Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(9):980-988. doi:10.7150/ijbs.7251 This issue Cite
Research Paper
Diabetes Inhibits Cerebral Ischemia-Induced Astrocyte Activation - an Observation in the Cingulate Cortex
Li Jing1, Li Mai1, Jian-Zhong Zhang1 ![]() , Jian-Gang Wang1, Yue Chang1, Jian-Da Dong1, Feng-Ying Guo1, P. Andy Li2,3
, Jian-Gang Wang1, Yue Chang1, Jian-Da Dong1, Feng-Ying Guo1, P. Andy Li2,3 ![]()
1. Department of Pathology, Ningxia Medical University; Ningxia Key Laboratory of Cerebrocranial Diseases, Incubation Base of National Key Laboratory, Yinchuan, Ningxia, P. R. China.
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technological Enterprise (BRITE), North Carolina Central University, Durham, North Carolina, USA.
3. Department of Neurology, the General Hospital of Ningxia Medical University, Ningxia Key Laboratory of Cerebrocranial Diseases, Incubation Base of National Key Laboratory, Yinchuan, Ningxia, P. R. China.
Received 2013-7-24; Accepted 2013-9-12; Published 2013-9-25
Abstract
The objective of this study was to study the effect of diabetic hyperglycemia on astrocytes after forebrain ischemia. Streptozotocin (STZ)-injected hyperglycemic and vehicle-injected normoglycemic rats were subjected to 15 minutes of forebrain ischemia. The brains were harvested in sham-operated controls and in animals with 1 and 6 h of recirculation following ischemia. Brain damage was accessed by haematoxylin and eosin (H&E) staining, cleaved caspase-3 immunohistochemistry and TdT-mediated-dUTP nick end labeling (TUNEL). Anti-GFAP antibody was employed to study astrocytes. The results showed that the 15-minute ischemia caused neuronal death after 1 and 6 h of reperfusion as revealed by increased numbers of karyopyknotic cells, edema, TUNEL-positive and active caspase-3-positive cells. Ischemia also activated astrocytes in the cingulated cortex as reflected by astrocyte stomata hypertrophy, elongated dendrites and increases in the number of dendrites, and immunoreactivity of GFAP. Diabetic hyperglycemia further enhanced neuronal death and suppressed ischemia-induced astrocyte activation. Further, diabetes-damaged astrocytes have increased withdrawal of the astrocyte end-foot from the cerebral blood vessel wall. It is concluded that diabetes-induced suppression and damages to astrocytes may contribute to its detrimental effects on recovery from cerebral ischemia.
Keywords: astrocyte, blood-brain barrier, cleaved caspase-3, cerebral ischemia, diabetes, hyperglycemia, TUNEL.
Introduction
It is well known that hyperglycemia induced by diabetes mellitus (DM), glucose infusion or stress worsens the prognosis of stroke patients by increasing mortality and morbidity [1-6 ]. It is reported that hyperglycemia presented in 28% of stroke patients without a previous known history of DM [3]. Several studies have demonstrated that hyperglycemic ischemia induces more production of oxidative stress markers, cerebral energy metabolism disturbance, neuronal death, and neurologic behavioral impairment than those observed following normoglycemic ischemia and reperfusion [3-8]. While injury to, and death of, neurons has been the focus of ischemic brain injury research, evidence suggests that astrocytes also undergo dysfunction and delayed death after global cerebral ischemia [9-11].
Astroglial cells are the most abundant cells in the central nervous system (CNS) and are believed to play a major role in maintaining the normal functions of the brain and spinal cord. Although never proved directly, astrocytes, the most dominant astroglial cells, are thought to exert a neuroprotective effect in stroke by shielding neurons from oxidative stress [10]. Astrocytes play a major role in transport and metabolism of a range of molecules, particularly those utilized in nutrition, cell-to-cell signaling, and neurotransmission. A failure to accomplish these functions may constitute a major pathogenic component in stroke and other CNS pathologies [10]. When malfunction occurs in astrocytes, extracellular potassium and glutamate rise rapidly and neurons become highly vulnerable to ischemic injury and to acidosis that inevitably occurs during ischemia due to anaerobic glycolysis [10].
