Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Current view of residual...
Contemporary pathways of...
Novel derivatives of adrenal...
Significance of DOC and 11OH-AED...
Rationale for targeting...
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):602-613. doi:10.7150/ijbs.8844 This issue Cite
Review
Beyond T and DHT - Novel Steroid Derivatives Capable of Wild Type Androgen Receptor Activation
Elahe A Mostaghel ![]()
Division of Clinical Research, Fred Hutchinson Cancer Research Center, Seattle WA, USA.
Received 2014-2-15; Accepted 2014-4-23; Published 2014-6-3
Abstract
While androgen deprivation therapy (ADT) remains the primary treatment for metastatic prostate cancer (PCa), castration does not eliminate androgens from the prostate tumor microenvironment, and residual intratumoral androgens are implicated in nearly every mechanism by which androgen receptor (AR)-mediated signaling promotes castration-resistant disease. The uptake and intratumoral (intracrine) conversion of circulating adrenal androgens such as dehydroepiandrosterone sulfate (DHEA-S) to steroids capable of activating the wild type AR is a recognized driver of castration resistant prostate cancer (CRPC). However, less well-characterized adrenal steroids, including 11-deoxcorticosterone (DOC) and 11beta-hydroxyandrostenedione (11OH-AED) may also play a previously unrecognized role in promoting AR activation. In particular, recent data demonstrate that the 5α-reduced metabolites of DOC and 11OH-AED are activators of the wild type AR. Given the well-recognized presence of SRD5A activity in CRPC tissue, these observations suggest that in the low androgen environment of CRPC, alternative sources of 5α-reduced ligands may supplement AR activation normally mediated by the canonical 5α-reduced agonist, 5α-DHT. Herein we review the emerging data that suggests a role for these alternative steroids of adrenal origin in activating the AR, and discuss the enzymatic pathways and novel downstream metabolites mediating these effects. We conclude by discussing the potential implications of these findings for CRPC progression, particularly in context of new agents such as abiraterone and enzalutamide which target the AR-axis for prostate cancer therapy.
Keywords: intracrine steroidogenesis, adrenal androgen, deoxycorticosterone, 11beta-hydroxyandrostenedione, 11ketodihydrotestosterone, castration resistant prostate cancer, androgen receptor.
Introduction
While androgen deprivation therapy (ADT) remains the primary treatment for metastatic prostate cancer (PCa), treatment is uniformly marked by progression to castration-resistant prostate cancer (CRPC) over a period of about 18 months, with an ensuing median survival of 1 to 2 years [1]. Importantly, castration resistant tumors are characterized by elevated tumor androgens that are well within the range capable of activating the androgen receptor (AR), and by steroid enzyme alterations which may potentiate de novo androgen synthesis or utilization of circulating adrenal androgens [2-5]. The dependence of CRPC on intratumoral androgen metabolism has been modeled in vitro and in vivo, and residual intratumoral androgens are implicated in nearly every mechanism by which AR-mediated signaling promotes castration-resistant disease [6].
The uptake and intracrine conversion of weak circulating adrenal androgens such as dehydroepiandrosterone sulfate (DHEA-S) to steroids capable of activating the wild type AR is a recognized driver of castration resistant prostate cancer (CRPC)[7]. However, recent data suggest that additional, less well-characterized steroids of adrenal origin, including 11beta-hydroxyandrostenedione (11OH-AED) and 11-deoxcorticosterone (DOC) may also be converted to downstream metabolites that mediate AR activation[8-11]. Herein we briefly discuss the currently recognized role and metabolic pathways underlying the generation and activity of residual tumor androgens in CRPC. We review the emerging data that suggests a role for alternative steroids of adrenal origin in activating the AR, and discuss the enzymatic pathways and novel downstream metabolites mediating these effects. We conclude by discussing the potential implications of these findings for understanding resistance and optimizing response to new agents targeting the AR-axis for prostate cancer therapy.
Current view of residual androgens in castration resistant prostate cancer
The efficacy of ADT is routinely based on achieving castrate levels of serum testosterone (T), defined as < 20 ng/dl. However, incomplete suppression of prostate tissue T and dihydrotestosterone (5α-DHT) has been confirmed in numerous studies of short and long term castration therapy, with levels remaining at ~25% of pre-castration levels[12]. Adrenal androgens have also been detected at significant levels in prostate tissue of castrate men. Prostatic levels of dehydroepiandrosterone (DHEA), DHEA-sulfate (DHEA-S), and androstenedione (AED) were decreased by about 50% in castrate patients and far exceeded values of T and 5α-DHT in recurrent tumor [2]. No decrease in prostatic levels of 5-androstenediol were found after castration [13], which is of particular significance as this androgen has been shown to bind wild type AR without being inhibited by flutamide or bicalutamide [14].
Two recent studies demonstrate that the addition of adrenal-androgen synthesis inhibitors to castration therapy substantially lowered prostate androgens below that achieved with standard testicular androgen suppression, strongly suggesting a role for circulating adrenal androgens in contributing to post-castration tissue androgen levels. The addition of dutasteride and ketoconazole to combined androgen blockade (CAB) prior to prostatectomy lowered prostate 5α-DHT from 0.92 ng/g (in the CAB arm) to 0.03ng/g [15], while the addition of the potent CYP17A1 inhibitor abiraterone decreased prostate tissue 5α-DHT from 1.3 ng/g (in men treated with LHRH agonist therapy alone) to 0.18 ng/g and also decreased prostate levels of AED and DHEA [16].
Locally recurrent and metastatic castration resistant tumors also demonstrate significant levels of residual androgens. Testosterone levels in locally recurrent tumors from castrate patients were equivalent to those of patients with benign prostatic hypertrophy, and 5α-DHT levels were only reduced 80%, to about 0.4 ng/g [2]. Compared to primary prostate tumors from untreated patients (T 0.25ng/g, 5α-DHT 2.75ng/g) androgen levels in metastatic CRPC tumors obtained via rapid autopsy showed 3-fold higher T levels and easily detected levels of 5α-DHT (T 0.74 ng/g; 5α-DHT 0.25ng/g) [3]. The source of residual androgens within prostate tumors of castrate men has not been fully elucidated, but is generally attributed to the uptake and conversion of circulating adrenal androgens [4, 13], and somewhat more controversially, to de novo biosynthesis of androgens from progesterone or cholesterol precursors [5].
These findings clearly demonstrate that achieving castrate levels of circulating T does not eliminate androgens from the prostate tumor microenvironment. Residual tissue androgens are implicated in driving the majority of mechanisms whereby persistent AR-mediated signaling drives castration resistant disease. These mechanisms include AR overexpression, AR mutations that broaden ligand specificity and/or confer sensitivity to adrenal androgens, alterations in AR coactivators and/or corepressors that modulate AR stability and ligand sensitivity, and activation of the AR or downstream regulatory molecules by “cross talk” with other signaling pathways. The ability of 5α-DHT in the range observed in castrate tumors (~1nm, 0.5 to 1.0 ng/g) to activate the AR, stimulate expression of AR-regulated genes, and promote androgen mediated tumor growth has been convincingly demonstrated in both in vitro and in vivo studies [2, 17-20], and is evidenced by the nearly universal rise in serum PSA that accompanies CRPC progression.
The clinical relevance of intratumoral androgens in promoting CRPC tumor growth is confirmed by the clinical responses to agents targeting residual androgen pathway activity. These include the striking clinical response observed with novel ligand synthesis inhibitors such as abiraterone, and potent AR inhibitors such as enzalutamide [21, 22]. Perhaps most importantly, emerging studies suggest that response and resistance to abiraterone is associated with tumoral evidence of upregulated androgen synthesis, clearly demonstrating the importance of intratumoral androgen metabolism in CRPC tumor survival [23-25].
