Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(4):358-366. doi:10.7150/ijbs.8634 This issue Cite
Research Paper
Adenovirus-Mediated Prothymosin α Gene Transfer Inhibits the Development of Atherosclerosis in Apoe-Deficient Mice
Meng-Ya Chang1,2, Yu-Shan Yang3, Mei-Ling Tsai4, Che-Hsin Lee5, Chih-Jui Chang6, Ai-Li Shiau7 ![]() , Chao-Liang Wu3
, Chao-Liang Wu3 ![]()
1. Institute of Medical Sciences, Tzu Chi University, Hualien, Taiwan;
2. Department of Medical Research, Buddhist Tzu-Chi General Hospital, Hualien, Taiwan;
3. Department of Biochemistry and Molecular Biology, National Cheng Kung University Medical College, Tainan, Taiwan;
4. Department of Physiology, National Cheng Kung University Medical College, Tainan, Taiwan;
5. Department of Microbiology, China Medical University Medical College, Taichung, Taiwan;
6. Department of Molecular Biology and Human Genetics, Tzu Chi University, Hualien, Taiwan;
7. Department of Microbiology and Immunology, National Cheng Kung University Medical College, Tainan, Taiwan.
Received 2014-1-21; Accepted 2014-2-20; Published 2014-3-15
Abstract
Prothymosin α (ProT) is involved in regulating expression of the oxidative stress-protective genes and it also exerts immunomodulatory activities. In this study, we investigated the therapeutic effects of ProT gene transfer on atherosclerosis in endothelial cells and in ApoE-deficient mice. Adenoviruses encoding mouse ProT (AdProT) were used for the management of atherosclerosis. In vitro, the effects of ProT on antioxidant gene expressions and the protection effect against oxidant-mediated injury in endothelial cells were examined. In vivo, AdProT were administered intraventricularly into the heart of ApoE-/- mice. Histopathological and immunohistochemical assessments of the aortic tissues were performed. Expressions of HO-1 and antioxidant genes in the aortic tissues were also determined. Our results demonstrated that ProT gene transfer increased antioxidant gene expressions, eNOS expression and NO release, as well as reduced the reactive oxygen species production in endothelial cells. Intraventricular administration of AdProT reduced the lesion formation, increased expressions of HO-1 and SOD genes, and reduced infiltrating macrophages in the aorta of ApoE-/- mice. This study suggests that ProT gene transfer may have the therapeutic potential for the management of atherosclerosis via inducing antioxidant gene expressions, eNOS expression and NO release, reducing ROS production and macrophage infiltration in endothelium.
Keywords: atherosclerosis, gene transfer, antioxidant gene expressions, prothymosin α.
Introduction
Cardiovascular disease is the leading cause of death and disability in the world. The majority of cardiovascular disease results from complications of atherosclerosis. Atherosclerosis is an inflammatory disease in which lipid deposition in the arterial wall. It is believed that oxidation and inflammation are two crucial events involved in the development of atherosclerotic lesions [1, 2].
Oxidative stress results from the reduction of antioxidant agents or overproduction of reactive oxygen species (ROS), and is considered a major mechanism involved in the pathogenesis of endothelial dysfunction, and the initiation and progression of atherosclerosis. The defense mechanisms to scavenge ROS in the vasculature and result in inhibition of nitric oxide (NO) degradation include enzymatic superoxide dismutase (SOD), catalase and glutathione peroxidase, and non-enzymatic low-molecular-mass molecules [1]. NO that is the principal vasodilator and is synthesized via the action of endothelial NO synthase (eNOS). Superoxide promotes cellular damage by reacting with and reducing vascular NO bioavailability [3]. As the vascular level of superoxide is regulated by the SOD, a role of SOD genes in the cardiovascular disease is of substantial interest [4].
Keap1 (Kelch-like ECH-associated protein 1) and Nrf2 (nuclear factor-erythroid 2-related nuclear factor 2) constitute a crucial cellular sensor for oxidative stress. In unstressed cells, Nrf2 appears to be sequestered in the cytoplasm via association with an inhibitor protein, Keap1, and then degraded via the proteasome system [5]. Upon exposure to electrophilic or oxidative stresses, Nrf2 is released from Keap1 and then translocates to the nucleus, forms heterodimers with Maf, and binds to the antioxidant response elements (AREs) to regulate ARE-mediated gene expression [6]. In the previous report, prothymosin α (ProT) was shown to liberate Nrf2 from the Nrf2-Keap1 inhibitory complex and regulate the expression of oxidative stress-protecting genes [7]. ProT is a highly acidic small protein and is distributed in the nuclei of a wide variety of tissues and is highly conserved among mammals [8]. Many studies have showed the immunoregulatory functions of ProT, including enhancing MHC class II surface antigen expression on antigen presenting cells, and human natural killer cell cytotoxicity [8, 9]. Moreover, ProT is likely to have a broader effect on transcription. It affects the activity of specific transcription factors, including the estrogen receptor [10] and Stat3 [11].