Astrocytes become active (reactive) in response to CNS injuries, including brain infection, inflammation, ischemia, trauma and neurodegenerative diseases [9-11]. Reactive astrocytes undergo changes in morphology and in their expression of a wide range of molecules, including glial fibrillary acidic protein (GFAP) [9-11]. Increased expression of GFAP, enlarged cell body, and increased number and length of dendrites are hallmarks of astrocyte activation. We have previously reported that 30 min of middle cerebral artery occlusion activates astrocytes and causes astrocyte infiltration into ischemic penumbra and core areas; whereas diabetes inhibits such ischemia-induced astrocyte activation and enhances astrocyte cell death [12]. The objective of this study was to study the influence of diabetic hyperglycemia on astrocytes following global cerebral ischemia. Diabetic hyperglycemia was induced in rats by injection of streptozotocin (STZ). Fifteen minutes of forebrain ischemia was induced by clamping bilateral common carotid arteries and hypotension achieved by blood withdrawal [13]. After 1 and 6 h of recirculation, brain samples were processed for Haematoxylin and eosin (H&E) staining, TdT-mediated-dUTP nick end labeling (TUNEL) staining, and immunohistochemistry for cleaved caspase-3 and GFAP. Cingulated cortex, a structure with distinctive damage patterns for normoglycemic and hyperglycemic ischemia, was selected as focus of this study. The results showed that diabetic hyperglycemia, in addition to augmenting neuronal damage, inhibited ischemia-induced astrocyte activation, reduced the number of GFAP-positive astrocytes and impaired the integrity of the blood-brain barrier (BBB).
Materials and Methods
Animals and reagents
Male Sprague-Dawley rats weighting 240 to 350g were provided by the Experimental Animal Center at Ningxia Medical University for in vivo experiments. All animal use and procedures were in strict accordance with the Chinese Laboratory Animal Use Regulations. Efforts were made to minimize animal stress and to reduce number of rats used for this study. Reagents were purchased from Boster Biotechnology Co (Wuhan, China), including monoclonal anti-GFAP antibody (Cell Signaling), horseradish peroxidase-conjugated anti-mouse secondary antibody (Sigma), the TdT-mediated-dUTP nick end labeling kit (Zymed) and STZ (Calbiochem).
Animal treatments
The rats were fasted overnight, injected intra-peritoneally with STZ (55 mg/kg) freshly dissolved in 0.1 M citrate buffered saline (pH 4.5). Age-matched rats receiving the same volume of 0.1 M citrate-buffered saline served as normoglycemic controls. Diabetes was confirmed by measurements of blood glucose levels 2 days after STZ injection using an OneTouch glucometer. Animals with blood glucose level higher than 16.7 mmol/L were designated the diabetic group. Cerebral ischemia was induced 7 days after STZ or citrate buffer injection.
Ischemiic model and experimental groups
Forebrain ischemia was introduced under anesthesia (4% sodium pentobarbital, 30 mg/kg) by bilateral clamping of the common carotid arteries and exsanguination through a cannulation into the right jugular vein for 15 minutes, maintaining blood pressure at 40 to 50 mmHg and yielding an isoelectric EEG. Circulation was resumed by re-infusing the shed blood and by releasing the ligatures placed around the carotid arteries [13].
Rats were randomly divided into four groups, 1) sham-operated, non-diabetic (normoglycemic) control group (n=5); 2) sham-operated diabetic (hyperglycemic) control group (n=5); 3) non-diabetic, normoglycemic ischemia (n=10); 4) diabetic hyperglycemic ischemia (n=10). Upon 1 and 6 h of reperfusion after 15 minutes ischemia, animals were reanesthetized, transcardially perfusion-fixed with 4% PBS-buffered paraformaldehyde, and then post-fixed in the same solution for 48 h. A 2 mm thick coronal block, at a level of -0.3 mm to the bregma, was obtained and embedded in paraffin. The brain samples were sectioned at 5 μm intervals and used for histopathology, TUNEL, immunohistochemistry and immunofluorescence studies.
Histology and TUNEL staining
H&E staining was used to observe cell morphology. Damaged neurons were defined as karyolysis in which reticular formation of chromatin and nuclear fading were evident, with karyopyknosis and void space surround the nucleus. Tissue edema is defined as increased small vacuoles in tissue. The numbers of swollen and shrunk neurons were counted and the data were presented as number per high power filed (HPF, 400X).