Contemporary pathways of androgen metabolism
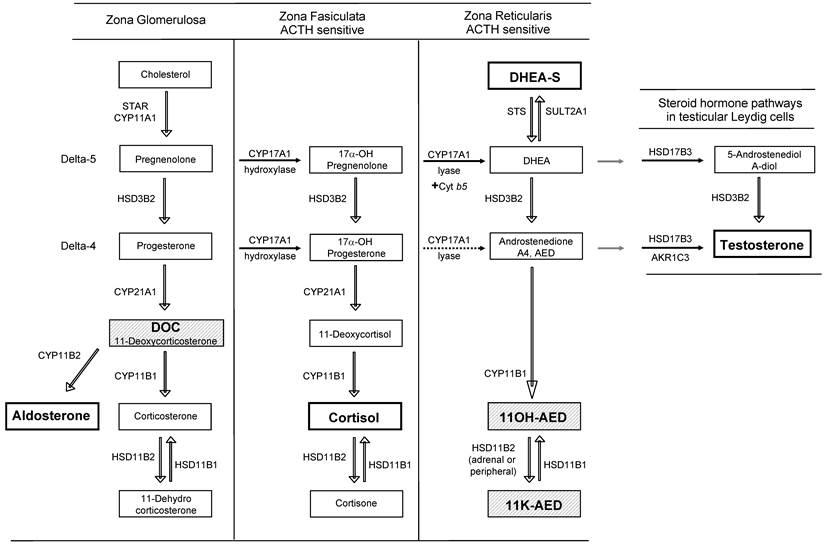
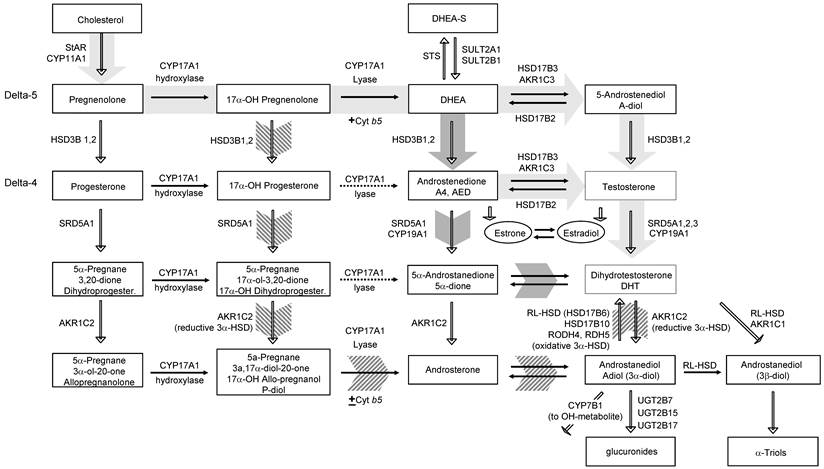
The uptake of circulating androgens and the local conversion of active steroids in steroid-responsive peripheral target tissues such as breast, prostate, skin and hormonal tumors have been termed intracrinology [26]. A general outline of androgen synthesis in adrenal and peripheral tissues is provided in Figure 1 (reviewed in [27] and [28]). Here we briefly review the adrenal (Figure 1), classical, 5α-dione and backdoor pathways of androgen metabolism (Figure 2) to provide context for understanding adrenal generation and downstream metabolism of the adrenal steroid intermediates DOC and 11OH-AED (hatched background, Figure 1).
Steroid hormone synthesis pathways in the adrenal gland and testis. Steroid synthesis in the adrenal gland occurs in three zones, each with a specific complement of enzymes. The zona glomerulosa contains the enzymes necessary to produce aldosterone. The zona fasciculata and reticularis additionally express CYP17A1. The hydroxylase activity of CYP17A1 is active in the zona fasiculata resulting in the production of cortisol. Due to tissue-specific expression of the cytochrome b5 coregulator, the lyase activity of CYP17A1 is only present in the zona reticularis and drives efficient production of DHEA which is then sulfated to DHEA-S (dotted arrows indicate low activity). 17α-OH progesterone is a poor substrate for CYP17A1 lyase (dotted arrow) and thus androstenedione is formed at lower levels from this substrate than 17α-OH pregnenolone. Testicular androgen synthesis follows a similar pathway to DHEA formation as that in the zona reticularis, but due to the absence of SULT2A1, and the presence of HSD3B2 and HSD17B3, DHEA is efficiently converted to testosterone. The primary product of each zone is denoted within the darker squares (aldosterone, cortisol, DHEA-S in the adrenal gland, and testosterone from the testis). The adrenal steroids involved in the generation of novel 5α-reduced androgens with activity against the AR are depicted in the hatched boxes (DOC, 11OH-AED, 11-K-AED).
Classical and non-classical pathways of androgen biosynthesis. Cholesterol is converted to C21 precursors (pregnenolone and progesterone) by the action of StAR and CYP11A. In the classical pathway (light gray arrows) C21 steroids are converted to the C19 adrenal androgens DHEA and androstenedione (AED) by the sequential hydroxylase and lyase activity of CYP17A1. The lyase activity of CYP17A1 (which requires the cytochrome b5 cofactor) favors production of DHEA rather than 17-OH pregnenolone (dotted arrows indicate low activity). DHEA (from intrinsic or circulating sources depending on the tissue) is subsequently acted on by HSD3B to form AED and HSD17B3 (or AKR1C3) to form testosterone which is converted to 5α-DHT via SRD5A. In tissues with expression of CYP19A1, estrone and estradiol can be generated from AED and T, respectively. In the backdoor pathway (hatched arrows) the progesterone intermediates are acted on first by the activity of SRD5A and the reductive activity of AKR1C2 prior to the lyase activity of CYP17A1. Androsterone is then acted on by HSD17B3 (or AKR1C3) and must undergo an oxidative step mediated by RL-HSD (or others) to generate 5α-DHT. In a third pathway, termed the 5α-Androstanedione pathway (dark gray arrows) DHEA and AED are produced as in the classical pathway. However, instead of conversion of AED to T followed by the activity of SRD5A to produce 5α-DHT, the enzymatic sequence is reversed such that AED is converted first by SRD5A to 5α-Androstanedione and then by HSD17B3 (or AKR1C3) to 5α-DHT. Modified from Best Practice & Research Clinical Endocrinology & Metabolism, Volume 22, Issue 2, Elahe A. Mostaghel and Peter S. Nelson. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Pages No. 243-258, Copyright (2008), with permission from Elsevier.
Adrenal androgen synthesis
Steroid synthesis begins with transfer of cholesterol from the outer to inner mitochondrial membrane by steroidogenic acute regulatory protein (StAR), followed by side chain cleavage to pregnenolone via CYP11A1 (side-change cleavage enzyme) [27, 29]. Subsequent metabolism to progesterone, mineralocorticoids, glucocorticoids (all C-21steroids), androgens (C-19) or estrogens (C-18) is dictated in a tissue-specific manner, driven by the expression of specific enzymes and catalytic cofactors[27].
CYP17A1 catalyzes the sequential but independent hydroxylase and lyase reactions required to convert pregnenolone and progesterone to androgens either along the delta-5 pathway from pregnenolone or the delta-4 pathway from progesterone. The hydroxylase activity of CYP17A1 is similar for pregnenolone and progesterone, but its lyase activity requires the presence of cytochrome b5, and is approximately 50 times more efficient for converting delta-5 17-OH pregnenolone to DHEA than delta-4 17-OH progesterone to AED [27].
HSD3B enzymes catalyze the conversion of delta-5 to delta-4 steroids. HSD3B2 is the primary isoform expressed in adrenal, testis and ovary, while HSD3B1 (10 fold more efficient) is the isoform expressed in peripheral tissues such as skin, breast, prostate, placenta and brain [27]. In adrenal steroidogenesis the zona glomerulosa lacks CYP17A1 activity and thus produces corticosterone and aldosterone via the sequential activity of HSD3B2, CYP21A1, and CYP11B on pregnenolone [27, 30].
Both the zona fasiculata and zona reticularis express CYP17A1, but the zona fasiculata does not express cytochrome b5 required to augment the lyase activity of CYP17A1, thereby channeling precursors to production of glucocorticoids. The differential expression of cytochrome b5 in the zona reticularis catalyzes the lyase activity of CYP17A1 10-fold, leading to robust production of DHEA, followed by conversion to DHEA-S via the sulfotransferase activity of SULT2A1. The zona reticularis is characterized by low expression of HSD3B2, favoring conversion of pregnenolone to DHEA and DHEA-S, although small amounts are still converted to AED [31].