In this study, we employed an adenovirus expressing ProT (designated AdProT) to examine the effect of ProT gene transfer on antioxidant gene expressions and consequently the protection of ProT on endothelial cells against oxidative-mediated injury in vitro and in vivo. Our results demonstrated that ProT gene transfer enhanced antioxidant gene expressions, such as heme oxygenase-1 (HO-1) and SOD, and reduced H2O2-induced oxidative damage in HUVECs, and inhibited the atherosclerosis progression in ApoE-/- mice via inducing the antioxidant gene expressions, reducing the reactive oxygen species production, and decreasing the macrophage infiltration.
Methods
Cells
Human umbilical vein endothelial cells (HUVECs) (Bioresource Collection and Research Center, HsinChu, Taiwan) were cultured in M199 (GIBCO, Grand Island, NY) supplemented with endothelial cell growth supplement (Upstate) and 10% fetal bovine serum. RAW 264.7 (mouse leukaemic monocyte macrophage cells) were cultured in HyQ-DMEM/High Glucose (HyClone, Logan, UT) supplemented with 10% cosmic calf serum (Hyclone). Cells were maintained at 37°C in an atmosphere of 5% CO2. This study conforms with the principles outlined in the Declaration of Helsinki.
Amplification of recombinant adenoviruses
AdProT, an adenovirus containing mouse ProT gene, was generated as previously described [12]. Control adenoviruses AdLacZ with β-glatosidase gene or AdLuc with firefly luciferase gene were generated with the same method described. All adenoviruses were propagated in 293 cells, purified by CsCl density gradient centrifugation, and quantified by plaque assay.
Reverse transcription-polymerase chain reaction (RT-PCR)
RNA was isolated using the Trizol (Invitrogen, CA) according to the manufacturer's instructions. The REVERSE-IT™ 1st strand synthesis kit (ABgene, Epsom, UK) was used for cDNA synthesis. Appropriate PCR primers were used, and the β-actin gene amplifications were used as an internal standard. PCR amplification was performed for 30 cycles, and the cycling conditions were as follows: 94°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec.
Reactive Oxygen Species (ROS) Production
Intracellular oxidative products were detected by 2,7-dichlorodihydrofluorescein diacetate (H2DCFDA, Sigma, MO). Briefly, 2 × 105 HUVECs were seeded in 6-well plates and infected with AdLacZ or AdProT at a MOI (multiplicity of infection) of 100 for 72 h, and sequentially rinsed twice with PBS and treated with 10 μM of H2DCFDA for 30 min. Cells were then collected and analyzed immediately by FACSCalibur (BD Biosciences, CA), and the results were analyzed using Cell Quest software (BD Biosciences). H2O2-treated cells were served as the positive control.
Cell viability
104 HUVECs were seeded in 96-well plates, and infected by AdLacZ or AdProT for 48 h, followed by a H2O2 (250 μM) incubation for 4 h. The cell viability was determined by MTT assay as previously described [13].
Determination of NO release
104 HUVECs or RAW264.7 were seeded in 96-well plates and infected by AdLacZ or AdProT for 72 h. NO2- in culture supernatant was measured to assess the NO production in HUVECs. 50 μl of samples were mixed with 50 μl of Griess reagent (1% sulfanilamide/0.1% naphthylethylene diamine dihydrochloride/2% phosphoric acid) and incubated at 25°C for 10 min. The absorbance at 540 nm was measured on a microplate reader (Anthos Labtec Instruments GmbH, Salzburg, Austria). NaNO2 was used as the standard to calculate NO2- concentrations.
Western blots
Total cells lysates were harvested for detection of protein expression by immunoblot analysis with HO-1 antibody (Stressgen Bioreagents, Ann Arbor, MI), eNOS antibody (Cell Signaling Technology, MA), iNOS antibody (R&D Systems, MN) or mouse β-actin monoclonal antibody (Sigma, MO). Appropriate as secondary antibodies were used and protein-antibody complexes were visualized by the ECL system (Amersham Biosciences, NJ).