In situ detection of DNA fragmentation was performed using TUNEL staining according to the manufacturer's instruction. In brief, after being washed 3 times in Tris-HCl (pH 7.7), sections were treated with 2% H2O2 for 10 minutes at room temperature to quench endogenous peroxidase activity. The sections were then incubated with terminal deoxynucleotidyl transferase enzyme solution at 37°C for 1 h. The sections were dipped in 300 mmol/L NaCl and 30 mmol/L sodium citrate for 15 minutes at room temperature to terminate the reaction. The sections were washed 3 times in Tris-HCl (pH 7.7) and subsequently blocked with PBS (pH 7.4) containing 10% normal goat serum and 0.3% Triton X-100. Biotinylated-16-dUTP was visualized by the ABC (avidin-biotin complex) method with 0.05% 3,3'-diaminobenzidine (DAB) tetrahydrochloride and 0.005% H2O2. Number of damaged neurons that were detected by H&E staining, TUNEL staining and cleaved caspase-3 immunolabeling was counted in 2 HPFs per slice in the cingulated cortex and the data were presented as number per HPF.
Immunohistochemistry and immunofluorescent labeling
For immunostaining, sections were treated with 3% H2O2 for 10 minutes at room temperature to quench endogenous peroxidase activity. The sections were submerged in citrate solution and heated in a microwave for antigen retrieval before nonspecific binding sites were blocked by 5% bovine serum albumin (BSA) in PBS/0.2% TX-100 for 30 minutes. The sections were incubated with anti-GFAP (1:400) or anti-cleaved caspase-3 (1:200) antibodies at 4°C overnight. The sections were washed and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:300). The reaction was visualized with DAB and the sections were counter-stained with haematoxylin. The sections were coverslipped and analyzed under a light microscope.
For GFAP immunofluorescent labeling, the sections were blocked with 3% bovine serum albumin and incubated overnight with primary antibody against GFAP at 1:100. The sections were then incubated with TRITC labeled anti- rabbit secondary antibody (1:100). Vectashield Hardset Mounting Media (H-1200, Vector) containing 4', 6 - diamidino- 2 - phenylindole (DAPI) was used to label nuclei. The sections were examined using a fluorescence confocal scanning microscope (Nikon Eclipse C1).
The number of dendrites per astrocyte was counted in 50 astrocytes per animal test group and presented as the mean value per astrocyte. The number of GFAP positive cells was counted in 3 sections per animal and presented as number of positive cells per HPF. For GFAP density measurement, visual images (400×) were acquired in each slice from the cingulate cortex. The images were processed using an IPP image-analysis system to analyze the optical density (OD) value. The mean optical density [integral optical density (IOD)/SUM AREA] was used for statistical analysis.
Statistical analysis
The data for figures 1G, 2G, 3G, 4G and 5D were presented as mean±SEM. For these parametric data, comparison of recirculation endpoints with sham-operated controls within the diabetic group or non-diabetic group were made by ANOVA followed by post hoc Scheffe's test. Comparison between diabetic and non-diabetic animals at an identical reperfusion endpoint was made by the Student t test. For non-parametric data in Figure 4H, comparison was made by Mann-Whitney's U test. A p value equal or less than 0.05 was considered statistically significant.
Representative H&E-stained photomicrographs demonstrating changes in the cingulate cortex. A&D, sham-operated controls, neurons are healthy; B&C, non-diabetic ischemic sample at 1 and 6 h reperfusion. Few pyknotic, swollen neurons (filled arrows) and glial cells were detected in the ischemic brain. E&F, diabetic animals at 1 and 6 h of reperfusion. Number of damaged neurons as well as tissue swelling are enhanced. Inserts are enlarged neurons. G, Mean numbers of damaged neuronal cells/HPF in the cingulate cortex. Filled arrows indicate neurons and open arrows indicate glial cells, non-DM, non-diabetes; DM, diabetes; I/R, ischemia and reperfusion. * P < 0.01 vs. sham control within the same glycemic group and # p<0.05 vs. non-DM samples at an identical time point. Bar = 50 µm.
