist binding pharmacology and the second messenger system that is activated upon Glu binding to each receptor (see [8] for a review). Activation of Group I mGluRs (mGluR1 and 5) and coupled Gq G-proteins initiates signaling cascades involving Phospholipase C/Inositol 1,4,5-triphosphate/diacylglycerol (PLC/IP3/DAG). Groups II and III both reduce cAMP levels by signaling through the inhibitory G-protein, Gi. Group II receptors include mGluR2 and 3 while Group III includes mGluR4, 6, 7 and 8 (Figure 2A). Within each group, amino acid sequence homologies are more than of 70%, while between groups, homology is only, on average, 45% (Figure 3).
The amino acid sequence homology among various metabotropic glutamate receptors. The phenogram in panel A illustrates the relationship between the three mGluR subgroups (Group I, II and III) and the individual receptors within each group. Branch length is indicative of amino acid identity between the protein sequences. The table in panel B illustrates the percent amino acid identity between all receptor pairwise combinations. Note that the highest similarity is observed between members of the same group (i.e. mGluR1 vs. mGluR5). The phenogram in panel A and the table in panel B were generated using the DNAStar software suite (DNASTAR, Inc., Madison, WI, USA) and amino acid sequences obtained from NCBI. Accession numbers: mGluR1a = NP_000829.2, mGluR2 = EAW65150.1, mGluR3 = NP_000831.2, mGluR4 = NP_000832.1, mGluR5a = NP_001137303.1, mGluR6 = AAB82068.1, mGluR7a = EAW63933.1, and mGluR8a = EAW83621.1.
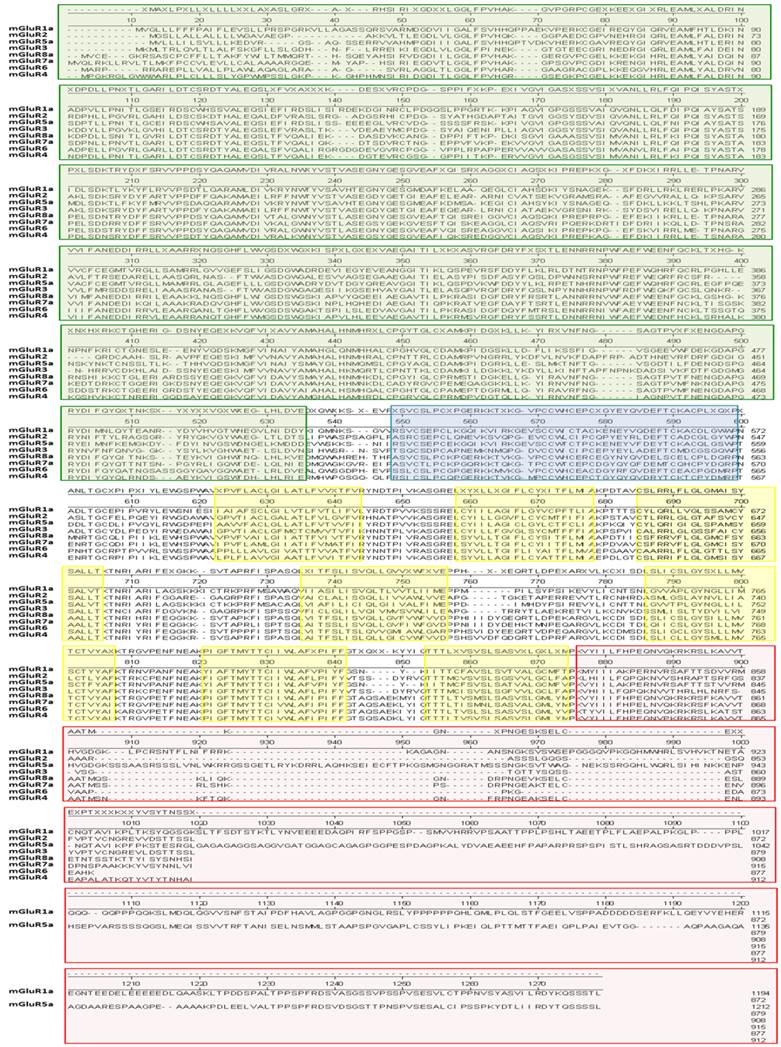
Amino acid sequence alignment of the eight mGluR genes. In this analysis, the “a” isoform of genes encoding multiple isoforms was used for simplicity. This figure was generated using the DNAStar software suite (DNASTAR, Inc., Madison, WI, USA) using the NCBI data base. NCBI accession numbers: mGluR1a = NP_000829.2, mGluR2 = EAW65150.1, mGluR3 = NP_000831.2, mGluR4 = NP_000832.1, mGluR5a = NP_001137303.1, mGluR6 = AAB82068.1, mGluR7a = EAW63933.1, and mGluR8a = EAW83621.1. Colored blocks indicate the following domains: Green, amino-terminal domain (ATD); blue, cysteine-rich domain (CRD); yellow, transmembrane-spanning domains (TMD); red, carboxy-terminal domain (CTD).