Luciferase assay
Hearts and aorta of mice were isolated. Tissues were homogenized for measuring luciferase activities using the Dual-Light Luciferase Reporter Assay System (Tropix, MA). Total protein concentrations were determined using the BCA kit (Pierce Biotechnology, IL) to normalize the luciferase signal.
Animal studies
Apolipoprotein E-deficient (ApoE-/-) mice from a C57BL/6J background that develop a spontaneous atherosclerosis that mimics most of the features of human atherogenesis were obtained from the Laboratory Animal Center of the National Cheng Kung University. Animals were anesthetized with pentobarbital intraperitoneally (90 μg/g body weight). Adenoviruses (2 × 108 PFU) in 50 μl of PBS were injected directly into the left ventricle of the heart. 4 weeks after virus administration, hearts with attached aortas and other tissues were removed for further examinations. All animal experiments were performed following the guidelines approved by the Laboratory Animal Care and Use Committee of the National Cheng Kung University.
Quantification of atherosclerotic lesion formation and macrophages in the lesions
Hearts isolated from the mice were fixed, dehydrated, and embedded in paraffin. Serial tissue sections of 5 μm thickness were collected in every 25 μm interval. Eight sections were obtained for each sample. Sections were stained with Mayer's hematoxylin and eosin (H&E) for histological assessment of lesion size in the aortic sinus under a microscopy. To detect macrophages within atherosclerotic lesions, sections were subjected to immunohistochemical staining using the monoclonal anti-Mac-3 antibody (BD PharMingen, CA). After sequential incubated with the appropriate secondary antibody, aminoethyl carbazole (AEC, Taipei, Taiwan) was used as a substrate chromogen. The quantitative data was calculated by Image Pro Plus (Media Cybernetics, MD).
Statistical analysis
Data were expressed as mean ± SEM. Statistical significance between groups was assessed by the unpaired Student's t-test. Values of p less than or equal to 0.05 were considered statistical significant.
Results
Effect of ProT gene transfer on HO-1 expression in HUVECs
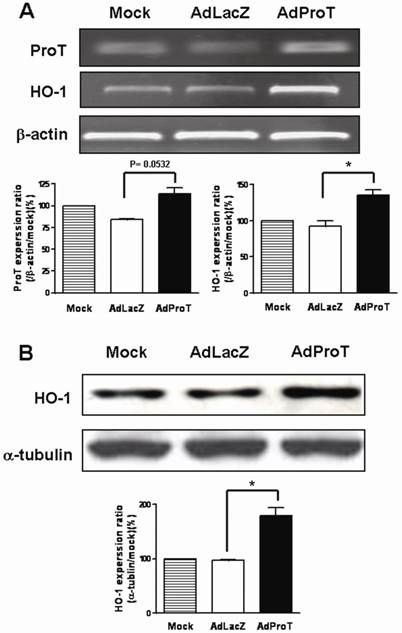
To examine the effect of ProT on HO-1 expression in HUVECs, cells were infected with AdProT or AdLacZ at a MOI of 100 for 48 h, and the HO-1 expression was detected both by RT-PCR and immonoblot analysis. Our results showed that ProT overexpression induced a marked increase of HO-1 both in mRNA and protein level (Figs. 1A&B).
Expressions of ProT and HO-1 in HUVECs after ProT gene transfer. HUVECs were infected with AdLacZ or AdProT at a MOI of 100 for 48 h. (A) ProT and HO-1 mRNA expressions were detected by RT-PCR. (B) HO-1 expression was detected by Western blotting.
Effect of ProT on intracellular ROS generation and H2O2-induced cell injury in HUVECs
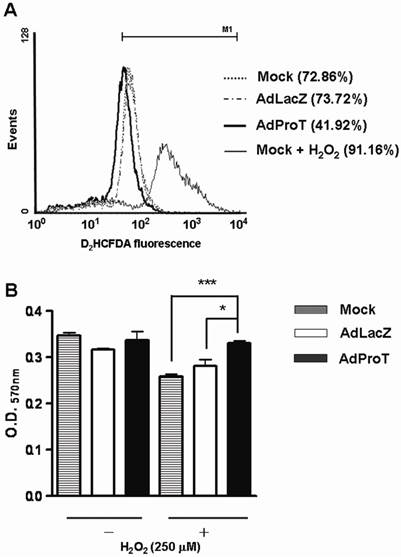
To investigate whether ProT has any influence on ROS generation and oxidative stress-induced endothelial cell injury, HUVECs were infected with AdProT or AdLacZ at a MOI of 100. For ROS detection, 72 h post-infection, cells were stained with a fluoresce probe D2HCFDA, and the ROS generation was evaluated using a flow cytometry. The result revealed that ProT overexpression decreased the ROS generation by 30% in HUVECs, while the H2O2-treated cells served as a positive control (Fig. 2A). For oxidative stress-induced cell injury, 48 h post-infection, cells were exposed to H2O2 for 4 h, and the cell viability was measured by MTT assay (Fig. 2B). The data showed that H2O2 decreased the cell viability, while ProT overexpression significantly restored cells from H2O2-induced damage. These results suggested that ProT overexpression increased intracellular antioxidant capacity and protected HUVECs from oxidative stress-mediated cell injury.