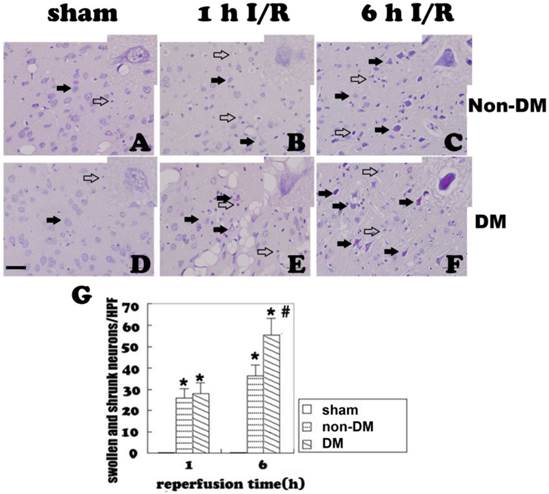
TUNEL staining in the cingulate cortex at 1 and 6 h of reperfusion. A&D, sham operated non-diabetic and diabetic controls. No obvious TUNEL staining was detected. B&C, non-diabetic ischemia at 1 h and 6 h reperfusion. TUNEL-positive cells were detected after 1 h and further increased after 6 h of recirculation (filled arrows). E&F, diabetic animals at 1 h and 6 h of reperfusion. Number of TUNEL-positive neurons significantly increased comparing to non-diabetic counterparts at identical reperfusion endpoints (filled arrows). G, mean numbers of TUNEL positively stained neuronal cells/HPF. Open arrows indicate TUNEL negative and filled arrows denote TUNEL positive cells. * p<0.05 vs. sham group; # p<0.05 vs. non-DM samples at an identical time point. Haematoxylin counterstain. Bar = 10 μm.
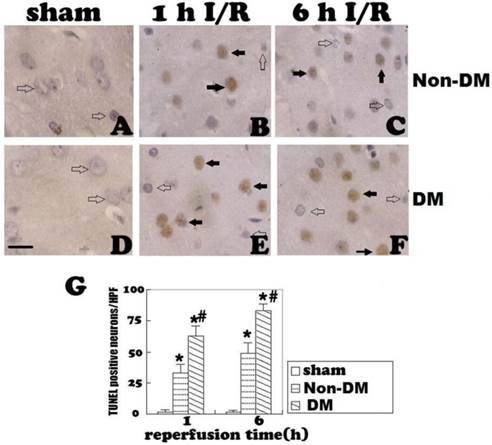
Cleaved caspase-3 immunostaining in the cingulate cortex at 1 and 6 h of reperfusion. A&D, non-diabetic and diabetic controls. No caspase-3 immunoreactivity was detected in the cingulate cortex. B, non-diabetic rats with 1 h of reperfusion. Close to 25 caspase-3 positive neurons were detected per HPF per section. C, non-diabetic rats with 6 h of reperfusion. Number of caspase-3 reactive neurons further increased. E&F, diabetic rats with 1 and 6 h of reperfusion. Number of caspase-3 positive neurons significantly increased comparing to non-diabetic counterpart. G, a summary of active caspase-3 positive neurons in the each experimental group/HPF. Open arrows indicate normal neurons and filled arrows denote active caspase-3 immunoreactive neurons. * p<0.05 vs. sham; #p <0.05 vs. non-DM samples at an identical endpoint. Haematoxylin counterstain. Bar = 40um.
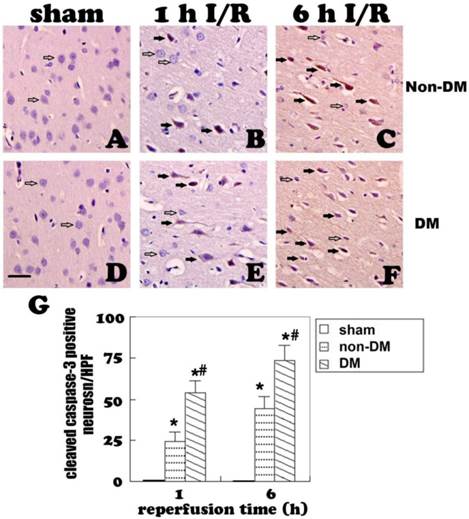
GFAP fluorescent staining (A-C) and immunohistochemistry (D-F) at 1 and 6 h of recovery in the cingulate cortex of non-diabetic animals. A&D, normal astrocyte morphology in sham controls. B,C,E, ischemia plus 1 and 6 h of recovery in non-diabetic rats demonstrating morphology of activated astrocytes. F, activated astrocytes surround a damaged neuron with pyknotic nuclei. G&H, Bar graph showing increases of dendrite branches and of GFAP immunoreactivity (measured by optical density), respectively. Open arrows indicate GFAP positive astrocytes, filled arrows in F indicate damaged neurons. * p<0.05 vs. sham at an identical endpoint. Bar = 10 μm.