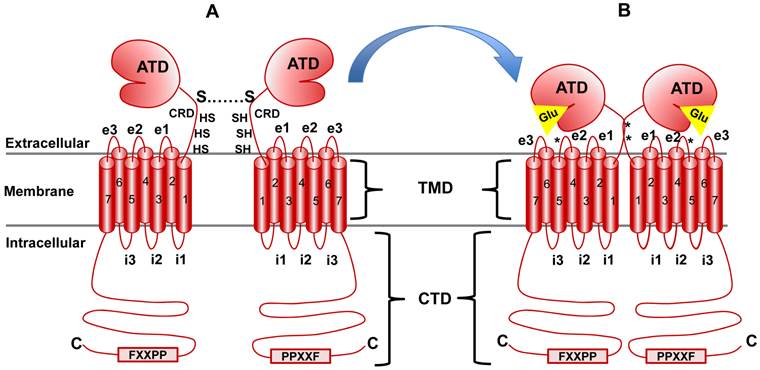
All mGluRs share a basic architecture (Figure 4 and 5). At the N-terminus exists a large bi-lobed extracellular domain known as the amino terminal domain (ATD), which in the protein crystallography literature is also referred to as the venus fly trap domain (VFT) because of its unique shape [16]. Following the ATD is a cysteine-rich domain (CRD) which is critical for dimerization and activation [17]. C-terminal to the CRD, the classic seven alpha-helical transmembrane domains (TMD) shared by all GPCRs are found and finally, an intracellular cytoplasmic tail domain (CTD) (reviewed in [18]). The receptor isoforms are divergent in structure within the CTD (Figure 5). Some isoforms of the Group I mGluRs (mGluR1a and 5a) contain a very long proline-rich domain which has been studied for its intricate protein-protein interactions and complex formations [19]. This domain, called the Homer1 binding motif, facilitates coupling of mGluRs with NMDA receptors among other proteins and will be described in detail below.
mGluR structure and conformational activation. mGluR family members share the same basic structure: Beginning at the N terminus (N), the amino terminal domain (ATD) is followed by the cysteine rich domain (CRD), the seven transmembrane domains (TMD; numbered) and the C-terminal domain (CTD). i1, i2, etc., indicate intracellular loops between membrane-spanning regions while e1, e2, etc., indicate extracellular loops. The star at loop e2 denotes the cysteine residue that is important in transmitting activation information to the docked G protein complex (see text for detail). Panel A illustrates the native resting conformation of mGluR1a. Cysteine residues in the CRD aid in dimerization, as indicated by the S-S bond. Upon Glu binding (Panel B), a conformational change in the ATD involving the CRD (stars indicate disulfide bridges created by Glu binding) causes activation of bound G proteins (see text for details).
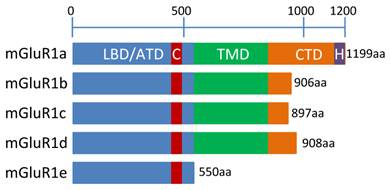
mGluR1 isoforms. This schematic represents the 5 human mGluR1 isoforms including relevant protein domains and amino acid (aa) lengths. Information on isoforms c, d and e were obtained from the literature [1-4, 39-44], while domain information for isoforms “a” and “b” was obtained using the NCBI “conserved domain search” function at: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi. NCBI accession numbers: mGluR1a = NP_000829.2 and mGluR1b = NP_001107801.1. Domain names are as follows: LBD/ATD = ligand binding domain/amino terminal domain, C = cysteine rich domain, TMD = transmembrane domain, CTD = C-terminal domain, H = Homer 1-binding motif.
Aside from the TMD and G-protein coupling, mGluRs are highly dissimilar at the structural and functional levels to any other classical GPCR groups studied to date and are therefore referred to as atypical GPCRs. In general, GPCRs are organized into five groups and virtually all of them belong to groups A, B, D and E. mGluRs, GABA receptors, two groups of taste receptors, and a Ca2+-sensing receptor (CaSRs) class belong to group C - a clearly divergent group based on amino acid sequence, size and structure [20, 21]. Interestingly, CaSR receptors are not sensitive to Glu or mGluR agonists, but share amino acid identity of 28% with Group I mGluRs [22]. CaSRs are mainly involved in Ca2+ sensing and homeostasis in the parathyroid gland and detection of L-amino acid levels [20]. Group C GPCRs diverge from other GPCR groups in a number of ways [23]. First, both mGluRs and CaSRs form homodimers in vivo through disulfide bridging between CRDs and this event is critical for function [17, 24, 25]. In addition, unlike other GPCRs which typically have a short extracellular ligand binding domain, the ATD is a large two-lobed structure allowing for Glu binding between the two lobes (Figure 4). The ATD in human mGluRs is, on average, 56 kDa (470-510 amino acids) which is far larger than the extracellular domain of other GPCRs that usually measure about 5 kDa, or 50 amino acids and correspond in size with ligand size [26]. For example, GPCRs which are activated by small molecules such as cations generally have a very small extracellular loop, while larger ligands require a larger LBD.
This does not hold true for Group C GPCRs since Ca2+ and Glu are small molecules, indeed. The ATD is related to bacterial periplasmic amino acid binding proteins, the structure of which has been extensively modeled and provides an excellent basis for models of the mGluR ATD [27]. Interestingly, some allosteric modifiers do not bind to mGluRs in Glu-binding pocket within the ATD. Instead, they typically bind to the small loop structure connecting the ATD to the TMD or within the TMD itself which is similar to activation of other GPCRs [25]. Reports have suggested that Ca2+ (which can activate both mGluRs and CaSRs) and L-amino acids (which can activate CaSR signaling) may bind to the extracellular loops connecting the seven transmembrane domains, further separating Group C GPCRs from the other groups [28, 29]. Finally, despite the canonical role of the third intracellular loop (i3; Figure 4) in G-protein activation, Group C GPCRs appear to use the second loop for this function (i2; [30-33]). The downstream signaling effects of Glu binding and the pharmacology pharmacodynamics of agonist/antagonist binding to mGluRs has been studied extensively due to this receptor group's clear implications in both benign and malignant disease progression.
mGluRs are localized throughout the CNS and PNS and have been shown to play a role in homeostasis in many organ systems [34]. Groups II and III receptors are generally found on presynaptic cells in the nervous system which correlates well with their function in regulating ion channel opening, while Group I receptors are generally found on postsynaptic cells ([35] and reviewed in [4]). This is not a rule, however, since all three receptor groups have been found on both pre-and postsynaptic cells. With regards to non-neural tissues, mGluRs have been found in all of the major organ systems including epithelium, connective and muscle tissues. More specifically, one or more mGluRs are expressed in the gastrointestinal system, immune system, genitourinary tract, bone, and hepatosplenic system (reviewed in [6]). In many cases, studies have established a functional role for mGluRs in organ homeostasis.
