Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(6):627-642. doi:10.7150/ijbs.8756 This issue Cite
Research Paper
Conversion of Androgen Receptor Signaling From a Growth Suppressor in Normal Prostate Epithelial Cells to an Oncogene in Prostate Cancer Cells Involves a Gain of Function in c-Myc Regulation
Donald J. Vander Griend1,3, Ivan V. Litvinov1,2, John T. Isaacs1,2,3 ![]()
1. Chemical Therapeutics Program, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins.
2. Cellular and Molecular Medicine Graduate Program at Johns Hopkins.
3. The Brady Urological Institute, Johns Hopkins.
Received 2014-2-4; Accepted 2014-3-12; Published 2014-6-10
Abstract
In normal prostate, androgen-dependent androgen receptor (AR) signaling within prostate stromal cells induces their secretion of paracrine factors, termed “andromedins” which stimulate growth of the epithelial cells. The present studies demonstrate that androgen-dependent andromedin-driven growth stimulation is counter-balanced by androgen-induced AR signaling within normal adult prostate epithelial cells resulting in terminal G0 growth arrest coupled with terminal differentiation into ΔNp63-negative, PSA-expressing secretory luminal cells. This cell autonomous AR-driven terminal differentiation requires DNA-binding of the AR protein, is associated with decreases in c-Myc m-RNA and protein, are coupled with increases in p21, p27, and SKP-2 protein expression, and does not require functional p53. These changes result in down-regulation of Cyclin D1 protein and RB phosphoryation. shRNA knockdown documents that neither RB, p21, p27 alone or in combination are required for such AR-induced G0 growth arrest. Transgenic expression of a constitutive vector to prevent c-Myc down-regulation overrides AR-mediated growth arrest in normal prostate epithelial cells, which documents that AR-induced c-Myc down-regulation is critical in terminal growth arrest of normal prostate epithelial cells. In contrast, in prostate cancer cells, androgen-induced AR signaling paradoxically up-regulates c-Myc expression and stimulates growth as documented by inhibition of both of these responses following exposure to the AR antagonist, bicalutamide. These data document that AR signaling is converted from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells during prostatic carcinogenesis and that this conversion involves a gain of function for regulation of c-Myc expression.
Keywords: Androgen Receptor, Human Prostate Cancer, Tumor Suppressor, Oncogene, MYC.
Introduction
Androgen is the major growth factor for the stratified epithelium of the prostate. Androgen deprivation induces the regression of the prostatic epithelium, which can be regenerated by androgen replacement [1]. More than 20 years ago, it was demonstrated that the prostate can undergo more than 30 successive cycles of androgen deprivation and replacement without diminishing its ability for continued epithelial regeneration [2]. Since then, a large number of independent groups have clarified how the prostate epithelium is organized into adult prostate epithelial stem cell units and how this organization allows such profound cyclic regenerative growth capacity [3-6]. In these adult prostate epithelial stem units, AR-negative adult prostate epithelial stem cells are located in the basal layer of the epithelium in niches that control their survival and self-renewal [7, 8]. The androgen independence of the adult prostate epithelial stem cells is supported by the tissue recombination studies of Gerry Cunha, which documented that epithelial morphogenesis occurs even when AR protein is not expressed by prostate epithelial cells as long as there is expression and signaling of AR in the supporting stromal cells [5].
The mechanism for epithelial morphogenesis without AR expression in the epithelial cells is related to the hierarchical expansion/ maturation of adult prostate epithelial stem cells and their progeny. Adult prostate epithelial stem cells divide giving rise to progeny that differentiate into either non-proliferating AR-negative neuroendocrine cells or ΔNp63-positive/AR-negative transient amplifying (TA) cells. TA cells undergo a limited number of amplifying rounds of proliferation before maturing into ΔNp63-negative/prostate stem cell antigen (PSCA)-positive intermediate cells [4, 9, 10]. Such TA proliferation requires the androgen regulated production and secretion of diffusible stromal-derived peptide growth factors collectively termed “andromedins.” These paracrine secreted andromedins diffuse from the stroma into the epithelial compartment where their binding to cognate receptors stimulates TA cell proliferation and maturation into intermediate cells [10]. Intermediate cells express AR protein and migrate from the basal to the luminal layer where they terminally differentiate into AR and prostate specific antigen (PSA) positive secretory-luminal cells whose survival depends on adequate levels of andromedins. Due to the hierarchically expanding nature of this process, the secretory-luminal cell is the most numerous cell type within an adult prostate epithelial stem cell unit, even though they eventually differentiate and stop proliferating (i.e. they terminally arrest in G0). Castration results in a rapid loss of AR-dependent transcription within prostate stromal cells [11] and thus cessation of stromal-derived paracrine andromedin production [12], which result in the lack of proliferation of TA cells and concomitant apoptosis of secretory-luminal cells [13]. Thus, castration induces prostate regression. Such regression is reversible, however, since re-administering androgen restores the critical level of stromal andromedins that allow TA cells to proliferate and mature into secretory-luminal cells, thereby regenerating the full complement of epithelial cells. Once regenerated, continuous growth ceases and the gland maintains a steady-state without regression or overgrowth.