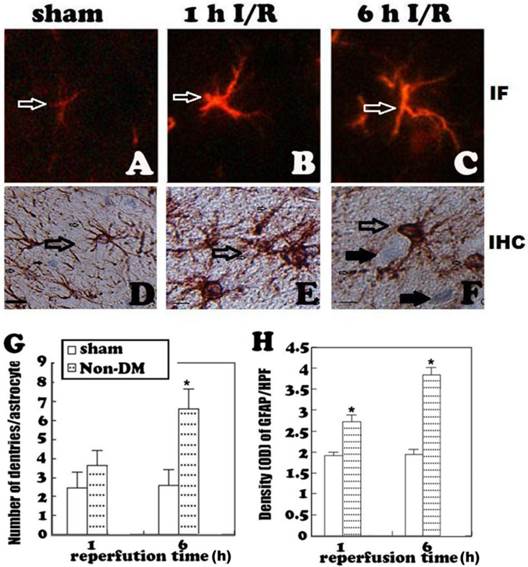
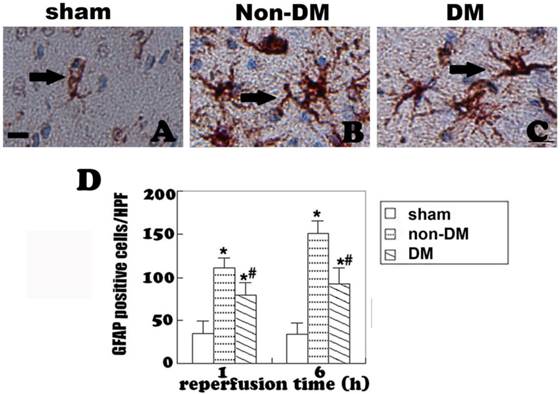
GFAP immunohistochemistry in the cingulate cortex at 1 h and 6 h of reperfusion. A, quiescent astrocytes in sham controls. B, non-diabetic ischemia plus 6 h of recovery. C, Diabetic ischemia plus 6 h of recovery. D: Bar graph summarizes number of GFAP positive astrocytes in each experimental group. Arrows indicate GFAP-positive astrocytes. * p<0.05 vs. sham; #p <0.05 vs. non-DM samples at an identical endpoint. Bar = 10 μm.
Results
Histopathology
H&E staining showed no obvious cell morphologic change in sham-operated controls (Fig 1A & 1D). Mild brain edema and cellular swelling could be detected in the cingulated cortex at 1 h reperfusion after 15 minutes of brain ischemia. Reticulum formation of chromatin and disappearance of the nucleolus were observed in few scattered neurons in the cingulated cortex at this early reperfusion stage (Fig. 1B). A few glial cells were detected around the degenerating neurons. The number of damaged neurons increased at 6 h of reperfusion as did tissue edema. About 20% of the total neurons in the cingulated cortex were observed as having pyknosis (Fig. 1C). In the diabetic group, tissue swelling was evident in the cingulated cortex at 1 h of reperfusion, but the number of damages neurons in the cingulated cortex was not increased compared to the non-diabetic counterpart (Fig. 1E). However, the number of damaged neurons was significantly higher in diabetic animals than that in non-diabetic animals at 6 h of reperfusion (Fig. 1F).
TUNEL staining
Positive TUNEL staining (brown color) suggests DNA fragmentation, which could be the result of either apoptosis or necrosis. TUNEL-positive cells were occasionally detected in non-diabetic and diabetic control animals (Fig. 2A & 2D). In the non-diabetic ischemic group, TUNEL-positive neurons appeared after 1 h of reperfusion following 15 minutes of ischemia in the cingulate cortex. The TUNEL-positive neurons increased along with the reperfusion time. At 6 h of reperfusion, a large of number of TUNEL stained cells were detected (Fig. 2B & 2C). Comparing to the non-diabetic animals, diabetes significantly increased the number of TUNEL-positive cells at 1 h and 6 h of reperfusion (p<0.05, Fig. 2E & 2F). A summary bar graph is given in Fig. 2G.
Cleaved caspase-3 staining
Cleaved caspase-3 immunoreaction was observed in both the nucleus and cytoplasm of diabetic and non-diabetic cells. No caspase-3-positive cells were detected in the cingulate cortex of the sham groups (Fig. 3A & 3D). In non-diabetic ischemic group, cleaved caspase-3 positive neurons were detected at 1 h of reperfusion (p<0.05 vs. sham, Fig. 3B). The positive neurons increased along with the reperfusion time (Fig. 3C). Compared with the non-diabetic group, the number of cleaved caspase-3 positive cells was significantly increased in diabetic animals at 1 and 6 h of reperfusion following 15-minute brain ischemia (p<0.05, Fig. 3E & 3F).