Structure and function of the mGluR1 gene
The first member of the human mGluR gene superfamily, mGluR1 (sometimes referred to as the Glutamate receptor metabotropic 1; GRM1) encodes five alternatively spliced isoforms (Figure 5). A complete literature review on this gene is stymied by multiple confusing nomenclatures that have been used interchangeably throughout the decades. For example, the first isoform of mGluR1 is synonymously referred to as the following: GRM1a, mGluR1a, mGluR1α, GPRC1a, and hmGlu1a. Surprisingly, a search for any of the synonyms excludes articles using the others, necessitating many searches to ensure completeness in collecting reliable and informative data in a review on this topic. We will use the simplest nomenclature which is the most utilized in recent studies, mGluR1a, for this review and will always be referring to the Homo sapiens gene unless explicitly noted. Adding to this complexity is the fact that many of the studies on the mGluR gene family have been performed using orthologues from the Norway rat, Rattus norvegicus. In fact, a large body of experimental data exists on the pharmacology, expression, mutational analysis and drug targeting of rodent mGluR1s. This phenomenon is so pervasive that it is difficult to find a study in the last decade that has used the human gene or protein. The rodent orthologues have certainly facilitated our understanding of mGluR signaling, but their translational value when interpreted in the context of non-malignant or malignant human diseases may remain questionable - at least in some cases.
Many of the initial studies on the human mGluR1 gene cloning and isoform identification were published in the mid-1990s, before the human genome sequence became available and was regularly annotated with newly published information [36-41]. As a result, despite numerous publications on the human mGluR1c and mGluR1d isoforms, there are no references to them in the NCBI human genome database at either the mRNA or protein levels. A nucleotide BLAST search with rat mGluR1d found similarity with the human mGluR1 and mGluR5 precursor gene sequences, but since isoforms c and d were not annotated in the human genome databases, and human mGluR1c and mGluR1d do not exist there as either mRNA or protein sequences. This issue is highlighted by the fact that annotation of isoforms a, b, c and d exists for the Rattus norvegicus and Mus musculus homologues of mGluR1 in the NCBI databases while the human gene shows annotation of only isoforms a and b.
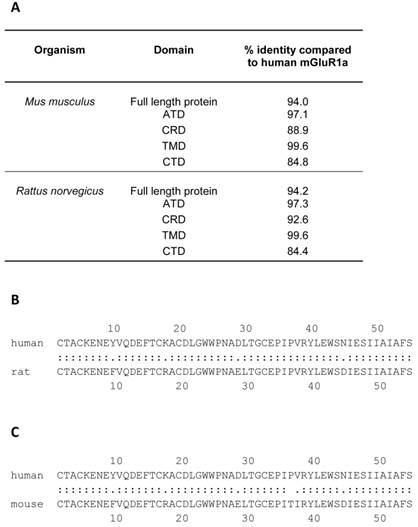
The rat mGluR1a protein sequence is 94% identical to the human mGluR1a protein at the amino acid level (Figure 6) and is certainly an excellent model for studies on mGluR1a function, however, it is a detriment to the field that genes with such a critical role in many organ systems, in NDDs and in benign and malignant diseases have been far better characterized with modern bioinformatics in rodents than their human orthologues. Figure 6 details the percent amino acid identity when the mouse and rat proteins are compared to the human protein. This analysis was performed using William Pearson's lalign program with Huang and Miller's algorithm which can be found at http://www.ch.embnet.org/software/LALIGN_form.html [42]. The highest homology between the mouse and rat protein and the human protein exists in the TMD, while the lowest percent identity is found in the CRD (Figure 6). Although all of the cysteine residues in the CRD are conserved, it is unclear whether or not the changes in the surrounding amino acids change the three dimensional structure of the domain or influence how it interacts with other mGluR1a proteins during homodimerization. Human mGluR1a was first expressed and characterized in vitro in 1998 by Hermans and colleagues using an inducible expression system [43, 44]. The localization (diffuse cytoplasmic), size (145 kDa) and induction of receptor activity were assayed in the common rodent model, Chinese hamster ovary (CHO) cells [43]. Subsequent studies have utilized this inducible rodent cell line system, known in the field as CHO-Lac-hmGlu1a. One such study noted that the induction of high levels of mGluR1a expression in CHO cells leads to a notable morphology change which is modulated by changing amounts of Ca2+ in the culture medium, but follow up studies in which the consequences of this change on cell motility or wound healing were investigated were not found [28].
A comparison of rodent and human mGluR1a at the amino acid level. The table in panel A illustrates the results of an lalign analysis of mGluR1a from Mus musculus (mouse) and Rattus norveticus (Norway rat). William Pearson's lalign program was used with default settings, which can be found at http://www.ch.embnet.org/software/LALIGN_form.html. The percent amino acid identity is shown. The highest amino acid identity is found in the transmembrane domain (TMD) and the lowest in the cytoplasmic tail domain (CTD). CRD = cysteine rich domain, ATD = amino terminal domain. Panels B and C display the lalign result of a comparison between human and rat CRD and human and mouse CRD, respectively. Note that all cysteine amino acids are conserved, but that a number of differences are found in surrounding amino acids. Between the two proteins, a “:” indicates completely identical while “.” represents a different amino acid.
It would be a great service to the field for a study to be undertaken in which full-length rodent and human mGluR1a are expressed in a human cell line and downstream signaling and biological assays are shown to yield identical results in side-by-side experiments. We were unable to find such a study, however, recent work by Esseltine and colleagues showed the result of human mGluR1a expression in human embryonic kidney 293 (HEK293) cells [45]. The mGluR1a protein that was expressed in in Esseltine and colleagues' study began at amino acid 19, therefore removing the amino-terminal end of the protein to include their own localization domain [45]. Although this study confirmed that the truncated mGluR1a localized to the plasma membrane and was able to initiate downstream signaling cascades upon stimulation, no biological effects such as cell growth, migration, morphology, etc. were examined [45].