Reductions of ROS formation and H2O2-induced cell damage in HUVECs after ProT gene transfer. (A) HUVECs were treated with AdLacZ or AdProT at a MOI of 100 for 72 h, and incubated with H2DCFDA for 30min. The intracellular ROS was measured by flow cytometry. (B) HUVECs were treated AdLacZ or AdProT at a MOI of 100 for 48 h prior to a 4 h H2O2 (250 μM) incubation. Cell viability was determined by MTT assay. Results are expressed as mean ± SEM (n = 4). *, p<0.05; ***, p<0.001.
Effects of ProT on SOD gene expression, eNOS expression and intracellular NO level in HUVECs
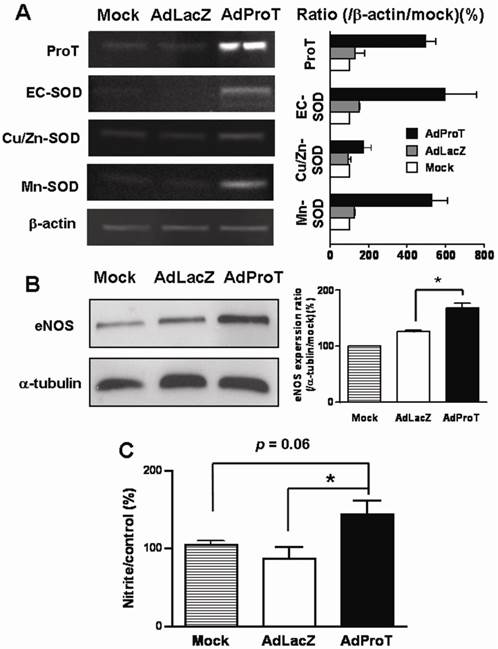
Blood vessels express 3 isoforms of SOD: copper/zinc SOD (Cu/Zn-SOD), manganese SOD (Mn-SOD), and extracellular SOD (EC-SOD) [4, 14, 15]. We examined the effect of ProT overexpression on SOD gene expressions in HUVECs infected with AdProT or AdLacZ at a MOI of 100 for 48 h by RT-PCR. As shown in Fig. 3A, ProT overexpression upregulated all three isoforms of SOD gene expressions. To investigate the endothelial function following ProT overexpression, eNOS expression was determined by Western blotting (Fig. 3B), and the NO release in conditioned medium were also determined indirectly by the content of nitrite and nitrate (Fig. 3C). Our data revealed that ProT gene transfer notably enhanced the eNOS expression and increased the intracellular level of NO.
Effects of ProT overexpression on the antioxidant gene expressions, eNOS expression and NO production in HUVECs. HUVECs were infected with AdLacZ or AdProT at a MOI of 100 for 48 h. The SOD expressions were detected with RT-PCR (A), eNOS expression was detected by Western blotting (B) and the accumulation of nitrite in the supernatants was measured with Griess reagent (C). Data are expressed as mean ± SEM. *, p< 0.05.
Effects of ProT on iNOS expression and intracellular NO level in RAW264.7 cells
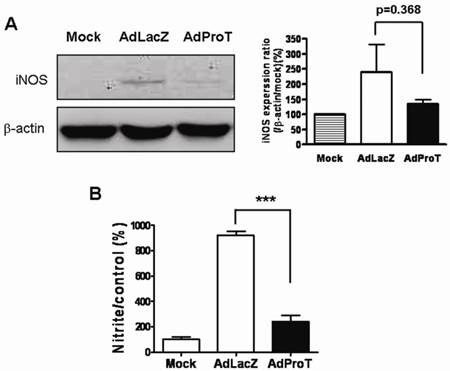
As macrophages produce NO to stimulate inflammatory reaction, to investigate the effect of ProT overexpression on NO production in macrophages, the iNOS expression and NO release in macrophages were determined. RAW264.7 cells were infected with AdProT or AdLacZ at a MOI of 400. 48 h post-infection, the iNOS expression was detected by Western blot analysis (Fig. 4A), and the level of NO in the conditioned medium was determined indirectly as the content of nitrite and nitrate (Fig. 4B). These results revealed that ProT overexpression reduced both the iNOS expression and the intracellular NO level in macrophages.