Changes of astrocytes
Astrocytes were revealed by GFAP immunohistochemistry and active astrocytes were defined as GFAP-positively stained cells that undergo stomata hypertrophy, dendrite extension, with an increased number of dendrite processes and increased GFAP immunoreactivity. Astrocytes with slim bodies and slender processes were detected in the sham controls (Fig. 4A & 4D). Fifteen minutes of cerebral ischemia and 1 h of reperfusion resulted in enlargement of the astrocyte cell body, increases in the number of astrocytes and astrocyte dendrites, and immunoreactivity of GFAP in the cingulate cortex (Fig. 4B & 4E). Activation of astrocytes was much more prominent after 6 h of reperfusion (Fig. 4C). It was also noticed that dying neurons were surrounded by activated astrocytes (Fig. 4F). Comparing to non-diabetic animals, diabetic hyperglycemia significantly reduced number of GFAP positively stained astrocytes at 1 and 6 h of recovery following a 15-minute ischemia (Fig 5).
Influence of diabetes on the blood-brain barrier after ischemia
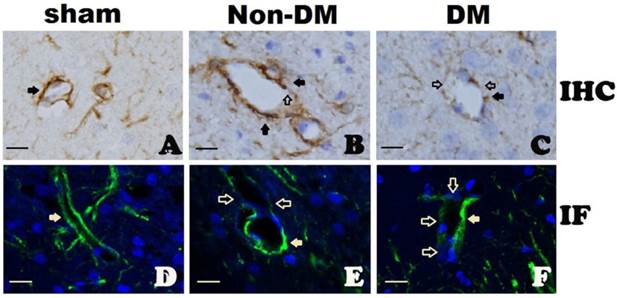
In sham-operated animals with either non-diabetic or diabetic conditions, cerebral blood vessel walls were completely ensheathed by astrocyte end-foot (Fig. 6A & 6D). At 6 h of reperfusion in non-diabetic animals, blood vessels were almost completely ensheathed by astrocyte dendrite processes as shown in Fig. 6B & 6E. However, in diabetic animals, the astrocyte end-foot failed to completely ensheath the vascular walls. Only a small proportion of cerebral blood vessels were encircled by astrocyte end-foot in the diabetic animals after 6 h of reperfusion (Fig. 6C & 6F).
GFAP immunohistochemistry (A-C) and immunofluorescent staining (D-F) in the cingulate cortex at 6 h of reperfusion following 15 minutes of forebrain ischemia. A&D, Complete encircling of blood vessels by astrocyte end-foot in controls. B&E, non-diabetic ischemia plus 6 h of recovery showing incomplete encircling of blood vessels by astrocyte end-foot. C&F, diabetic ischemia plus 6 h recovery showing exacerbated damage to the end-foot. Filled arrows indicate astrocyte end-foot surround blood vessel walls and open arrows indicate lack of astrocytes end-foot on vascular wall. Bar = 10 μm.
Discussion
Hyperglycemia increases neuronal damage caused by transient global or focal cerebral ischemia, particularly increasing neuronal injury in ischemic resistant areas such as cingulated cortex and the hippocampal CA3 and dentate gyrus subregions [1,12,14]. This study observed the morphological changes of the brain at 1 and 6 h of reperfusion after 15 minutes of forebrain ischemia in both non-diabetic normoglycemic and diabetic hyperglycemic rats. In the non-diabetic rats, brain edema, cellular swelling, reticulum formation of chromatin, and disappearance of the nucleolus were observed in the cingulate cortex (Fig. 1-3), dentate gyrus and caudate putamen (data not shown) at 1 h of reperfusion following the ischemia. Neuronal damages were observed after 1 h and slightly increased after 6 h of reperfusion. Diabetic hyperglycemia significantly augmented neuronal death after 6 h reperfusion. The exacerbative effect of diabetes on ischemic brain damage was further confirmed by increased numbers of TUNEL-positive and of caspase-3 positive neurons at 1 and 6 h of reperfusion. We have previously shown that diabetes accelerates and enhances ischemic brain damage induced either by focal ischemia or forebrain ischemia [14-17]. The present study further confirmed the aggravating effects of diabetes on ischemic brain damage. The mechanisms underlying hyperglycemia-enhanced ischemic brain damage may involve tissue acidosis, energy depletion, free radical production, loss of ionic homeostasis, mitochondrial dysfunction, mitochondrial dynamic imbalance, neuroinflammatory processes, and increased activation of autophagy after transient cerebral ischemia [17-22]. All these factors can cause activation of cell death pathways and damage to the BBB and neuroglial cells, eventually inducing increased neuronal death and enhanced brain edema.