Many studies have been aimed at determining how many isoforms were produced by the mGluR1 gene and the function and localization of each (see [46, 47] for reviews). The longest isoform, and most homologous to other Group I mGluRs is mGluR1a. The shortest and least well-characterized, mGluR1e, is truncated before the TMD, leading to the predicted expression of only the ATD (Figure 5) [46]. Follow up studies on a possible function for this isoform are not available. Excluding mGluR1e, the human mGluR1 isoforms are virtually identical except for alternative splicing changes near the 3' end of the mRNA which result in much smaller cytoplasmic domains at their c-termini for the mGluR1b, c and d proteins than the mGluR1a isoform (Figure 5). This difference in size is dramatic: An amino acid sequence alignment of human mGluR1a and mGluR1b are 100% identical until amino acid 886. Afterwards, a small exon containing a stop codon is incorporated in mGluR1b that is not included in mGluR1a, explaining the difference in size between the two isoforms (Figure 5). Alternative splicing for human mGluR1a results in a significantly longer protein than the other isoforms and includes the proline-rich Homer1 binding motif at the c-terminus. Figure 5 depicts a model of human mGluR1 isoforms. The extended length of the mGluR1a isoform prompts the question of why such a large cytoplasmic tail is required: Studies have shown that extensive protein-protein interactions are facilitated by the proline-rich region which contains the Homer-binding motif [19]. This phenomenon is not limited to mGluR1a since mGluR5a (a member of Group I) also possesses this domain, however, GPCR Group C member, CaSR does not [21].
The Homer-binding motif found in the CTD of mGluR1a and mGluR5a is a poly-proline region that has been shown to interact with proteins containing a Homer Ena/VASP Homology 1 domain (Homer EVH1) [19, 48-51]. Homer1, in particular, has been well-characterized for its role in the regulation of mGluR1a and mGluR5a activity. Binding of full-length Homer1isoforms to the consensus site found in the Homer-binding motif of mGluR1a/5a, PPXXF (where X is any amino acid), has been shown to influence not only the trafficking of the receptor between the cytoplasm and the membrane, but also receptor clustering within the membrane through its scaffolding activity (Figure 4). Further, Homer1 proteins can form homodimers and therefore link mGluRs to other Homer binding motif-containing proteins. This has been shown to be the case for PLC signaling where Homer1 homodimers create a scaffold which brings mGluR1a/5a in close proximity to the Inositol triphosphate receptor (IP3R). Activation of mGluR1a/5a results in IP3 production and rapid, localized activation of intracellular Ca2+ release through IP3R - the speed of which is facilitated by the close physical proximity of IP3R to active mGluR1a/5a via Homer1 scaffolding [52].
In addition to the previously described domains within the mGluR proteins, a cysteine-rich domain referred to as the 9-Cystein domain in some studies or the Cysteine-rich domain (CRD) in others is found between the ATD and TMD (Figure 4) [24, 34]. This domain, unique to group C GPCRs, has been crystallized and studied in Rat group II and III mGluRs. This short domain (only 69 amino acids in mGluR1) consists of three β-pleated sheets and nine cysteine residues - all nine of which are very well-conserved in the other mGluR proteins, irrespective of subgroup classifications and are also conserved in the human orthologues. Although the exact structure and function of this domain is still under investigation, it is clear from mutational analyses that certain cysteine's are critical for receptor clustering and for facilitating the interaction between the ATD and the TMD regions during ligand binding and activation [17]. Activation of the mGluR dimer has been shown to elicit a conformational change which brings the C-terminal regions of the CRD in closer proximity to one another (represented by a * in Figure 4) and to involve a cysteine-cysteine interaction in the e2 loop of the TMD. Crystal structures have indicated that this conformation change also involves a shift in the TMD, therefore coupling the ATD with G-protein activation [25]. The mutational analyses and crystallography that was used to obtain these models were all completed using the Rat orthologues of the mGluR family members and although all of the cysteine's in the CRD are conserved in the human protein, as shown in Figure 6, studies will need to be performed to confirm the residues of importance in the human orthologues.
mGluR1 signaling in the nervous system
Ligand binding to mGluR1 results in a wide array of downstream signaling effects. Ligands are diverse and include not only Glu, but also Ca2+ and neurotransmitters. Glu binding and aforementioned conformational changes can precipitate a number of varied signaling responses including: activation of coupled Ca2+, K+ and Na+ channels, activation of Gq and consequent downstream signaling cascades, activation of Extracellular-signal-regulated kinase (ERK) 1/2 (also known as mitogen activated protein kinase; MAPK) and retrovirus AK thymoma [AKT; also known as protein kinase B (PKB)] signaling and even stimulation of Sarcoma (Src) kinase signaling (Figure 7) [4, 15, 53-56]. Interestingly, mGluR1 differs from fellow group I mGluR family member, mGluR5, in that its activation results in a single transient rise in intracellular Ca2+ while active mGluR5 causes recurrent Ca2+ oscillations of unknown function [57]. Each of these second messenger systems and their functions in specific tissues will be expanded upon below.
G-protein signaling pathways activated by mGluR1a. This drawing represents an overview of G-protein dependent signaling pathways that are activated upon Glu binding to mGluR1a. Following receptor activation and downstream of G-protein dissociation, the following second messenger systems are activated in the nervous system: PKB, PLCγ, PI3K/AKT/mTorC1 and 2, IP3/DAG, NFκB and CaM. Please see the text for detailed description on these signaling pathways.