Effects of ProT overexpression on iNOS expression and NO production in RAW264.7 cells. RAW264.7 cells were infected with AdLacZ or AdProT at a MOI of 400. (A) 48 h post-infection, the iNOS expression was detected by Western blotting, and (B) the accumulation of nitrite in the supernatants was measured with Griess reagent.
Transgene expression and effect of ProT on antioxidant gene expressions in arterial walls in ApoE-/- mice after intraventricular virus administration
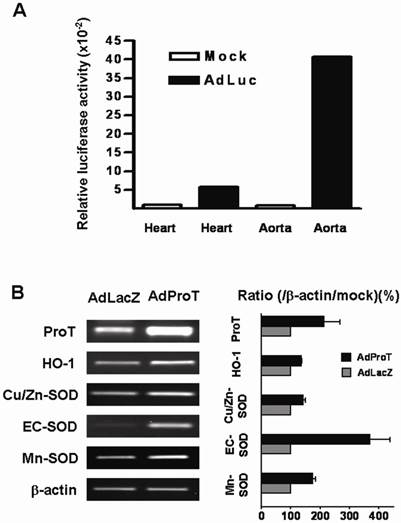
To evaluate the transgene expression in aortic tissues of animals, AdLuc (2 × 108 PFU) was administered directly into the heart of anesthetized ApoE-/- mice, and the luciferase activity in heart and aorta were examined 3 days post-administration. The result showed that higher luciferase activity was detected in the aortic tissues compared to that in the hearts (Fig. 5A). Effects of ProT on antioxidant gene expressions in aorta of ApoE-/- mice were also evaluated in this study. We injected AdProT or AdLacZ (2 × 108 PFU) intraventricularly to the heart of anesthetized ApoE-/- mice. 3 days post-virus injection, the expressions of HO-1 and SOD genes in the aortic tissues of mice were examined by RT-PCR (Fig. 5B). A notably increase of ProT mRNA was observed in aorta of ApoE-/- mice receiving AdProT administration compared to the AdLacZ control. Moreover, consistent with our in vitro observations, the upregulation of HO-1 and SOD gene expressions by ProT overexpression were seen in the aorta of AdProT-treated ApoE-/- mice.
AdLuc expression and effects of ProT overexpression on HO-1 and SOD expressions in the aorta of ApoE-/- mice intraventricularly receiving AdLuc or AdProT. 9-week-old ApoE-/- mice were received 2 × 108 PFU AdLuc (n=2) or AdProT (n=2). Tissues were collected 3 days after injection. (A) Luciferase activity in the heart and aorta of AdLuc-treated mice determined by luciferase assay. (B) Effects of ProT overexpression on HO-1 and SOD mRNA expressions in the aortic tissues of ApoE-/- mice detected by RT-PCR.
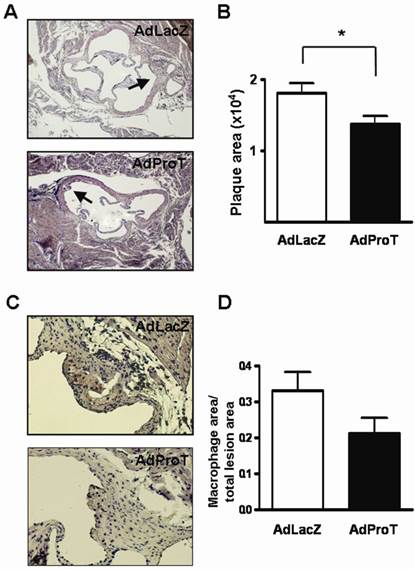
Effect of ProT on atherosclerotic lesion formation and macrophages infiltration in ApoE-/- mice
To assess the effect of ProT gene transfer on lesion formation in the early phase of atherosclerosis in vivo, we administered AdProT or AdLacZ intraventricularly to ApoE-/- mice. Four weeks post-administration, the aortic tissues of mice were excised, sectioned and stained with H&E. The extent of atherosclerotic lesions developed in aortic sinus was assessed under a microscopy (Fig. 6A), and the quantitative data was shown as mean ± SEM in Fig. 6B. The result demonstrated that the lesion area developed at the aortic sinus of AdProT-treated mice was significantly smaller compared to those in AdLacZ-treated group. Furthermore, macrophage infiltration in the lesions was also detected by immohistochemistry with anti-MAC-3 antibody (Fig. 6C), and the quantitative data was presented in Fig. 6D. Although no significant difference was determined, more macrophages were observed in the atherosclerotic lesions of mice receiving AdLacZ than those receiving AdProT. Taken together, our data showed that intraventricularly administration of AdProT reduced the macrophage filtration, and significantly reduced the formation of atherosclerotic lesions in the early stage of atherosclerosis in vivo.