The human zona reticularis also expresses HSD17B5 (called AKR1C3), which mediates the final step in T synthesis from AED, with several studies of adrenal vein sampling demonstrating adrenal to peripheral venous T gradients [32, 33]. Leydig cells of the testis express a similar metabolic machinery, but lack SULT2A1 (preventing conversion to DHEA-S) with the final steps in T biosynthesis being catalyzed by HSD17B3 (rather than AKR1C3 which mediates production of T and 5α-DHT in other peripheral tissues) [20].
Classical, backdoor and 5α-dione pathways of androgen metabolism
In the classical pathway of androgen synthesis (Figure 2, light gray arrows), cholesterol is converted to C-21 precursors (pregnenolone and progesterone), which are converted to the C-19 steroids DHEA (via the sequential hydroxylase and lyase activity of CYP17A1) and AED (through the action of HSD3B1,2 on DHEA), and then acted on by AKR1C3 (or potentially HSD17B3) to generate T, with peripheral conversion of T to 5α-DHT carried out by SRD5A1 or 2 in target tissues [34]. AKR1C2 mediates the reduction of 5α-DHT to 3α-diol (a metabolite with weak/low androgenic activity), whereas AKR1C1 catalyzes the conversion of 5α-DHT to 3β-diol [35]. The glucuronidating enzymes UGT2B15 and UTG2B17 irreversibly terminate the androgen signal by glucuronidation of 3α-diol (as well as T, 5α-DHT and other metabolites) [36-38].
However, in steroidogenic tissues in which both CYP17A1 and SRD5A are co-expressed, an alternate route to 5α-DHT, called the 'back-door' pathway (Figure 2, hatched arrows) is possible wherein C-21 steroids are acted on by the reductive activity of SRD5A and AKR1C2 prior to the lyase activity of CYP17A1 [29]. In fact, 17-OH progesterone is a better substrate for SRD5A (especially SRD5A1) than either AED or T [30]. Since 17-OH dihydroprogesterone (the 5α-reduced product of 17-OH progesterone) is a poor substrate for the lyase activity of CYP17A1, synthesis proceeds via ARK1C2, which yields 17-OH allo-pregnanolone, an excellent substrate for CYP17A1 lyase activity that is minimally dependent on cytochrome b5 [39]. Androsterone generated by the lyase activity of CYP17A1 is then acted upon by HSD17B3 or AKR1C3 to generate 3α-diol followed by, a reverse oxidative step (not required in the classical pathway) to generate 5α-DHT [27]. Candidate enzymes mediating the reverse conversion of 3α-diol to 5α-DHT include RL-HSD (17BHSD6), 17BHSD10, RODH4, RDH5, and DHRS9 [35, 40, 41]. RL-HSD also acts as an epimerase to convert 3α-diol to 3β-diol, although at much higher substrate concentrations [42], and can directly catalyze conversion of physiologic levels of 5α-DHT to 3β-diol [43].
In a third pathway, termed the 5α-androstanedione (5α-dione) pathway (Figure 2, dark gray arrows) DHEA and AED are produced as in the classical pathway. However, AED is converted first by SRD5A to 5α-dione and then by AKR1C3 (or potentially HSD17B3) to 5α-DHT, rather than conversion of AED to T followed by the activity of SRD5A to produce DHT. Consistent with prior findings that SRD5A activity in PCa cells has a preference for AED rather than T [44-46], Sharifi recently demonstrated that SRD5A1 mediated 5α-reduction of AED to5α-dione is required for 5α-DHT synthesis in PCa cells, and that T and AED are actually negligible substrates for SRD5A2 [47] (possibly related to the altered redox environment of tumor cells as SRD5A1 and 2 have different pH optima). The subtotal loss of SRD5A2 and upregulation of SRD5A1 observed in the transition from benign prostate to CRPC may thus reflect selection of tumors cells capable of efficiently synthesizing 5α-DHT via this pathway [4, 48-50]. The classical pathway primarily refers to the production of T by the testes. After castration, the backdoor and 5α-dione pathways allow for the production of DHT from steroids of adrenal origin. Importantly, regardless of which pathway is operative in CRPC tissues, inhibition of steroidogenesis via the backdoor and 5α-dione pathways requires inhibition of the same enzymatic conversions which produce 5α-DHT via the conventional pathway.
Novel derivatives of adrenal steroids capable of activating the androgen receptor
While the well-understood products of the adrenal gland are mineralocorticoids (aldosterone, corticosterone), glucocorticoids (cortisol), and the adrenal androgens (DHEA-S, AED and small amounts of T), the activity of several other intermediates with potential relevance to CRPC have recently been elucidated. These include DOC, the immediate precursor to corticosterone in the mineralocorticoid pathway, and 11OH-AED, derived from the hydroxylase activity of adrenal CYP11B1 on AED (Figure 1) [8-11]. In particular, recent data demonstrate that the 5α reduced metabolites of DOC and 11OH-AED can mediate wild type AR activation [8-11]. Given the well-recognized presence of SRD5A activity in CRPC tissue, these observations suggest that in the low androgen environment of CRPC, alternative sources of 5α-reduced ligands may supplement AR activation normally mediated by the canonical 5α-reduced agonist 5α-DHT. Moreover, whereas a number of non-canonical ligands, including pregnenolone, glucocorticoids, AED, androstenediol, and estradiol can cause promiscuous activation of mutated AR, the 5α-reduced metabolites of 11OH-AED discussed herein have nM activity toward the wild type AR.
11deoxycorticosterone (DOC) and 5α-dihydro-deoxycorticosterone (5α-DH-DOC)
In a screen designed to identify novel ligands capable of initiating AR signaling, Uemura et al identified the immediate precursor of corticosterone, DOC, as a novel substrate for SRD5A1, and showed that DOC and 5α-DH-DOC could stimulate both wild type and mutated AR activity in CRPC cell lines [8]. Although aldosterone is considered the final step and primary product of adrenal mineralocorticoid synthesis, serum levels of its precursor DOC (2-19 ng/dl, 0.06-6 nM) are equivalent to that of aldosterone (3-16 ng/dl, 0.08-0.4 nM), and in the range of serum concentrations of T in castrate men (20ng/dl or 0.69 nM). Moreover, in primary adrenal cultures, DOC occupies a similar percentage of the basal steroid output as AED (3% and 4%, respectively) and shows a 12.8 fold increase upon ACTH stimulation [51]. In adrenal vein sampling of corticosteroids, DOC showed an 82-fold post-ACTH increase [52].
Both DOC and 5α-DH-DOC showed trans-activation of mutant AR (T877A in LNCaP, H874Y in 22RV1 cells) and of wild type AR (PC3 cells transfected with wild-type AR). There was no activity in mock-transfected PC3 cells, and transactivation was blocked by the AR antagonist bicalutamide. Whereas DOC was more effective than 5α-DH-DOC in activation of mutant AR (activation achieved at 10nM vs 100nM), the 5α-reduced metabolite was more effective than DOC against wild-type AR (activation achieved at 10nM vs 100nM, Table 1) [8].