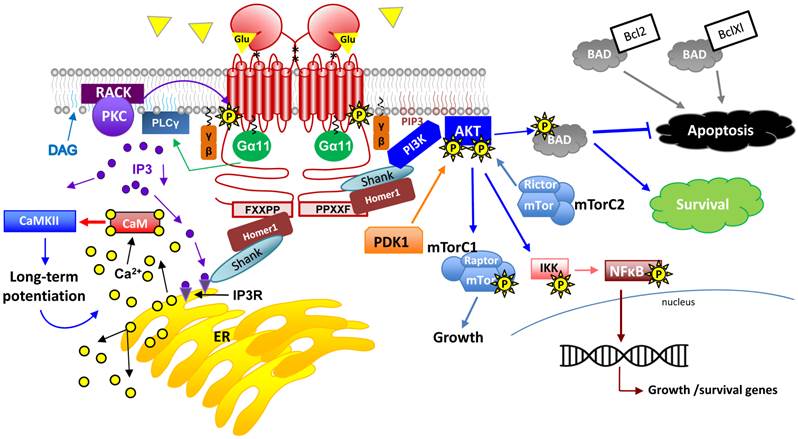
Glu stimulation of both mGluRs and iGluRs in neural cells results in increased intracellular Ca2+ levels via differing signaling mechanisms. Unlike iGluR signaling, which directly involves the opening of an ion channel upon Glu-binding, mGluR signaling can lead to Ca2+ influx by indirect ion channel opening and/or by second messenger signaling systems. Homer1 binding to the Homer binding motif can recruit the scaffold protein, Shank, which facilitates the direct interaction of mGluRs and NMDARs resulting in Ca2+ influx [58]. Further, activation of PLCs by Gα11 results in the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into two products: the phosphate head group (IP3) and the fatty acid chains (e.g., DAG). DAG remains in the plasma membrane while IP3 diffuses freely in the cytosol and binds to its cognate receptor, IP3R. Each of these two products not only initiates a signaling cascade of its own but can also work cooperatively to achieve further protein activation (Figure 7). Protein kinase C (PKC) is recruited to the plasma membrane by cooperatively binding DAG and Ca2+ and is maintained there in its active state by membrane-bound receptor for activated protein kinase C proteins (e.g., receptor for activated C kinases (RACKs)). For excellent reviews on neural PKC signaling, see [54, 59]. Moreover, active IP3R results in further cytosolic increases in Ca2+ through the release of intracellular Ca2+ stores from the endoplasmic reticulum (ER). PKC activation plays a role in receptor desensitization through phosphorylation of mGluR1a itself, a critical mechanism allowing for return to resting membrane potential after action potential propagation [60].
In addition to coupling of mGluR1 to IP3 and to NMDARs, Homer1 proteins also appear to couple active mGluR1 to PI3K, initiating signaling which has been shown to have potent effects on cell survival, metabolism, proliferation and tumorigenesis/cancer progression [61, 62]. Activated PI3K leads to the accumulation of phosphoinsitol 3,4,5-triphosphate (PIP3) and consequent recruitment of pleckstrin homology (PH) domain-containing proteins such as Akt1. Native Akt1 is recruited to the membrane via its PH domain where it is subsequently phosphorylated first at serine 473 by mammalian Target of rapamycin complex 2 (mTorC2) and this event triggers a second phosphorylation at threonine 308 by phosphoinositide dependent kinase 1 (PDK1) [63]. Fully active Akt1 participates in a number of important downstream signaling events that certainly occur in neurons but are not limited to this cell type (reviewed in [64, 65]).
Akt1 and Akt2 activation by mGluR1 is not only neuroprotective but is also pro-proliferation in many neuronal cell types [61, 66, 67]. It should be noted, however, that activation of mGluR1 is also considered a pro-apoptosis signal in subsets of neural cells and in certain experimental paradigms [68] (Reviewed in [69]). Therefore, the link between survival and mGluR1 signaling may be complex. As a survival signal, Akt1 can phosphorylate Bcl2-associated death protein (BAD) resulting in BAD's dissociation from the Bcl-2/Bcl-X complex and therefore leading to its anti-apoptotic and cell survival activities [70, 71]. In addition, nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) activation following Akt1-mediated phosphorylation of IκB kinase (IKK) results in active transcription of survival and growth promoting genes [72, 73]. Further, phosphorylation of mTorC1 complex members by Akt1 results in increased translation of mRNAs required for cell growth [74] (Figure 7). Therefore, mGluR1 activation is responsible for regulating cell cycle and proliferation in many cell types. Glu signaling through NFκB and IκB kinase (IKK) has been shown to play a role in many neuronal processes including learning, memory and other processes where synaptic remodeling and plasticity are critical [75]. In addition, NFκB target genes are implicated in the growth of dendritic spines and other neural processes [76, 77]. Interestingly, not only is NFκB activated by this mechanism, but it can also be phosphorylated by PKC downstream of Gq signaling through PLC.
Intracellular calcium signaling has been well-studied for its role in neural transmission. Active GluRs (both mGluR and iGluR families) stimulate release of intracellular calcium stores via multiple previously-discussed mechanisms. In signal transduction, calcium can act as either second messenger (when influx or release is downstream of G-protein signaling) or as direct effector of enzyme activity (where influx occurs as a direct or indirect result of ion-channel opening). As outlined above, Ca2+ has an established role in PLC signaling [4, 78]. Further, Ca2+ binds and activates calmodulin (CaM), the main second messenger that transduces Ca2+-related signals [79]. The binding of 4 Ca2+ molecules in CaM's 4 EF-hand motifs results in the activation of Ca2+/CaM-dependent kinases (CaMK) and subsequent signaling has effects on learning and memory and long-term potentiation of action potentials (Figure 7) [4, 78].