While this adult stem cell model provides a framework for understanding both the paracrine mechanism of androgen action in the normal prostate and how the gland maintains its ability to regenerate through multiple cycles of androgen deprivation/restoration, only recently has there been an understanding of what restricts the continuous overgrowth of normal adult prostate epithelial stem cell units in the continuous presence of high levels of andromedins in the prostate of non-castrated adult males. Immunocytochemical studies document that in the human prostate the small fraction of proliferating normal prostate epithelial cells is located in the basal compartment and do not express AR protein while the AR positive secretory-luminal cells are proliferative quiescent [14]. It has also been demonstrated experimentally that AR signaling activated by androgen binding in prostate epithelial cells induces their growth arrest and eventual differentiation into secretory-luminal cells [15-17]. Likewise, transgenic mouse studies have documented that when the AR gene is knocked out selectively in secretory-luminal cells within the prostate, then only these AR deficient cells become hyper-proliferative and do not terminally differentiate [18-20]. These data document that AR-induced epithelial cell growth arrest limits the positive feed-forward proliferative stromal-driven paracrine loop, which prevents continuous prostatic epithelial hyperplasia in the presence of high levels of stromal andromedins chronically maintained by physiological levels of androgen in an intact male [21]. These studies document that the mechanism for such growth suppression is due to AR signaling down-regulating the transcription of c-Myc. During prostatic carcinogenesis, there is a molecular conversion from stromal dependent paracrine to cell autonomous autocrine AR signaling pathways in which prostate cancer cells become “addicted” to such cell autonomous AR oncogenic signaling [6, 22]. This oncogenic conversion involves a differential switch from AR signaling suppressing c-Myc expression in normal prostate epithelial cells to stimulating c-Myc expression in prostate cancer cells.
Materials and Methods
Materials and Cell Lines: The synthetic androgen R1881 was purchased from Perkin Elmer (Boston MA). The AR antagonist, Casodex (bicalutamide) was purchased from LKT laboratories (St Paul, MN). All other chemicals were purchased from JT Baker (Phillipsburg NJ) or Sigma-Aldrich (St. Louis MO). LNCaP, PrEC, and 957E/hTERT human prostate lines were obtained and grown as previously described [23]. Human normal prostate epithelial cells (PrECs) from young donors were obtained commercially from Lonza (Lonza/Cambrex, Walkersville MD) and grown in serum free defined PrEGM growth media containing Insulin (I), EGF, Bovine pituitary extract (BPE), triiodothyronine (T3), transferin, hydrocortisone (HC), epinephrine, and retinoic acid as supplied by manufacture (Lonza/Cambrex, Walkersville MD). PrECs and human normal prostate stromal cells (PrSC) from older donors undergoing radical prostatectomy were established as previously described [24]. 957E/hTERT cells were grown in Keratinoctye Serum Free defined media supplemented with growth factors (GFs) as supplied by manufacture (Invitrogen Life Technologies). LNCaP cells were grown in RPMI-1640 media containing 10% fetal bovine serum (FBS). LNCaP cells were grown on Poly-D-Lysine coated dishes when treated with Casodex to prevent detachment [22]. BPH-1 cells were provided by Simon Hayward and grown in RPMI-1640 media containing 10% FBS as previously described [25]. HPr-1AR cells were generously provided by Dr. CK Choo [16]. All cells were routinely screened for the absence of mycoplasma contamination.