Relative predicted potency of canonical and non-canonical AR ligands against wild type and mutated AR*.
| Levels in castrate tissue** | wild type AR | LnCaP (T877A) | 22Rv1 (H874Y) | ||
|---|---|---|---|---|---|
| Canonical Ligands | |||||
| Dihydrotestosterone (5α-DHT) | 0.86 nM [3] | 0.01-0.1 nM [13, 64, 66] | 0.1-1 nM [66-68] | 0.01 nM [66] | |
| Testosterone | 2.57 nM [3] | 0.1-1 nM [13, 66] 60% of DHT at 1nM [10] | 1 nM [13] | 0.01-0.1 nM [66] | |
| Non-canonical Ligands | |||||
| Pregnenolone (P5) | 20.82 nM [16] | 0.2-2 nM [65] | |||
| Progesterone (P4) | 0.35 nM [16] | 10-100 nM [8, 66, 68] | 1-10 nM [66-68] | 10 nM [66] | |
| Aldosterone | >10 nM [68] | >10 nM [68] | |||
| Cortisol | >10 nM [68] | >10 nM [68] | |||
| DHEA | 88.8 nM [16] | >100 nM [13] | 100 nM [66] | 10-100 nM [66] | |
| Androstenedione (AED) | 0.94 nM [16] | 1-10 nM [13, 66] | 10 nM [65, 66] | 1-10 nM [66] | |
| Androstenediol (A5-diol) | 9.66 nM [13] | 0.1-1 nM [14] | 0.1 nM[13] | ||
| Estradiol | >10 nM [66, 68] | 1-10 nM [66-68] | 0.1-1 nM [66] | ||
| Flutamide | >10 nM [66, 68] | 1-10 nM [66-68] | 10 nM [66] | ||
| Novel derivatives of DOC and AED | |||||
| 11-deoxycorticosterone (DOC) | 100 nM [8] | 10 nM [8] | 10-100 nM [8] | ||
| 5αDH-DOC | 0.01-0.05 nM [8] | 10-100 nM [8] | 100 nM [8] | 1 uM [8] | |
| 11OH-AED | 0% of DHT at 1nM [10] | ||||
| 11OH-T | 10-100nM [64] 30% of DHT at 1nM [10] | ||||
| 11K-T | 1-10nM [64] 62% of DHT at 1nM [10] | ||||
| 11OH-5α-DHT | 47% of DHT at 1nM [10] | ||||
| 11K-5α-DHT | 96% of DHT at 1 nM [10] | ||||
*By necessity, estimates of agonist activity are drawn from multiple different studies with differing methods of measuring predicted potency. Values are intended only to provide a reasonable likelihood for the range of activity.
**Values for T, DHT and 5αDH-DOC are from CRPC metastases, remaining androgen values are from prostate tissue resected after neoadjuvant medical castration therapy.
CRPC is characterized by a shift away from SRD5A2 (the principle isoform expressed in benign prostate tissue [48]) to increased expression of SRD5A1 [4, 49, 50] and the more recently described SRD5A3 [53, 54]. To investigate whether the intra-cellular conversion of DOC to 5α-DH-DOC could be mediated by SRD5A isoforms present in CRPC, Uemura evaluated conversion of DOC to 5α-DH-DOC in COS cells transfected with SRD5A1 or SRD5A3, demonstrating robust conversion with SRD5A1 but none with SRD5A3. Conversion could be blocked by dutasteride, a dual SRD5A1 and 2 inhibitor, or by knockdown of basal SRD5A1 expression in 22RV1 cells by siRNA, confirming the endogenous ability of PCa cells to generate 5α-DH-DOC from DOC.
Additional support for the potential clinical relevance of these observations was provided by LC-MS/MS studies demonstrating the presence of measurable 5α-DH-DOC levels in CRPC tissue. While 5α-DHT levels were comparable to those previously reported (100-800pg/g tissue) [2], 5α-DH-DOC, although at much lower levels, was also detected in 8 of 13 fresh-frozen tumor samples (in concentrations ranging from 2-16pg/g, 0.01-0.05nM). Intriguingly, there was a strong inverse correlation between tissue levels of 5α-DHT and 5α-DH-DOC (Pearson r= -0.57, p<0.05), suggesting DOC and its conversion to 5α-DH-DOC may contribute to facilitating AR signaling in cells with relatively lower levels of 5α-DHT.
11beta-hydroxyandrostenedione (11OH-AED) and its 5α-reduced metabolites
In addition to producing the C-19 steroids DHEA-S, DHEA and AED, the adrenal gland secretes 11OH-AED, derived from the CYP11B1 mediated hydroxylation of AED (Figure 1) [9, 11]. The low androgenic potential of this metabolite (with 35-fold less androgen activity than 5α-DHT) [55] had historically rendered it of little biologic interest, and its production from AED was primarily viewed as a mechanism to inactivate AED and regulate adrenal androgen output [56]. However, subsequent work has demonstrated that 5α reduced metabolites of 11OH-AED are produced in prostate cancer cell lines, and can activate wild type AR, some with a predicted potency similar to 5α-DHT, suggesting these metabolites may have an unappreciated role in maintaining AR-axis activity in CRPC [10, 11].
Recent studies (employing ultra-sensitive mass spectrometry techniques) in adrenal cell-line models, primary adrenal cultures, and in adrenal vein sampling studies have confirmed that 11OH-AED is a major product of adrenal steroidogenesis [51, 57, 58]. Circulating levels of 11OH-AED (4.4 nM) are similar to that of AED (1.5-6.5 nM) [57], and its output is elevated in response to ACTH [51]. Forskolin stimulation of adrenal H295R cells (which produce steroids from all three zones of the adrenal gland, but are insensitive to ACTH) demonstrated a 3.4 fold increase in pregnenolone (to 799 nM), 2 fold increase in DHEA (to 465 nM), 1.4 fold increase in AED (to 1338 nM), and a 3.5 fold increase in 11OH-AED (to 330 nM) [51]. In primary adrenal cultures, while glucocorticoids are the predominant steroids produced (45%), AED (4%) and 11OH-AED (9%) are the two most abundant androgens produced under basal conditions, and upon ACTH stimulation showed a 25 fold and 16.6 fold increase in production, respectively[51]. Evaluation of adrenal vein samples after ACTH stimulation likewise showed adrenal production of 11OH-AED (811 nM) similar to that of AED (585 nM) [58].
The metabolic pathways downstream of 11OH-AED have also been the subject of recent investigation. A schematic of the pathways mediating adrenal conversion of AED to 11OH-AED by CYP11B1, and the conversion (likely peripheral) of 11OH-AED to 11K-AED by HSD11B2, and the downstream metabolites of each is presented in Figure 3. Notably, adrenal output of 11K-AED is low [58], whereas expression of HSD11B2 has been observed in prostate cancer cells [59, 60], in prostate tissue [61, 62], and in CRPC metastases (Mostaghel, unpublished data), suggesting conversion of 11OH-AED to 11K-AED may be peripherally mediated. Accordingly, Swart et al demonstrated conversion of 11OH-AED to 11K-AED in LNCaP cells, confirming the intracellular activity of HSD11B2 [11].
Conversion of AED to 11OH-AED and subsequent conversion to 5α-reduced metabolites. AED is converted to its 11hydroxy (11OH-AED) and 11keto (11K-AED) derivatives (hatched squares) via the activity of CYP11B1 and HSD11B2. Testosterone can be acted on by either CYP11B1 and CYP11B2. 11OH-AED and 11K-AED (as well as 11OH-T and 11K-T) are then converted to their 5α-reduced metabolites via pathways that essentially parallel the conversion of AED to 5α-DHT. The activity of 17BHSD2 favors conversion of the 11OH derivatives (11OH-T and 11OH-5α-DHT) to 11OH-AED and 11OH-5α-dione, whereas activity of 17BHSD3/AKR1C3 favors conversion of the 11K (11K-AED and 11K-5α-dione) to11K-T and 11K-5α-DHT (unfavored reaction denoted by dashed horizontal arrows). 11BHSD1 can mediate conversion of the keto-derivatives back to the hydroxy forms, but is not expressed in LNCaPs (long dashed arrows).