G-protein independent signaling mechanisms involving mGluRs have been documented in rodents [54, 56]. Indeed, an additional consequence of active mGluRs appears to be the activation of Src tyrosine kinase and downstream messengers such as ERK [56]. The functional role for G-protein independent glutamatergic signaling clearly involves long-term potentiation in the CNS, since certain tyrosine kinase inhibitors have negative effects on this process in rats [54, 56, 80]. Studies have also demonstrated that activation of ERK/MAPK signaling in the CNS is also critical for synaptic plasticity and memory formation - two processes that require long-term potentiation [56, 81, 82].
Canonical neural pathways involving mGluRs have been well-established by a large body of literature (see [4] for example). However, other functions for mGluR1s are suggested by work in in vitro systems. First, mGluR1a expressed in baby hamster kidney (BHK) cells inhibits adenylate cyclase (AC) activity via signaling through Gα1/o, suggesting that mGluR1a can couple to inhibitory G proteins [83]. In contrast, mGluR1a in Chinese hamster ovary (CHO) cells does not affect AC or appear to couple with Gα1/o [84]. Several overexpression models including CHO, Human embryonic kidney (HEK 293) and Xenopus oocytes demonstrated that mGluR1a can couple with Gαs to stimulate cAMP production [30, 85, 86]. Therefore, there is no established rule for which G proteins mGluR1s couple with - only that in the nervous system, they most commonly couple with G protein complex q and more specifically, Gα11.
mGluR1 signaling in malignant diseases
A large body of work has linked mGluRs to the development and progression of cancer. Rodent mGluR1 expression is sufficient for cellular transformation of melanocytes in culture, indicating that mGluR genes can function as oncogenes [87, 88]. Furthermore, recent work has shown that not only is Glu a serum biomarker for prostate cancer aggressiveness, but also that mGluR1 is overexpressed in prostate tumors [89]. A detailed review on the involvement of mGluRs in malignant disease has been recently published by our group [90].
In addition to the described role for mGluR signaling in normal nervous system function, glutamatergic signaling through the mGluR family has been implicated in NDDs and learning and memory disorders [91]. For example, in severe autism and Alzeimer's disease, secretion of amyloid precursor protein-α (sAPPα) is a known contributor to disease severity [92, 93]. Many reports have postulated that targeting mGluR signaling may provide symptomatic relief from Alzheimer's and autism [90, 91, 94]. The mechanism underlying this phenomenon has not been documented as of this review, but it clearly involves antagonizing or inhibition of the PI3K/AKT/mTOR pathway which is a known target of active mGluR signaling [91].
Concluding Remarks
Decades of studies have clearly established that glutamatergic signaling is critical for normal nervous system function. Much of the research done to characterize this complex network of receptors and second messenger systems has been performed using rodent models. This has led to a critical lack of information on the human orthologues of the mGluR family - a fact which may have far-reaching impacts on drug discovery for NDDs and other nervous system disorders as well as for anti-cancer therapies. For example, it is clear from an analysis of the literature on the crystal structure of the mGluR proteins that much of this work has been done on the rodent orthologues. In fact, when analyzing publications on this subject, a protein BLAST search is often necessary to determine the species of origin for the currently discussed peptides or proteins. The exact amino acid sequence of the human mGluR that are involved in the disulfide bridging between members of the receptor dimer are not known and are inferred from these studies. Although it is true that in the majority of cases, the information obtained from rodent crystallography may be applicable to the human proteins, however, without mutational analysis, the question will remain. Furthermore, no studies were found that provide an analysis of the rodent and human mGluR proteins expressed in the same cell line to allow a comparison between the orthologues in terms of expression level and function. Hopefully, future work will address these issues and the field will gain insight into exactly how identical the rodent and human proteins are in terms of function and therapeutic targeting potential.
Acknowledgements
This work was supported by Roswell Park Cancer Institute and National Cancer Institute (NCI) grant #P30 CA016056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Cancer Institute.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kelly A, Stanley CA. Disorders of glutamate metabolism. Mental retardation and developmental disabilities research reviews. 2001;7:287-95
2. Alix JJ, Domingues AM. White matter synapses: form, function, and dysfunction. Neurology. 2011;76:397-404
3. Fairman WA, Amara SG. Functional diversity of excitatory amino acid transporters: ion channel and transport modes. The American journal of physiology. 1999;277:F481-6
4. Ribeiro FM, Paquet M, Cregan SP, Ferguson SS. Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS & neurological disorders drug targets. 2010;9:574-95
5. Prickett TD, Samuels Y. Molecular pathways: dysregulated glutamatergic signaling pathways in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:4240-6
6. Julio-Pieper M, Flor PJ, Dinan TG, Cryan JF. Exciting times beyond the brain: metabotropic glutamate receptors in peripheral and non-neural tissues. Pharmacological reviews. 2011;63:35-58
7. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK. et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacological reviews. 2010;62:405-96
8. Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annual review of pharmacology and toxicology. 1997;37:205-37
9. Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochimica et biophysica acta. 2012;1822:675-84
10. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta pharmacologica Sinica. 2009;30:379-87
11. Jaiswal MK, Zech WD, Goos M, Leutbecher C, Ferri A, Zippelius A. et al. Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC neuroscience. 2009;10:64
12. Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. Journal of neurotrauma. 2004;21:754-74
13. Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. European journal of pharmacology. 2013;698:6-18
14. Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R. et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. Journal of cellular physiology. 2011;226:2484-93
15. Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. The Biochemical journal. 2001;359:465-84
16. Beqollari D, Kammermeier PJ. Venus fly trap domain of mGluR1 functions as a dominant negative against group I mGluR signaling. Journal of neurophysiology. 2010;104:439-48
17. Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3759-64
18. Seebahn A, Sticht H, Enz R. Expression, purification, and structural analysis of intracellular C-termini from metabotropic glutamate receptors. Methods in enzymology. 2013;520:257-79
19. Shiraishi-Yamaguchi Y, Furuichi T. The Homer family proteins. Genome biology. 2007;8:206
20. Brown E, Quinn S, Vassilev P. The Plasma Membrane Calcium Sensor. In: Carafoli E, Klee CB, editors. Calcium as a Cellular Regulator. Oxford, NY: Oxford University Press. 1993:295-310
21. Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Current drug targets. 2007;8:169-84
22. Magno AL, Ward BK, Ratajczak T. The calcium-sensing receptor: a molecular perspective. Endocrine reviews. 2011;32:3-30
23. Kroeze WK, Sheffler DJ, Roth BL. G-protein-coupled receptors at a glance. Journal of cell science. 2003;116:4867-9
24. Romano C, Yang WL, O'Malley KL. Metabotropic glutamate receptor 5 is a disulfide-linked dimer. The Journal of biological chemistry. 1996;271:28612-6
25. El Moustaine D, Granier S, Doumazane E, Scholler P, Rahmeh R, Bron P. et al. Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16342-7
26. Iwazumi T. Myofibril tension fluctuations and molecular mechanisms of contraction. Advances in experimental medicine and biology. 1988;226:595-608
27. Lampinen M, Pentikainen O, Johnson MS, Keinanen K. AMPA receptors and bacterial periplasmic amino acid-binding proteins share the ionic mechanism of ligand recognition. The EMBO journal. 1998;17:4704-11
28. Kubo Y, Miyashita T, Murata Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science. 1998;279:1722-5
29. Jiang Y, Huang Y, Wong HC, Zhou Y, Wang X, Yang J. et al. Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1{alpha} (mGluR1{alpha}) that controls receptor activation. The Journal of biological chemistry. 2010;285:33463-74
30. Francesconi A, Duvoisin RM. Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. The Journal of biological chemistry. 1998;273:5615-24
31. Pin JP, Joly C, Heinemann SF, Bockaert J. Domains involved in the specificity of G protein activation in phospholipase C-coupled metabotropic glutamate receptors. The EMBO journal. 1994;13:342-8
32. Gomeza J, Joly C, Kuhn R, Knopfel T, Bockaert J, Pin JP. The second intracellular loop of metabotropic glutamate receptor 1 cooperates with the other intracellular domains to control coupling to G-proteins. The Journal of biological chemistry. 1996;271:2199-205
33. Pin JP, Gomeza J, Joly C, Bockaert J. The metabotropic glutamate receptors: their second intracellular loop plays a critical role in the G-protein coupling specificity. Biochemical Society transactions. 1995;23:91-6
34. Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annual review of pharmacology and toxicology. 2010;50:295-322
35. Mateos JM, Benitez R, Elezgarai I, Azkue JJ, Lazaro E, Osorio A. et al. Immunolocalization of the mGluR1b splice variant of the metabotropic glutamate receptor 1 at parallel fiber-Purkinje cell synapses in the rat cerebellar cortex. Journal of neurochemistry. 2000;74:1301-9
36. Berthele A, Laurie DJ, Platzer S, Zieglgansberger W, Tolle TR, Sommer B. Differential expression of rat and human type I metabotropic glutamate receptor splice variant messenger RNAs. Neuroscience. 1998;85:733-49
37. Hiltscher R, Seuwen K, Boddeke HW, Sommer B, Laurie DJ. Functional coupling of human metabotropic glutamate receptor hmGlu1d: comparison to splice variants hmGlu1a and -1b. Neuropharmacology. 1998;37:827-37
38. Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O'Hara PJ, Mulvihill ER. et al. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991;252:1318-21
39. Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760-5
40. Desai MA, Burnett JP, Mayne NG, Schoepp DD. Cloning and expression of a human metabotropic glutamate receptor 1 alpha: enhanced coupling on co-transfection with a glutamate transporter. Molecular pharmacology. 1995;48:648-57
41. Laurie DJ, Boddeke HW, Hiltscher R, Sommer B. HmGlu1d, a novel splice variant of the human type I metabotropic glutamate receptor. European journal of pharmacology. 1996;296:R1-R3
42. Huang X, Miller M. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337-57
43. Hermans E, Challiss RA, Nahorski SR. Effects of varying the expression level of recombinant human mGlu1alpha receptors on the pharmacological properties of agonists and antagonists. British journal of pharmacology. 1999;126:873-82
44. Hermans E, Young KW, Challiss RA, Nahorski SR. Effects of human type 1alpha metabotropic glutamate receptor expression level on phosphoinositide and Ca2+ signalling in an inducible cell expression system. Journal of neurochemistry. 1998;70:1772-5
45. Esseltine JL, Willard MD, Wulur IH, Lajiness ME, Barber TD, Ferguson SS. Somatic mutations in GRM1 in cancer alter metabotropic glutamate receptor 1 intracellular localization and signaling. Molecular pharmacology. 2013;83:770-80
46. Costantino G, Pellicciari R. Homology modeling of metabotropic glutamate receptors. (mGluRs) structural motifs affecting binding modes and pharmacological profile of mGluR1 agonists and competitive antagonists. Journal of medicinal chemistry. 1996;39:3998-4006
47. Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1-26
48. Beneken J, Tu JC, Xiao B, Nuriya M, Yuan JP, Worley PF. et al. Structure of the Homer EVH1 domain-peptide complex reveals a new twist in polyproline recognition. Neuron. 2000;26:143-54
49. Luo P, Li X, Fei Z, Poon W. Scaffold protein Homer 1: implications for neurological diseases. Neurochemistry international. 2012;61:731-8
50. Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M. et al. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717-26
51. Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD. et al. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron. 1998;21:707-16
52. Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends in neurosciences. 2000;23:80-8
53. Wang JQ, Fibuch EE, Mao L. Regulation of mitogen-activated protein kinases by glutamate receptors. Journal of neurochemistry. 2007;100:1-11
54. Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735-40
55. Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H. et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. The Journal of clinical investigation. 2008;118:3051-64