AdProT reduced the atherosclerotic lesion formation in ApoE-/- mice. 9-week-old ApoE-/- mice were received intraventricularly 2 × 108 PFU AdLacZ (n=6) or AdProT (n=6). Tissues were fixed, dehydrated, and embedded in paraffin. Serial sections (n=8) of 5 μm thickness were collected. (A) Histological assessment of lesion size in the aortic sinus. Sections were stained with H&E and observed under a microscopy (magnification × 200). The quantitative data were shown in (B). (C) Immunostaining for macrophages in the lesion (magnification × 400). (D) Quantitative comparison of macrophage composition in AdLacZ- and AdProT-treated groups (n = 6). Data are expressed as mean ± SEM. *, p<0.05.
Discussion
Antioxidant and anti-inflammatory strategies are considered to have the therapeutic potential for the treatment of atherosclerosis [16]. ProT has been reported to have immunomodulatory activities and function as a biological response modifier, which may be beneficial in clinical applications [17]. Moreover, ProT was demonstrated to disrupt the Nrf2-Keap1 inhibitory complex, and consequently increased the transcriptional activity of antioxidant response elements (AREs) [7]. As AREs have been found in the 5′-flanking region of many genes involved in cytoprotection from oxidative stress, such as γ-glutamylcysteine synthetase, and HO-1 [18], therefore, the potential therapeutic effect of ProT gene transfer on atherosclerosis is of our interest.
In this study, we demonstrated that adenovirus-mediated ProT gene transfer reduced the H2O2-induced ROS generation and prevented the oxidative-induced cell damage in HUVECs. In addition, ProT overexpression induced eNOS expression and thereafter the NO production in HUVECs. Knowing that NO is synthesized by eNOS, and overproduction of ROS may result in reduced availability of NO [19], these results suggested the protective effect of ProT on maintaining the endothelium function. In the previous studies, estrogen was shown to rapidly activate eNOS and promote NO release in endothelial cells [20, 21] and have a protective effect on vascular endothelial cells in the prevention of atherosclerosis [22]. Interestingly, ProT was reported to enhance estrogen receptor transcriptional activity [23], therefore, this raised the possibility that ProT may be able to induce the eNOS expression and NO release to prevent atherosclerosis through estrogen receptor-mediated signal pathway.
Apoptosis of endothelial cells highly affects endothelium permeability and thus facilitates the development of atherosclerosis [24]. ROS overproduction causes endothelial cell damage even to apoptosis. In this study, ProT overexpression reduced the ROS production in HUVECs, and moreover, moreover, a previous study showed the anti-apoptotic effect of ProT [25]. These two observations implicate that ProT may be able to reduce the apoptosis of endothelium cells via reducing the ROS formation, and/or by its anti-apoptotic feature to further inhibit the atherosclerosis process.
To further assess the anti-atherosclerotic effects of ProT in vivo, we treated ApoE-/- mice with adenoviruses expressing ProT gene. ProT gene transfer increased the antioxidant gene expressions in the aortic tissues, and significantly reduced the formation of atherosclerotic lesions in ApoE-/- mice. There was no significant difference between the AdProT- and AdLacZ-treated groups in body weight and in levels of plasma cholesterol 4 weeks after virus treatment (data not shown). In the previous studies, inhibition of SOD leads to a significant impairment of NO bioavailability in an animal model [26], and adenovirus-mediated Cu/Zn-SOD gene transfer has been shown to decrease vascular superoxide levels in atherosclerosis [27]. In agreement with these studies showing the importance of SOD genes in the regulation of NO-dependent endothelial functions, in this study, we demonstrated that the ProT overexpression increased all 3 isoforms of SOD gene expressions both in vitro and in vivo, which suggested the protective effect of ProT against oxidative stress was associated with scavenging intracellular ROS formation via regulating antioxidant gene expressions. Moreover, EC-SOD is highly expressed in blood vessels and constitutes up to 70% of the SOD activity [4]. Importantly, we observed a dramatic increase of EC-SOD expression in aortic tissues of AdProT-injected mice in our study. This revealed an enhanced ability of ProT to inhibit NO degradation and protect the NO-mediated signaling. However, no ARE was found within the promoter regions of SOD genes. Hence, the regulation of ProT on the expression of SOD genes may be via an ARE-independent pathway.