Vectors and shRNA Knockdown: Creation of lentiviral-GFP control vector and the expression vector containing wild type AR flanked by loxP sites (lenti-GFP/AR vector), transduction of cells with control and Lenti-AR viruses, and AR protein expression and function were described previously [26]. CRE-Lox mediated excision was described previously [27]. The Lenti-AR(A573D) mutant vector was created by replacing the Bsu36I/Tth111I fragment with the same fragment from an A573D mutant vector generously provided by Dr. Hetty van der Korput [28]. The Lenti-AR(A573D) vector was sequenced to confirm the presence of the DNA-binding domain mutation. Lentivirus was produced by transfection of HEK293T cells according to Yu et al. [29], and GFP-positive expressing populations were isolated using a Becton Dickson FACS Aria machine. The pWZL-Blast-Myc plasmid was obtained from Addgene (Plasmid 10674) [30]. This c-Myc plasmid was packaged using the LinX retroviral expression system. This same system was used to package the V2HS_130611 shRNA construct coding for siRNA for RB that was purchased from Open Biosystems (Huntsville AL). Lentiviral shRNA vectors targeting p21 and p27 were constructed using the pRNATin-H1.4-Lenti vector according to the manufacturer's specifications (Genscript, Piscataway, NJ). Targeted sequences were p21 (AACTTCGACTTTGTCACCGAG) and p27 (AACCCGGGACTTGGAGAAGCA) as previously reported [31].
In Vitro Growth Assays: Cell growth was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay from Promega Corp. (Madison WI)) as previously described [26]. Time lapse fluorescence digital microscopy was performed using a TE2000 (Nikon) inverted microscope with a heated stage, the Live Cell (Pathology Devices) CO2 chamber, and a ELWD 20x objective and the Photometric CoolSnap ES digital camera; images were captured using Elements AR software program (Nikon). Clonogenic assays were performed by pre-treating cells in either K-SFM medium, or K-SFM supplemented with 1nM R1881. After 3 days, the cells are trypsinized, and a clonogenic assay was set up using 2000 cells in 3 dishes. The cells were given standard K-SFM or K-SFM supplemented with 1nM R1881 as above. After 6 days, the media was aspirated and the cells were washed with HBSS, stained with crystal violet, and counted.
Western Blotting: Western blotting was performed as previously described [24]. Whole-cell lysates collected from 100,000 cells were used per lane. Antibodies used were: anti-AR (N-20, Santa Cruz; Santa Cruz, CA); anti-Beta Actin (Cell Signaling; Beverly, MA); anti-ΔNp63 (4A4, Santa Cruz); anti-p21 (Cell Signaling); anti-p27 (BD Transduction Labs; San Diego, CA); anti-Rb (4H1, Cell Signaling); anti-phospho-Rb (Ser 608, Cell Signaling); anti-Skp2 (Zymed; San Francisco, CA); anti EGF receptor (#2232, Cell Signaling); anti-IGF-type 1 receptor (Cell Signaling); anti-Cdk-2 (H-298; Santa Cruz); anti-Cyclin D1(Upstate Biotechnology; Lake Placid, NY); and anti-c-Myc (Calbiochem; San Diego, CA). All secondary horseradish peroxidase-conjugated antibodies and chemiluminescent detection reagents (ECL) were purchased from Amersham Biosciences (Piscataway NJ).
Real Time PCR and RNAse Protection: RNA extraction and real-time PCR were performed as previously described [24]. AR and PSA mRNAs was normalized per unit 18S mRNA expressed. The following primers were synthesized by Invitrogen Life Technologies Custom Primers and used in RT-PCR: PSA-Forward (5`-AAAAGCGTGATCTTGCTGGG-3`); PSA-Reverse (5`-TCACAGCATCCGTGAGCTC-3`); AR-Forward (5`-CCACAGGCTACCTGGTCCTG-3`); AR-Reverse (5`- TCCTCGTCCGGAGGTGCTG-3`); h-18S-Forward (5`-GAGCGAAAGCATTTGCCAAG-3`; h-18S-Reverse (5`-AGACTTTGGTTTCCCGGAAG-3`). PCR to detect c-Myc mRNA transcripts was conducted using the Superscript III One-Step RT-PCR System (Invitrogen) per the manufacturer's instructions using the primers: c-Myc forward (5'-CCTACCCTCTCAACGACAGC-3'); and c-Myc Reverse (5'-CTCTGACCTTTTGCCAGGAG-3'). RNAse Protection was performed using BD Riboquant RNAse protection Assay System (BD Biosciences, San Deigo, CA). RNA was purified using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA) and quality tested using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). 4 μg of total RNA was hybridized to radio-labeled p21, p27 or AR probes, and of hybridized probes detected according to the manufacturer's specifications.