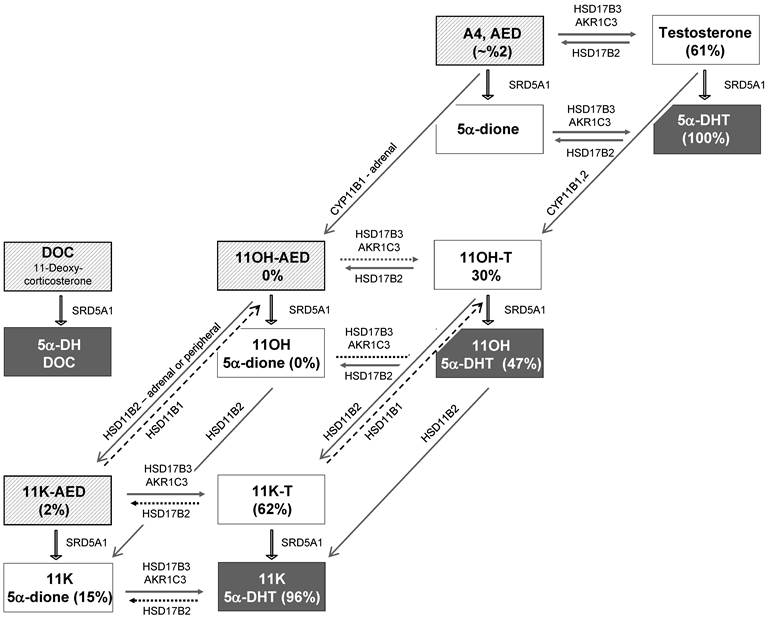
Storbeck el al further demonstrated that downstream metabolism of these two AED derivatives essentially parallels the intracrine metabolism of AED to T, 5α-dione, and 5α-DHT (Figure 3), ultimately resulting in production of the novel C-19 steroids, 11K 5α-dione, 11-OH 5α-DHT and 11-K-5α-DHT, which had not been previously described [10]. Overexpression of HSD17B3 in COS-1 cells resulted in 75% conversion of AED to T in 24 hours, 90% conversion of 11K-AED to 11K-T (consistent with a prior report), but demonstrated poor conversion of 11OH-AED (5%). Overexpression of AKR1C3 similarly yielded efficient conversion of 11K-AED to 11K-T (80%) and poor conversion of 11OH-AED (5%), but had less activity toward AED than did HSD17B3 (8%; consistent with prior findings that AED is a better substrate for HSD17B3 than AKR1C3) [63]. Both SRD5A1 and SRD5A2 mediated conversion of 11OH-AED, 11OH-T, 11K-AED, and 11K-T to their respective dihydro forms: 11OH-5α-dione, 11OH-5α-DHT, 11K-5α-dione and 11K-5α-DHT. Consistent with prior observations that AED is a better substrate for 5α-reduction than T [44, 46, 47], 11OH-AED and 11K-AED were metabolized more efficiently than 11OH-T and 11K-T. 11OH-T and 11OH-5α-DHT were metabolized to 11OH-AED and 11OH-5α-dione, consistent with the reductive activity of 17BHSD2 for T and 5α-DHT, while this enzyme exhibited poor activity towards 11K-T and 11K-5α-DHT.
The authors confirmed these findings in LNCaP cells, demonstrating conversion of the 11OH derivatives of AED, T, 5α-dione and 5α-DHT to the 11K derivatives, 11K-AED, 11K-T, 11K-5α-dione and 11K-5α-DHT, respectively, consistent with the known dehydrogenase action of 11BHSD2 in LNCaP cells (while 11BHSD1 mediates conversion of the keto-derivative back to the hydroxy form, this enzyme is not expressed in LNCaPs)[59]. Interestingly the activity of 17BHSD2 favored conversion of the 11OH derivatives (11OH-T and 11OH-5α-DHT) to 11OH-AED and 11OH-5α-dione, whereas activity of 17BHSD3/AKR1C3 favored conversion of the 11K derivatives (11K-AED and 11K-5α-dione) to11K-T and 11K-5α-DHT. Finally, 11OH-AED was converted to 11OH-5α-dione, and 11K-T was converted to 11K-5α-DHT, consistent with the activity of SRD5A.
Despite the negligible androgen activity of 11OH-AED, generation of the potent AR agonist 5αDHT, via 5α-reduction of the relatively weak AR agonist AED (Table 1) led Storbeck et al to question whether the 5α-reduced products of known 11OH-AED metabolites might be similarly potent AR agonists. In COS cells expressing full-length wild type AR and an ARE-driven luciferase reporter construct, 11OH-AED, 11OH-5α-dione, and 11K-AED at 1nM elicited no agonist activity, while 11K-5α-dione showed weak agonist activity (16% of 1nM 5α-DHT). 11OH-T demonstrated moderate activity (30%), while 11OH-5α-DHT (47%) and 11K-T (62%) demonstrated agonist activity comparable to 1nM T (61%), similar to earlier findings of Yazawa et al who found 11K-T to have similar agonist activity as T [64]. Notably, the agonist activity of 11K-5α-DHT at the wild-type AR was comparable to that of 5α-DHT (96%). Activity against a mutated AR was not tested, and to date, levels of these novel metabolites in prostate or CRPC tissue have not been reported, although quantitative evaluation is limited by the lack of commercially available standards.
Significance of DOC and 11OH-AED steroid derivatives in CRPC progression
The observation that circulating DOC and 11-OH-AED are significant products of adrenal steroidogenesis, that wild type AR can be activated by their 5α-reduced metabolites, and that SRD5A1 is highly expressed in CRPC tissue, all suggest that 5α-reduced ligands other than 5α-DHT may facilitate AR activation in the low androgen environment of CRPC. The significance of this hypothesis, however, is subject to a number of important questions. How does activation of wild-type or mutated AR by these ligands compare to AR activation by other non-canonical ligands? Are these ligands present in serum or tissue at levels likely to achieve AR activation? Is generation of these ligands inhibited by new ligand synthesis inhibitors such as abiraterone, or is their activity inhibited by new AR inhibitors such as enzalutamide? Is specific therapeutic targeting of these ligands achievable and/or likely to be clinically relevant?
Notably, the relative androgen activity of these derivatives at wild type AR (10nM for 5αDH-DOC, 1nM for the11OH-AED derivatives) is comparable to that of noncanonical ligands such as progesterone, estradiol and flutamide for activating mutated AR (summarized in Table 1) [14, 65-68]. As mutated AR is expressed in 10-30% of CRPC tumors [69, 70], this suggests the activity of these novel 5α-reduced derivatives may actually be relevant in the larger proportion of CRPC tumors in which wild type AR continues to be expressed.
Levels of DOC and 11OH-AED metabolites in CRPC tissue
Whether the 5α-reduced metabolites of DOC or 11OH-AED are present in CRPC tumors at levels capable of contributing to AR activation depends on several factors, including the serum and tissue levels of upstream precursors available for conversion, and tumor expression of enzymes mediating conversion of precursors to downstream metabolites. In particular, while the presence of SRD5A1, AKR1C3 and HSD17B3 in CRPC tissue (described above) is well established, the expression of CYP11B1/B2 and 11BHSD1 has not been rigorously evaluated.
Depending on factors such as the magnitude of AR amplification or overexpression, and residual levels of canonical ligands still present in the tumor, even low levels of these metabolites may serve to increase the overall androgenicity to a threshold capable of maintaining AR activity. From this perspective, the data of Uemura et al would suggest that given the predicted potency of 5α-DH-DOC for activating wild type AR (~10nM), the absolute level of 5α-DH-DOC detected in CRPC tissue (0.01-0.05nM) is not high enough (alone) to activate wild-type AR [8]. However, the strong inverse correlation noted between tissue levels of 5α-DHT and 5α-DH-DOC is consistent with the hypothesis that 5α-DH-DOC may serve to facilitate AR signaling in cells with relatively lower levels of 5α-DHT.
In contrast, the predicted potency of the 11OH-AED derivatives for activating wild type AR (1nM) is much higher than that of 5α-DOC (10-100nM), and serum levels of the key precursor, 11OH-AED, are higher than levels of DOC. Whereas serum levels of DOC are relatively low (0.06-0.6nM)[8], serum levels of 11OH-AED prior to castration are much higher (4.4nM)[57], and are similar to levels of AED (1.5-6.5 nM) and 5α-DHT (1.0-2.9 nM). While absolute tissue levels of 11OH-AED have not been reported, extrapolation from available data on AED suggests that serum and tissue levels of 11OH-AED are likely to be unchanged after castration. In particular, although serum and tissue levels of 5α-DHT decrease markedly following castration (to 0.18 nM in serum; 15 to 5.3 nM in tissue), serum and tissue levels of the adrenal androgens AED do not (AED: 1.2 nM in serum, 0.9 to 0.9 nM in tissue) [15, 16]. These observations suggest that serum and tissue levels of adrenally-derived 11OH-AED may also be preserved following castration.