HO-1 is a cytoprotective enzyme that is induced in response to oxidative stress. Accumulating evidence has shown the relationship of HO-1 and atherosclerosis [28, 29]. Here, we showed that ProT overexpression induced an enhanced HO-1 expression in vitro and in vivo. Moreover, based on our preliminary data, the ProT-induced upregulation of SOD gene expression was only partially inhibited by the HO-1 inhibitor ZnPP in HUVECs (data not shown). This leads to the speculation that ProT may induce antioxidant gene expressions both through HO-1-dependent and -independent pathways.
From early fatty-streak lesions to advanced plaques, macrophage-derived foam cells are integral to the development and progression of atherosclerosis [2, 30]. In this study, we intended to see the effect of ProT gene transfer on lesion formation in the early phase of atherosclerosis in vivo; therefore, 4 weeks post-infection were chosen to analyze the lesion formation and macrophage infiltration. We showed that ProT overexpression reduced the macrophage filtration, and significantly reduced the formation of atherosclerotic lesions. Although the effects were not dramatic, it still revealed the prevention role of ProT on atherosclerotic lesion formation. Furthermore, the iNOS and NO production in macrophages were notably inhibited by ProT gene transfer. It is known that overproduction of NO via the iNOS has cytotoxic effect and plays a role in the development of atherosclerosis [31]. Our results indicated that ProT overexpression inhibited the development of atherosclerosis could also via reducing of macrophage infiltration as well as the iNOS-mediated NO production in macrophages.
In conclusion, this study is the first to suggest that ProT gene transfer may have therapeutic potential for the management of atherosclerosis. In vitro, our results demonstrated that ProT enhanced eNOS expression and increased NO release to maintain endothelial function, and protected endothelial cells against H2O2-induced injury, which was probably associated with the increased antioxidant gene expressions, including HO-1, and three isoforms of SOD. In vivo, we showed that adenovirus-mediated ProT gene transfer ameliorated the lesion formation of atherosclerosis in ApoE-/- mice. The induction of a distinct set of antioxidant gene expressions may play an important role in inhibiting the endothelial impairment and atherosclerosis process in ProT gene therapy. Taken together, these results demonstrate that the delivery of adenoviral vectors encoding ProT may be an effective strategy for the treatment of atherosclerosis.
Conclusions
This study suggests that ProT gene transfer may have the therapeutic potential for the management of atherosclerosis via inducing the antioxidant gene expressions, reducing the reactive oxygen species production, and decreasing the macrophage infiltration.
Abbreviations
ProT, Prothymosin α; HO-1, heme oxygenase-1; ROS, reactive oxygen species; NO, nitric oxide; SOD, superoxide dismutase; eNOS, endothelial NO synthase; AdProT, an adenovirus expressing ProT; HUVECs, human umbilical vein endothelial cells
Acknowledgements
This work was supported by Grant NSC 100-2320-B-006-023-MY3 from the National Science Council, Taiwan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381-478
2. Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29-38
3. Yokoyama M. Oxidant stress and atherosclerosis. Curr Opin Pharmacol. 2004;4:110-5
4. Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367-73
5. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76-86
6. Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199-207
7. Karapetian RN, Evstafieva AG, Abaeva IS, Chichkova NV, Filonov GS, Rubtsov YP. et al. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: implications for expression of oxidative stress-protecting genes. Mol Cell Biol. 2005;25:1089-99
8. Gast K, Damaschun H, Eckert K, Schulze-Forster K, Maurer HR, Muller-Frohne M. et al. Prothymosin alpha: a biologically active protein with random coil conformation. Biochemistry. 1995;34:13211-8
9. Cordero OJ, Sarandeses CS, Lopez JL, Cancio E, Regueiro BJ, Nogueira M. Prothymosin alpha enhances interleukin 2 receptor expression in normal human T-lymphocytes. Int J Immunopharmacol. 1991;13:1059-65