56. Heuss C, Scanziani M, Gahwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nature neuroscience. 1999;2:1070-7
57. Kim CH, Braud S, Isaac JT, Roche KW. Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on Serine 839 regulates Ca2+ oscillations. The Journal of biological chemistry. 2005;280:25409-15
58. Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P. et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583-92
59. Ramakers GM, Pasinelli P, Hens JJ, Gispen WH, De Graan PN. Protein kinase C in synaptic plasticity: changes in the in situ phosphorylation state of identified pre- and postsynaptic substrates. Progress in neuro-psychopharmacology & biological psychiatry. 1997;21:455-86
60. Dhami GK, Ferguson SS. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacology & therapeutics. 2006;111:260-71
61. Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA. et al. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nature neuroscience. 2003;6:1153-61
62. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature reviews Molecular cell biology. 2012;13:283-96
63. Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends in biochemical sciences. 2009;34:620-7
64. Liu L, Parent CA. Review series: TOR kinase complexes and cell migration. The Journal of cell biology. 2011;194:815-24
65. Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chinese journal of cancer. 2012;31:8-18
66. Chong ZZ, Kang JQ, Maiese K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histology and histopathology. 2003;18:173-89
67. Allen JW, Knoblach SM, Faden AI. Activation of group I metabotropic glutamate receptors reduces neuronal apoptosis but increases necrotic cell death in vitro. Cell death and differentiation. 2000;7:470-6
68. Pshenichkin S, Dolinska M, Klauzinska M, Luchenko V, Grajkowska E, Wroblewski JT. Dual neurotoxic and neuroprotective role of metabotropic glutamate receptor 1 in conditions of trophic deprivation - possible role as a dependence receptor. Neuropharmacology. 2008;55:500-8
69. Maiese K, Chong ZZ, Shang YC, Hou J. Therapeutic promise and principles: metabotropic glutamate receptors. Oxidative medicine and cellular longevity. 2008;1:1-14
70. Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: the cell-signalling perspective. Drug discovery today. 2011;16:485-94
71. Schelman WR, Andres RD, Sipe KJ, Kang E, Weyhenmeyer JA. Glutamate mediates cell death and increases the Bax to Bcl-2 ratio in a differentiated neuronal cell line. Brain research Molecular brain research. 2004;128:160-9
72. Mantovani A. Molecular pathways linking inflammation and cancer. Current molecular medicine. 2010;10:369-73
73. Mantovani A, Garlanda C, Allavena P. Molecular pathways and targets in cancer-related inflammation. Annals of medicine. 2010;42:161-70
74. Alsina FC, Ledda F, Paratcha G. New insights into the control of neurotrophic growth factor receptor signaling: implications for nervous system development and repair. Journal of neurochemistry. 2012;123:652-61
75. Faissner A, Heck N, Dobbertin A, Garwood J. DSD-1-Proteoglycan/Phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. Advances in experimental medicine and biology. 2006;557:25-53
76. Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK. A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:5414-25
77. Gutierrez H, Hale VA, Dolcet X, Davies A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development. 2005;132:1713-26
78. Tabata T, Kano M. Calcium dependence of native metabotropic glutamate receptor signaling in central neurons. Molecular neurobiology. 2004;29:261-70
79. Stevens FC. Calmodulin: an introduction. Canadian journal of biochemistry and cell biology = Revue canadienne de biochimie et biologie cellulaire. 1983;61:906-10
80. Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121-34
81. Pelkey KA, Topolnik L, Lacaille JC, McBain CJ. Compartmentalized Ca(2+) channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52:497-510
82. Topolnik L, Azzi M, Morin F, Kougioumoutzakis A, Lacaille JC. mGluR1/5 subtype-specific calcium signalling and induction of long-term potentiation in rat hippocampal oriens/alveus interneurones. The Journal of physiology. 2006;575:115-31
83. Hermans E, Saunders R, Selkirk JV, Mistry R, Nahorski SR, Challiss RA. Complex involvement of pertussis toxin-sensitive G proteins in the regulation of type 1alpha metabotropic glutamate receptor signaling in baby hamster kidney cells. Molecular pharmacology. 2000;58:352-60
84. Selkirk JV, Price GW, Nahorski SR, Challiss RA. Cell type-specific differences in the coupling of recombinant mGlu1alpha receptors to endogenous G protein sub-populations. Neuropharmacology. 2001;40:645-56
85. Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8:757-65
86. Thomsen C. Metabotropic glutamate receptor subtype 1A activates adenylate cyclase when expressed in baby hamster kidney cells. Progress in neuro-psychopharmacology & biological psychiatry. 1996;20:709-26
87. Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nature medicine. 2001;7:1010-5
88. Teh JL, Chen S. Glutamatergic signaling in cellular transformation. Pigment cell & melanoma research. 2012;25:331-42
89. Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D. et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:5888-901
90. Willard SS, Koochekpour s. Glutamate signaling in benign and malignant disorders: Current status, future perspectives, and therapeutic implications. Int J Biol Sci. 2013;9(7):728-742 doi:10.7150/ijbs.6475
91. Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK. Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology. 2011;76:1344-52
92. Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA. et al. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. Journal of child neurology. 2006;21:444-9
93. Bailey AR, Giunta BN, Obregon D, Nikolic WV, Tian J, Sanberg CD. et al. Peripheral biomarkers in Autism: secreted amyloid precursor protein-alpha as a probable key player in early diagnosis. International journal of clinical and experimental medicine. 2008;1:338-44
94. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in neurosciences. 2004;27:370-7
Author contact
![]() Corresponding author: Dr. Shahriar Koochekpour, Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA, Telephone: 716-845-3345; Fax: 716-845-1698; Email: Shahriar.Koochekpourorg.
Corresponding author: Dr. Shahriar Koochekpour, Departments of Cancer Genetics and Urology, Center for Genetics and Pharmacology, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA, Telephone: 716-845-3345; Fax: 716-845-1698; Email: Shahriar.Koochekpourorg.