Whereas tissue levels of AED following castration are within the 1nM concentration range at which the 5α-reduced metabolites of 11OH-AED activated wild-type AR, it is unknown whether uptake and levels of 11OH-AED (or its metabolites) in tissue is similar to levels of AED. The relatively high serum level of 11OH-AED suggests tissue levels will be primarily contributed by adrenal sources. However, although primarily expressed in the adrenal gland, some expression of the CYP11B1 and CYP11B2 enzymes (which mediate conversion of AED and T to the 11OH derivatives) in CRPC tissue and cell lines has been reported [71, 72], suggesting uptake of adrenally-derived 11OH-AED could potentially be supplemented by intracrine generation of 11OH-AED and 11OH-T (from AED and T, respectively).
In contrast, serum levels of 11K derivatives are quite low [58], suggesting that clinically relevant tissue levels of these metabolites would likely be derived from peripheral HSD11B2-mediated conversion of the 11OH derivatives to 11K-AED and 11K-T. In this regard, expression of HSD11B2 has been observed in prostate cancer cells [59, 60], in murine and human prostate tissue [61, 62], and in CRPC metastases (Mostaghel, unpublished data). Moreover, peripheral expression of HSD11B2 in steroidogenic tissues, such as placenta, testis, ovary, skin, and kidney is well-recognized; suggesting its expression in the steroidogenic environment of CRPC is plausible.
These data suggest that a larger pool of residual androgens may be available to activate the AR in CRPC than previously anticipated, and that measurements of residual T and 5α-DHT may underestimate the contribution of ligand-mediated activation of wild type AR. Although these pathways remain to be more fully elucidated, including evaluation of tissue steroid levels and expression of the relevant enzymes, available data, particularly for 11OH-AED, are consistent with the proposed intracrine generation and activity of these novel 5α-reduced metabolites.
Rationale for targeting 5α-reduced metabolites of DOC and 11OH-AED
As the target of these novel 5α-metabolites remains the AR, and the enzymatic pathways mediating conversion of DOC and11OH-AED to their respective 5α-reduced metabolites essentially parallel the metabolism of AED to 5α-DHT, the activity of these metabolites should theoretically be abrogated by the potent AR, CYP17A1 and SRD5A1/2 inhibitors already in clinical practice (e.g. enzalutamide, abiraterone, and dutasteride). However, SRD5A inhibition has failed to demonstrate significant clinical efficacy in CRPC [73, 74], and while clinical responses to enzalutamide and abiraterone have been impressive, not all patients respond, the duration of response is variable, and a majority of men eventually progress with a rising PSA suggestive of AR axis reactivation [7, 75].
Although mechanisms of resistance have not been fully elucidated, the involvement of these alternative ligands in activating the AR has precedent in the activation of mutated AR by non-canonical ligands, and may even be more clinically relevant, given that the 5α-reduced metabolites of DOC and 11OH-AED are capable of activating wild type AR. While progression on abiraterone is not associated with an increase in serum levels of adrenal androgens, studies in xenograft models demonstrate that increased tissue androgens and steroidogenesis at least partially underlie resistance to abiraterone (16-18). Moreover, the ability of new AR antagonists such as enzalutamide to inhibit AR activation by these ligands cannot necessarily be assumed, as 5-androstenediol has been shown to activate wild type AR with nM potency without being inhibited by flutamide or bicalutamide [14].
Recent data evaluating serum and tissue steroid levels in abiraterone-treated patient suggests generation of these 5α-reduced metabolites would not be abrogated. In particular, although residual levels of DHEA-S in abiraterone-treated men are markedly decreased compared to eugondal levels (from ~5 uM to 424 nM in serum, and 1 uM to 42.4 nM in tissue) [16], a substantial reservoir of this precursor clearly remains available for adrenal or peripheral conversion to 11OH-AED. Likewise, residual prostate levels of 5α-DHT (0.65 nM) are consistent with ongoing activity in the AKR1C3/HSD17B3 and SRD5A pathways necessary for conversion of 11OH-AED to its 5α-reduced metabolites.
Elevated levels of DOC and its subsequent conversion to 5α-DH-DOC may also play a role in resistance if abiraterone is given without prednisone. Abiraterone, which inhibits both the lyase and hydroxylase activity of CYP17A1, results in an ACTH-stimulated increase in mineralocorticoids including a 10 fold increase in DOC [76]. In men who have progressed on abiraterone alone, addition of dexamethasone (which suppresses ACTH to less than baseline and DOC to < 5ng/dL) has been associated with secondary PSA responses [76]. This has been attributed to reversal of promiscuous activation of mutated AR by high levels of upstream steroids, but may potentially reflect reversal of wild type AR activation by 5α-reduced metabolites of DOC.
Targeting HSD11B2 and CYP11B1, enzymes unique to the generation of 11OH-AED
Collectively, these observations suggest that simultaneously targeting multiple nodes of androgen production will be required to truly abrogate generation of all AR activating downstream metabolites. Moreover, the significant activity of 11OH-5α-DHT and 11K-5α-DHT in activating the AR suggests that inhibiting the enzymes unique to the production of these metabolites might significantly decrease the overall androgenicity of the CRPC tumor microenvironment. Enzymatic targets specific to these metabolites include the adrenal activity of CYP11B1 in converting AED to 11OH-AED, and the (most likely) peripheral action of HSD11B2 in yielding the 11K-derivatives.
While endogenous selective inhibitors of 11BHSD isoforms have been described [77, 78], inhibition of HSD11B2, which metabolizes cortisol to its inactive form cortisone, may not be a viable therapeutic target. Aldosterone and cortisol have similar affinity for the mineralocorticoid receptor, and the presence of this enzyme in mineralocorticoid target tissues prevents excess stimulation of the receptor by cortisol [77, 78]. Notably, mutations in HSD11B2 which affect enzyme activity result in a rare autosomal dominant disorder, the syndrome of apparent mineralocorticoid excess (AME), and inhibition of this enzyme by chronic licorice ingestion results in pseudohyperaldosteronism [79].
In contrast, inhibition of CYP11B1 is likely to be a clinically feasible target; in fact, metyrapone is a CYP11B1 inhibitor already in use for treatment of hypercortisolism associated with Cushing's syndrome. Of more relevance to the treatment of prostate cancer however, is the recent development of dual CYP17A1 and CYP11B inhibitors [80, 81]. A dual CYP17A1/CYP11B2 inhibitor (Novartis, CFG920), intended to ameliorate the mineralocorticoid side effects of CYP17A1 inhibition, is currently under clinical evaluation (NCT01647789). Although this agent would not be anticipated to have efficacy in suppressing generation of 11OH-AED, specific inhibitors of CYP17A1 and CYP11B1 have also been reported [80, 81], and the potential androgenicity associated with the downstream metabolites of 11OH-AED would support clinical evaluation of these dual inhibitors as well.
Conclusions
Data regarding the molecular response of PCa to hormone therapy continues to emerge, providing critical insight into cellular growth and signaling pathways that may be exploited as therapeutic targets. The presence of residual androgens and persistent activation of the AR signaling axis in CRPC suggest that a multi-targeted treatment approach to ablate all contributions to AR signaling within the prostate tumor will be required for optimal anti-tumor efficacy. Emerging data demonstrate that novel 5α-reduced derivative of the adrenal steroids DOC and 11OH-AED can activate the wild type AR, and suggest that a larger pool of residual androgens may be available to activate the AR in CRPC than previously anticipated. Although these pathways remain to be more fully elucidated, available data are consistent with the proposed intracrine generation and activity of these novel 5α-reduced metabolites. Moreover, these data suggest that measurements of residual T and 5α-DHT may underestimate the contribution of ligand-mediated activation of wild type AR. Importantly, the significant activity of 11OH-5α-DHT and 11K-5α-DHT in activating the wild type AR suggests that inhibiting the enzymes unique to the production of these metabolites may significantly decrease the overall androgenicity of the CRPC tumor microenvironment and thereby improve clinical outcomes.