10. Martini PG, Delage-Mourroux R, Kraichely DM, Katzenellenbogen BS. Prothymosin alpha selectively enhances estrogen receptor transcriptional activity by interacting with a repressor of estrogen receptor activity. Mol Cell Biol. 2000;20:6224-32
11. Yang CH, Murti A, Baker SJ, Frangou-Lazaridis M, Vartapetian AB, Murti KG. et al. Interferon induces the interaction of prothymosin-alpha with STAT3 and results in the nuclear translocation of the complex. Exp Cell Res. 2004;298:197-206
12. Shiau AL, Chen SY, Chang MY, Su CH, Chung SY, Yo YT. et al. Prothymosin {alpha} Lacking the Nuclear Localization Signal as an Effective Gene Therapeutic Strategy in Collagen-Induced Arthritis. J Immunol. 2007;178:4688-94
13. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55-63
14. Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. 2002;55:239-49
15. Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M. et al. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288:H13-21
16. Lahera V, Goicoechea M, de Vinuesa SG, Miana M, de las Heras N, Cachofeiro V. et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins. Curr Med Chem. 2007;14:243-8
17. Low TL, Thurman GB, McAdoo M, McClure J, Rossio JL, Naylor PH. et al. The chemistry and biology of thymosin. I. Isolation, characterization, and biological activities of thymosin alpha1 and polypeptide beta1 from calf thymus. J Biol Chem. 1979;254:981-6
18. Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20:3906-17
19. Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454-6
20. Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y. et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276:3459-67
21. Sumi D, Ignarro LJ. Estrogen-related receptor alpha 1 up-regulates endothelial nitric oxide synthase expression. Proc Natl Acad Sci U S A. 2003;100:14451-6
22. Cid MC, Schnaper HW, Kleinman HK. Estrogens and the vascular endothelium. Ann N Y Acad Sci. 2002;966:143-57
23. Martini PG, Katzenellenbogen BS. Modulation of estrogen receptor activity by selective coregulators. J Steroid Biochem Mol Biol. 2003;85:117-22
24. Xu GL, Qian ZY, Yu SQ, Gong ZN, Shen XC. Evidence of crocin against endothelial injury induced by hydrogen peroxide in vitro. J Asian Nat Prod Res. 2006;8:79-85
25. Malicet C, Giroux V, Vasseur S, Dagorn JC, Neira JL, Iovanna JL. Regulation of apoptosis by the p8/prothymosin alpha complex. Proc Natl Acad Sci U S A. 2006;103:2671-6
26. Mugge A, Peterson T, Harrison DG. Release of nitrogen oxides from cultured bovine aortic endothelial cells is not impaired by calcium channel antagonists. Circulation. 1991;83:1404-9
27. Miller FJ Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298-305
28. Immenschuh S, Schroder H. Heme oxygenase-1 and cardiovascular disease. Histol Histopathol. 2006;21:679-85
29. Morita T. Heme oxygenase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1786-95
30. Rajavashisth TB, Andalibi A, Territo MC, Berliner JA, Navab M, Fogelman AM. et al. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature. 1990;344:254-7
31. Esaki T, Hayashi T, Muto E, Yamada K, Kuzuya M, Iguchi A. Expression of inducible nitric oxide synthase in T lymphocytes and macrophages of cholesterol-fed rabbits. Atherosclerosis. 1997;128:39-46
Author contact
![]() Corresponding author: Professor Chao-Liang Wu, Department of Biochemistry and Molecular Biology, National Cheng Kung University Medical College, 1 Dashiue Road, Tainan 701, Taiwan; Tel: 886-6-2353535 ext. 5536; Fax: 886-6-2741694; E-mail:wumolbioncku.edu.tw or Professor Ai-Li Shiau, Department of Microbiology and Immunology, National Cheng Kung University Medical College, 1 Dashiue Road, Tainan 70101, Taiwan. E-mail: alshiauncku.edu.tw.
Corresponding author: Professor Chao-Liang Wu, Department of Biochemistry and Molecular Biology, National Cheng Kung University Medical College, 1 Dashiue Road, Tainan 701, Taiwan; Tel: 886-6-2353535 ext. 5536; Fax: 886-6-2741694; E-mail:wumolbioncku.edu.tw or Professor Ai-Li Shiau, Department of Microbiology and Immunology, National Cheng Kung University Medical College, 1 Dashiue Road, Tainan 70101, Taiwan. E-mail: alshiauncku.edu.tw.