As the most abundant type of cells in the CNS, astrocytes participate in releasing neurotrophic factors, limiting the expansion of tissue injury and repairing damaged nerve tissues [9-11]. Astrocytes become activated (reactive) in response to many CNS injuries, including brain infection, inflammation, ischemia, neurodegenerative diseases, trauma and tumor [9-11]. Our previous study demonstrated that 5 min of forebrain ischemia caused astrocyte activation peak at 6 h to 1 day recovery. Diabetes significantly inhibited ischemia-activated astrocytes [21]. Similarly, in a recent study, we observed that 30 min of middle cerebral artery occlusion caused activation of astrocytes, which was observed after 30 min of reperfusion and peaked between 3 h to 1 day after reperfusion in ischemic penumbra and 7 days in ischemic core. Preischemic diabetic hyperglycemia significantly inhibited ischemia-induced astrocyte activation [12]. This study further examined the effects of diabetic ischemia on astrocytes following a moderate global ischemia. The results obtained were consistent with those published previously. Thus, a transient global ischemia of 15 minutes activated astrocytes after 1 h of recovery as reflected by somatic hypertrophy, dendrite elongation, increased number of dendrite processes and increased synthesis of GFAP. The observed activation of astrocytes may be protective by shielding neurons from ischemic brain damage because astrocytes have the ability to engage in spatial buffering, transporting and metabolizing amino acids, glucose, and other key molecules, and upregulating antioxidants and free radical scavengers in the ischemic region [9-11]. We also observed that astrocytes were initially activated in the periventricular zone, the nesting area of astrocytes. Activated astrocytes were then observed in cingulate cortex, corpus callosum and caudate putamen (data not shown). Comparing to the non-diabetic group, diabetes inhibited the ischemia-induced astrocyte activation and proliferation, Suppression of neuroprotective astrocytes by preischemic diabetes may contribute to the detrimental effects of hyperglycemia on ischemic brain damage. Astrocytes are especially sensitive to acidosis and die quickly in acidic and hypoxic conditions [23,24]. Diabetic ischemia enhances tissue acidosis during ischemia/hypoxia due to increased anaerobic glucose metabolism [16]. Other mechanisms underlying the suppression effects of hyperglycemia on astrocyte activation and proliferation remain to be explored.
Astrocytes participate in the formation of and maintaining the integrity of the BBB. We found in this study that the vast majority blood vessels in the brain were completely ensheathed by the end-foot of astrocytes with few exceptions at 6 h of recovery following a 15-minute ischemia in non-diabetic rats. In contrast, diabetic hyperglycemia caused damage to astrocytes at 6 h after reperfusion. As a result, many cerebral blood vessels were only partially ensheathed by astrocyte end-foot. Since astrocyte end-foot is a critical constituent of the BBB, incomplete ensheathment of blood vessel wall by the astrocyte end-foot will inevitably affect the integrity of the BBB. These results are consistent with the findings that hyperglycemic ischemia impairs the BBB [25-27]. We hypothesize that hyperglycemia induced astrocyte damage impairs the integrity of the BBB, which is one of the important pathophysiologies responsible for the overall deteriorative effects of hyperglycemia on ischemic outcome.
In summary, a 15-minute forebrain ischemia caused neuronal death and activated astrocytes in the brain. Diabetic hyperglycemia further enhanced neuronal death, inhibited ischemia-induced activation of astrocytes and caused damage to astrocytes, which likely impairs the integrity of the BBB. These changes may account for the detrimental effects of hyperglycemia on cerebral ischemia.
Acknowledgements
LJ was supported by National Science Foundation of China (81260184) and Ningxia Medical University Research Fund (XT2011009, KF20100-26); JZZ by the National Science Foundation of China (81060105); and PAL by the National Institute of Health (7R01DK075476-06). We thank Dr. Kevin P. Williams for proof reading.
Authors Contributions
Conceived and designed the experiments: LJ, JZZ, PAL. Performed the experiments: LM, JGW, YC, JDD, FYG. Wrote the paper: LJ, JZZ, PAL
Competing Interest
The authors have declared that no competing interest exists.