Abbreviations
11OH-AED: 11beta-hydroxyandrostenedione; ADT: androgen deprivation therapy; AED: androstenedione; AR: androgen receptor; CAB: combined androgen blockade; CRPC: castration resistant prostate cancer; DHEA: dehydroepiandrosterone; DHEA-S: dehydroepiandrosterone sulfate; 5α-DHT: dihydrotestosterone; DOC: 11-deoxcorticosterone; PCa: prostate cancer; T: testosterone.
Acknowledgements
This work was supported by Pacific Northwest Prostate Cancer SPORE P50 CA97186; Department of Defense CDMRP; Prostate Cancer Foundation; Damon Runyon Cancer Research Foundation (Damon Runyon-Genentech Clinical Investigator Award CI-40-08).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ross RW, Xie W, Regan MM, Pomerantz M, Nakabayashi M, Daskivich TJ. et al. Efficacy of androgen deprivation therapy (ADT) in patients with advanced prostate cancer: association between Gleason score, prostate-specific antigen level, and prior ADT exposure with duration of ADT effect. Cancer. 2008;112:1247-53
2. Mohler JL, Gregory CW, Ford OH 3rd, Kim D, Weaver CM, Petrusz P. et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440-8
3. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS. et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447-54
4. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM. et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815-25
5. Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L. et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217-27
6. Mostaghel EA, Plymate SR, Montgomery B. Molecular pathways: targeting resistance in the androgen receptor for therapeutic benefit. Clin Cancer Res. 2014;20:791-8
7. Mostaghel EA. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag Res. 2014;6:39-51
8. Uemura M, Honma S, Chung S, Takata R, Furihata M, Nishimura K. et al. 5alphaDH-DOC (5alpha-dihydro-deoxycorticosterone) activates androgen receptor in castration-resistant prostate cancer. Cancer Sci. 2010;101:1897-904
9. Bloem LM, Storbeck KH, Schloms L, Swart AC. 11beta-hydroxyandrostenedione returns to the steroid arena: biosynthesis, metabolism and function. Molecules. 2013;18:13228-44
10. Storbeck KH, Bloem LM, Africander D, Schloms L, Swart P, Swart AC. 11beta-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: a putative role in castration resistant prostate cancer? Mol Cell Endocrinol. 2013;377:135-46
11. Swart AC, Schloms L, Storbeck KH, Bloem LM, Toit T, Quanson JL. et al. 11beta-hydroxyandrostenedione, the product of androstenedione metabolism in the adrenal, is metabolized in LNCaP cells by 5alpha-reductase yielding 11beta-hydroxy-5alpha-androstanedione. J Steroid Biochem Mol Biol. 2013;138:132-42
12. Page ST, Plymate SR, Bremner WJ, Matsumoto AM, Hess DL, Lin DW. et al. Effect of medical castration on CD4+ CD25+ T cells, CD8+ T cell IFN-gamma expression, and NK cells: a physiological role for testosterone and/or its metabolites. Am J Physiol Endocrinol Metab. 2006;290:E856-63
13. Mizokami A, Koh E, Fujita H, Maeda Y, Egawa M, Koshida K. et al. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res. 2004;64:765-71
14. Miyamoto H, Yeh S, Lardy H, Messing E, Chang C. Delta5-androstenediol is a natural hormone with androgenic activity in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95:11083-8
15. Mostaghel EA, Nelson PS, Lange P, Lin DW, Taplin ME, Balk S. et al. Targeted androgen pathway suppression in localized prostate cancer: a pilot study. J Clin Oncol. 2014;32:229-37
16. Taplin M, Montgomery RB, Logothetis C, Bubley GJ, Richie JP, Dalkin BL. et al. Effect of neoadjuvant abiraterone acetate (AA) plus leuprolide acetate (LHRHa) on PSA, pathological complete response (pCR), and near pCR in localized high-risk prostate cancer (LHRPC): Results of a randomized phase II study. ASCO Annual Meeting. 2012 Chicago, Illinois: abstract 4521
17. Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A. et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999;81:242-51
18. Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892-8
19. Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL. et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718-24
20. Mohler JL, Morris TL, Ford OH 3rd, Alvey RF, Sakamoto C, Gregory CW. Identification of differentially expressed genes associated with androgen-independent growth of prostate cancer. Prostate. 2002;51:247-55
21. de Bono JS. Abiraterone acetate improves survival in metastatic castration-resistant prostate cancer: Phase III results. 2010 European Society for Medical Oncology. Milan. 2010
22. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E. et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437-46
23. Mostaghel EA, Marck B, Plymate S, Vessella RL, Balk SP, Matsumoto AM. et al. Resistance to CYP17A1 inhibition with abiraterone in castration resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011
24. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA. et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503-13
25. Efstathiou E, Titus M, Tsavachidou D, Tzelepi V, Wen S, Hoang A. et al. Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. J Clin Oncol. 2012;30:637-43
26. Labrie F, Luu-The V, Lin S, Simard J, Labrie C, El-Alfy M. et al. Intracrinology: role of the family of 17 beta-hydroxysteroid dehydrogenases in human physiology and disease. J Mol Endocrinol. 2000;25:1-16
27. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81-151
28. Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. Dissecting human adrenal androgen production. Trends in Endocrinology and Metabolism. 2002;13:234-9
29. Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15:432-8
30. Auchus RJ. Non-traditional metabolic pathways of adrenal steroids. Rev Endocr Metab Disord. 2009;10:27-32
31. Endoh A, Kristiansen SB, Casson PR, Buster JE, Hornsby PJ. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3 beta-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab. 1996;81:3558-65
32. Nakamura Y, Hornsby PJ, Casson P, Morimoto R, Satoh F, Xing Y. et al. Type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. J Clin Endocrinol Metab. 2009;94:2192-8
33. Sanford EJ, Paulson DF, Rohner TJ Jr, Santen RJ, Bardin CW. The effects of castration on adrenal testosterone secretion in men with prostatic carcinoma. J Urol. 1977;118:1019-21
34. Mostaghel EA, Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab. 2008;22:243-58
35. Penning TM, Bauman DR, Jin Y, Rizner TL. Identification of the molecular switch that regulates access of 5[alpha]-DHT to the androgen receptor. Molecular and Cellular Endocrinology. 2007;265-266:77-82
36. Guillemette C, Levesque E, Beaulieu M, Turgeon D, Hum DW, Belanger A. Differential regulation of two uridine diphospho-glucuronosyltransferases, UGT2B15 and UGT2B17, in human prostate LNCaP cells. Endocrinology. 1997;138:2998-3005
37. Chouinard S, Barbier O, Belanger A. UDP-glucuronosyltransferase 2B15 (UGT2B15) and UGT2B17 enzymes are major determinants of the androgen response in prostate cancer LNCaP cells. J Biol Chem. 2007;282:33466-74
38. Chouinard S, Pelletier G, Belanger A, Barbier O. Cellular specific expression of the androgen-conjugating enzymes UGT2B15 and UGT2B17 in the human prostate epithelium. Endocr Res. 2004;30:717-25
39. Gupta MK, Guryev OL, Auchus RJ. 5alpha-reduced C21 steroids are substrates for human cytochrome P450c17. Arch Biochem Biophys. 2003;418:151-60
40. Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the Major Oxidative 3{alpha}-Hydroxysteroid Dehydrogenase in Human Prostate That Converts 5{alpha}-Androstane-3{alpha},17{beta}-diol to 5{alpha}-Dihydrotestosterone: A Potential Therapeutic Target for Androgen-Dependent Disease. Mol Endocrinol. 2006;20:444-58
41. Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB. et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486-96