Statistics: All values are presented as means ± SE. Statistical analysis was performed using a one-way ANOVA with the Newman-Keuls test for multiple comparisons.
Results
Ligand-Dependent AR Signaling Induce Terminal Growth Arrest and Increase Differentiation of Non-Immortalized Normal Human Prostate Epithelial Cells
Normal human prostate epithelial cells (PrECs) can be cultured using a low-calcium (i.e.<300μM) serum-free defined (SFD) media devoid of prostate fibroblasts and smooth muscle cells for 8-10 serial passages [24]. Such PrEC cultures do not express a detectable level of AR protein, since they consist of mostly ΔNp63-positive TA cells, and minor populations of CD133-positive stem cells, PSCA-positive intermediate cells, and Chromogranin A-positive neuroendocrine cells [4, 23, 24]. The growth response of prostate epithelial cells to exogenous expression of wild-type AR with and without ligand was evaluated using PrEC cultures as the model system. PrEC cultures were transduced using a GFP-expressing lentiviral construct containing the full length AR cDNA flanked by loxP sites (PrEC-AR) or an empty control vector (PrEC-Control) (Figure 1A) [26]. Western blot analysis (Figure 1B) documented that PrEC-AR cells express AR protein at a level comparable to LNCaP prostate cancer cells [32]. When AR signaling is induced in these AR-expressing PrECs by the addition of a physiological level (i.e. 1nM) of the synthetic androgen R1881, a profound growth arrest was induced, which was not observed in PrEC-Control cells (Figure 1C). Monitoring of PrEC-AR cultures by time-lapse fluorescence microscopy documented that the PrEC-AR cells growth arrested within 1-2 days in the presence of R1881 but remained mobile and viable. The remaining non-infected PrECs within the culture continued to proliferate. The observed AR-induced growth arrest was observed consistently within a series (n=4) of PrEC cultures from both a commercial source (i.e. donors less than 40 years of age) and cultures derived within our institution from patients undergoing radical prostatectomy (i.e. donors greater than 50 years of age) [4].
AR Inhibits the Proliferation of Human Prostate Epithelial Cells (PrECs). Androgen Receptor (AR) was expressed via lentiviral infection of PrECs. (A) PrEC-Lenti-GFP/AR cells before R1881 treatment, right panel, phase contrast and left panel fluorescence microscopy showed ~80% GFP expression. (B) Expression of Lenti-AR protein in PrEC cultures (PrEC-AR). The LNCaP prostate cancer cell line was used as a positive control. PrEC-Vector-GFP cells did not express detectable AR protein (PrEC-Contol). Actin was used as a loading control.(C) Growth response of PrEC-Control (Lenti-GFP vector) versus PrEC-AR (Lenti-GFP/AR vector) cells when exposed to physiologic levels of R1881 (1nM). (D) Expression of CDK inhibitors p21 and p27 and basal marker ΔNp63 by PrEC-AR and PrEC-Control cells in response to 48 hr exposure to 1nM R1881. Actin was used as a loading control.
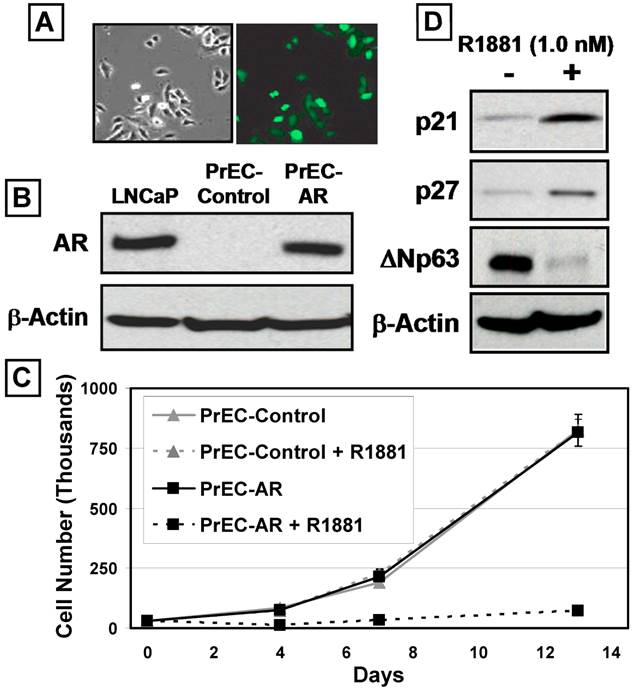
Time-lapse fluorescence microscopy demonstrated that engagement of AR signaling in PrECs resulted in irreversible proliferative quiescence (i.e. terminal growth arrest). After seven days of androgen exposure, none of the growth-arrested PrEC-AR cells were recruited back into active proliferation after replacement with fresh media lacking R1881 or media containing 10μM of the anti-androgen Casodex. Co-incident with this terminal growth arrest was an increase of Cyclin-dependent kinase (Cdk) inhibitors p21 and p27 proteins (Figure 1D). In contrast, expression of the TA specific marker, ΔNp63, decreased (Figure 1D). These findings are significant because ΔNp63 protein expression decreases as prostate TA cells differentiate into intermediate cells [33]. These combined results are consistent with AR signaling in prostatic epithelial cells inducing irreversible growth arrest coupled to enhanced differentiation.