References
1. Li PA, He Q. Mechanisms of hyperglycemia-enhanced ischemic brain damage. Transl Med Res. 2013;3:1-11
2. Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32:2426-2432
3. Rajesh G, Ajay C, Frederick M, Paresh D. Hyperglycemia, insulin, and acute ischemic stroke: A mechanistic justification for a trial of insulin infusion therapy. Stroke. 2006;37:267-273
4. Balan IS, Fiskum G, Hazelton J, Cotto-Cumba C, Rosenthal RE. Oximetry-guided reoxygenation improves neurological outcome after experimental cardiac arrest. Stroke. 2006;37:3008-3013
5. Cipolla MJ, Godfrey JA. Effect of hyperglycemia on brain penetrating arterioles and cerebral blood flow before and after ischemia/reperfusion. Transl Stroke Res. 2010;1:127-134
6. Stead LG, Gilmore RM, Bellolio MF, Mishra S, Bhagra A, Vaidyanathan L, Decker WW, Brown RD Jr. Hyperglycemia as an independent predictor of worse outcome in non-diabetic patients presenting with acute ischemic stroke. Neurocrit Care. 2009;10:181-186
7. Hannah R, Vitullo L, Cipolla MJ. Preexisting hyperglycemia during stroke is associated with enhanced reactive oxygen species (ROS) production and worsened stroke outcome. FASEB J. 2005;19:A1230
8. Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood brain barrier dysfunction. Stroke. 2007;38:1044-1049
9. Kraig R, Lascola C, Caggiano A. Glial response to brain ischemia. In: (ed.) Kettenmann H, Ransom BR. Neuroglia. New York: Oxford University Press. 1995:964-76
10. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7-35
11. Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Marngnkis N, Nilsson M, Pekny M. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468-81
12. Jing L, He QP, Zhang JZ, Li PA. Temporal profile of astrocytes and changes of oligodendrocyte-based myelin following middle cerebral artery occlusion in diabetic and non-diabetic Rats. Int J Biol Sci. 2013;9:190-199
13. Smith M-L, Kalimo H, Warner DS, Siesjo BK. Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385-401
14. Li PA, Shamloo M, Smith ML, Katsura KI, Siesjo BK. The influence of plasma glucose concentrations on ischemic brain damage is a threshold function. Neurosci Lett. 1994;177:63-65
15. Zhang JZ, Jing L, Ma Y, Guo FY, Chang Y, Li PA. Monosialotetrahexosy-l ganglioside attenuates diabetes-enhanced brain damage after transient forebrain ischemia and suppresses phosphorylation of EPK1/2 in the rat brain. Brain Res. 2010;16:200-8
16. Li PA, Shamloo M, Katsura K, Smith ML, Siesjo BK. Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiol Dis. 1995;2:97-108
17. Ding C, He Q, Li PA. Activation of cell death pathway after a brief period of global ischemia in diabetic and non-diabetic animals. Exp Neurol. 2004;188:421-29
18. Muranyi M, Fujioka M, He Q, Han A, Yong G, Csiszar K, Li PA. Diabetes activates cell death pathway after transient focal cerebral ischemia. Diabetes. 2003;52:481-86
19. Song EC, Chu K, Jeong SW, Jung KH, Kim SH, Kim M, Yoon BW. Hyperglycemia exacerbates brain edema and perihematomal cell death after intracerebral hemorrhage. Stroke. 2003;34:2215-20
20. Mehta SL, Lin Y, Chen W, Yu F, Cao L, He Q, Chan PH, Li PA. Manganese superoxide dismutase deficiency exacerbates ischemic brain damage under hyperglycemic conditions by altering autophagy. Transl Stroke Res. 2011;2:42-50
21. Muranyi M, Ding C, He Q, Lin Y, Li PA. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes. 2006;55:349-355
22. Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3:296-304
23. Giffard RG, Monyer H, Choi DW. Selective vulnerablility of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990;530:138-141
24. Bondarenko A. Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134-42
25. Ennis SR, Keep RF. Effect of sustained-mild and transient-severe hyperglycemia on ischemia-induced blood-brain barrier opening. J Cereb Blood Flow Metab. 2007;27:1573-1582
26. Cipolla MJ, Bullinger LV. Reactivity of brain parenchymal arterioles after ischemia and reperfusion. Microcirculation. 2008;15:495-501
27. Dietrich WD, Alonso O, Busto R. Moderate hyperglycemia worsens acute blood-brain barrier injury after forebrain ischemia in rats. Stroke. 1993;24:111-116
Author contact
![]() Corresponding author: Dr. P. Andy Li, Email: pliedu or Dr. Jian-zhong Zhang, zhangjzedu.cn.
Corresponding author: Dr. P. Andy Li, Email: pliedu or Dr. Jian-zhong Zhang, zhangjzedu.cn.