42. Huang XF, Luu-The V. Molecular characterization of a first human 3(alpha-->beta)-hydroxysteroid epimerase. J Biol Chem. 2000;275:29452-7
43. Muthusamy S, Andersson S, Kim HJ, Butler R, Waage L, Bergerheim U. et al. Estrogen receptor beta and 17beta-hydroxysteroid dehydrogenase type 6, a growth regulatory pathway that is lost in prostate cancer. Proc Natl Acad Sci U S A. 2011;108:20090-4
44. Negri-Cesi P, Motta M. Androgen metabolism in the human prostatic cancer cell line LNCaP. J Steroid Biochem Mol Biol. 1994;51:89-96
45. Thigpen AE, Cala KM, Russell DW. Characterization of Chinese hamster ovary cell lines expressing human steroid 5 alpha-reductase isozymes. J Biol Chem. 1993;268:17404-12
46. Samson M, Labrie F, Zouboulis CC, Luu-The V. Biosynthesis of dihydrotestosterone by a pathway that does not require testosterone as an intermediate in the SZ95 sebaceous gland cell line. J Invest Dermatol. 2010;130:602-4
47. Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ. et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728-33
48. Klein H, Bressel M, Kastendieck H, Voigt KD. Androgens, adrenal androgen precursors, and their metabolism in untreated primary tumors and lymph node metastases of human prostatic cancer. Am J Clin Oncol. 1988;11(Suppl 2):S30-6
49. Luo J, Dunn TA, Ewing CM, Walsh PC, Isaacs WB. Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: implications for finasteride therapy of prostate carcinoma. Prostate. 2003;57:134-9
50. Titus MA, Gregory CW, Ford OH, Schell MJ, Maygarden SJ, Mohler JL. Steroid 5{alpha}-Reductase Isozymes I and II in Recurrent Prostate Cancer. Clin Cancer Res. 2005;11:4365-71
51. Xing Y, Edwards MA, Ahlem C, Kennedy M, Cohen A, Gomez-Sanchez CE. et al. The effects of ACTH on steroid metabolomic profiles in human adrenal cells. J Endocrinol. 2011;209:327-35
52. Nakamura Y, Rege J, Satoh F, Morimoto R, Kennedy MR, Ahlem CN. et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein corticosteroids before and after adrenocorticotropic hormone stimulation. Clin Endocrinol (Oxf). 2012;76:778-84
53. Godoy A, Kawinski E, Li Y, Oka D, Alexiev B, Azzouni F. et al. 5alpha-reductase type 3 expression in human benign and malignant tissues: a comparative analysis during prostate cancer progression. Prostate. 2011;71:1033-46
54. Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Adv Urol. 2012;2012:530121
55. Belanger B, Fiet J, Belanger A. Effects of adrenocorticotropin on adrenal and plasma 11 beta-hydroxyandrostenedione in the guinea pig and determination of its relative androgen potency. Steroids. 1993;58:29-34
56. Goldzieher JW, de la Pena A, Aivaliotis MM. Radioimmunoassay of plasma androstenedione, testosterone and 11beta-hydroxyandrostenedione after chromatography on Lipidex-5000 (hydroxyalkoxypropyl Sephadex). J Steroid Biochem. 1978;9:169-73
57. Yokokawa A, Yamamoto K, Omori Y, Shibasaki H, Shinohara Y, Kasuya Y. et al. Simultaneous determination of androstenedione, 11beta-hydroxyandrostenedione, and testosterone in human plasma by stable isotope dilution mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:621-6
58. Rege J, Nakamura Y, Satoh F, Morimoto R, Kennedy MR, Layman LC. et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. J Clin Endocrinol Metab. 2013;98:1182-8
59. Dovio A, Sartori ML, De Francia S, Mussino S, Perotti P, Saba L. et al. Differential expression of determinants of glucocorticoid sensitivity in androgen-dependent and androgen-independent human prostate cancer cell lines. J Steroid Biochem Mol Biol. 2009;116:29-36
60. Page N, Warriar N, Govindan MV. 11 beta-Hydroxysteroid dehydrogenase and tissue specificity of androgen action in human prostate cancer cell LNCaP. J Steroid Biochem Mol Biol. 1994;49:173-81
61. Pelletier G, Luu-The V, Li S, Ouellet J, Labrie F. Cellular Localization of mRNA Expression of Enzymes Involved in the Formation and Inactivation of Hormonal Steroids in the Mouse Prostate. 10.1369/jhc.4A6311. J Histochem Cytochem. 2004;52:1351-6
62. Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol. 1994;105:R11-7
63. Dufort I, Rheault P, Huang XF, Soucy P, Luu-The V. Characteristics of a highly labile human type 5 17beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:568-74
64. Yazawa T, Uesaka M, Inaoka Y, Mizutani T, Sekiguchi T, Kajitani T. et al. Cyp11b1 is induced in the murine gonad by luteinizing hormone/human chorionic gonadotropin and involved in the production of 11-ketotestosterone, a major fish androgen: conservation and evolution of the androgen metabolic pathway. Endocrinology. 2008;149:1786-92
65. Grigoryev DN, Long BJ, Njar VC, Brodie AH. Pregnenolone stimulates LNCaP prostate cancer cell growth via the mutated androgen receptor. J Steroid Biochem Mol Biol. 2000;75:1-10
66. Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M. et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450-9
67. Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E. et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534-40
68. Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM. et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6:703-6
69. Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B. et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511-5
70. Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J. et al. Androgen receptor mutations in androgen-independent prostate cancer. J Clin Oncol. 2003;21:2673-8
71. Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK. et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142-52
72. Biancolella M, Valentini A, Minella D, Vecchione L, D'Amico F, Chillemi G. et al. Effects of dutasteride on the expression of genes related to androgen metabolism and related pathway in human prostate cancer cell lines. Invest New Drugs. 2007;25:491-7
73. Shah SK, Trump DL, Sartor O, Tan W, Wilding GE, Mohler JL. Phase II study of Dutasteride for recurrent prostate cancer during androgen deprivation therapy. J Urol. 2009;181:621-6
74. Sartor O, Nakabayashi M, Taplin ME, Ross RW, Kantoff PW, Balk SP. et al. Activity of dutasteride plus ketoconazole in castration-refractory prostate cancer after progression on ketoconazole alone. Clin Genitourin Cancer. 2009;7:E90-2
75. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187-97
76. Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S. et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563-71
77. Ge RS, Dong Q, Niu EM, Sottas CM, Hardy DO, Catterall JF. et al. 11{beta}-Hydroxysteroid dehydrogenase 2 in rat leydig cells: its role in blunting glucocorticoid action at physiological levels of substrate. Endocrinology. 2005;146:2657-64
78. Latif SA, Pardo HA, Hardy MP, Morris DJ. Endogenous selective inhibitors of 11beta-hydroxysteroid dehydrogenase isoforms 1 and 2 of adrenal origin. Mol Cell Endocrinol. 2005;243:43-50
79. Coeli FB, Ferraz LF, Lemos-Marini SH, Rigatto SZ, Belangero VM, de-Mello MP. Apparent mineralocorticoid excess syndrome in a Brazilian boy caused by the homozygous missense mutation p.R186C in the HSD11B2 gene. Arq Bras Endocrinol Metabol. 2008;52:1277-81
80. Hu Q, Jagusch C, Hille UE, Haupenthal J, Hartmann RW. Replacement of imidazolyl by pyridyl in biphenylmethylenes results in selective CYP17 and dual CYP17/CYP11B1 inhibitors for the treatment of prostate cancer. J Med Chem. 2010;53:5749-58
81. Pinto-Bazurco Mendieta MA, Hu Q, Engel M, Hartmann RW. Highly potent and selective nonsteroidal dual inhibitors of CYP17/CYP11B2 for the treatment of prostate cancer to reduce risks of cardiovascular diseases. J Med Chem. 2013;56:6101-7
Author contact
![]() Corresponding author: Elahe A Mostaghel MD PhD. Fred Hutchinson Cancer Research Center, 1100 Fairview Ave NE, MS D5-100, Seattle, WA 98109. emostaghorg phone: 206-667-3506 fax: 206-667-5456.
Corresponding author: Elahe A Mostaghel MD PhD. Fred Hutchinson Cancer Research Center, 1100 Fairview Ave NE, MS D5-100, Seattle, WA 98109. emostaghorg phone: 206-667-3506 fax: 206-667-5456.