AR Signaling in Immortalized Normal Human Prostate Epithelial Cells Induce Terminal G0 Growth Arrest and Differentiation
Immortalized PrECs were tested to confirm that ligand bound AR functions as a cell-context dependent growth suppressor of prostate epithelial cells. The 957E/hTERT human prostate epithelial cell line was derived from a PrEC culture immortalized by retroviral expression of human Telomerase Reverse Transcriptase (hTERT) [34]. p53, RB, c-Myc and other cell cycle regulatory pathways are not perturbed via hTERT immortalization, unlike SV40 or HPV immortalization. Similar to PrEC cultures, 957E/hTERT cells were maintained in low-calcium SFD media, had a normal karyotype, were non-transformed, and contained cells that expressed the basal markers ΔNp63, PSCA and Cytokeratin 5, but not the luminal markers AR (Figure 3A, upper panel) or PSA [34]. The growth of these 957E/hTERT cells was neither stimulated nor inhibited by androgen or anti-androgens.
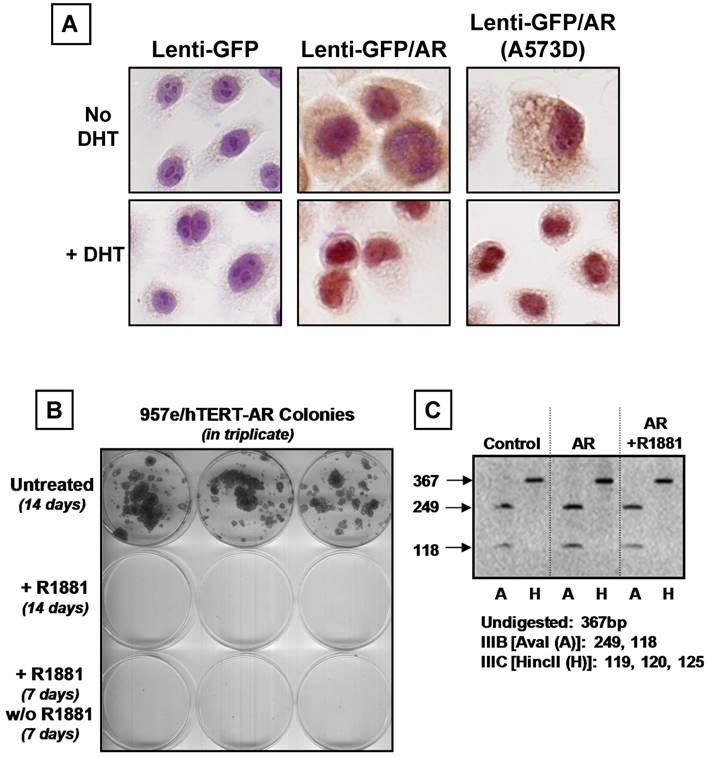
The same GFP-expressing lentiviral AR flanked by loxP sites and control constructs were used to establish 957E/hTERT-AR and 957E/hTERT-Control cultures. Cells were FACS sorted for GFP-positive cells to obtain pure populations. Analysis of AR expression using western blot documented that 957E/hTERT-AR cells expressed AR at a level comparable to LNCaP cells (Figure 2A). Similar to PrEC-AR cells, treatment of 957E/hTERT-AR cells with androgen (1nM R1881 or 1nM DHT) resulted in profound (p<0.05) growth inhibition compared to untreated cells when assayed by total cell number in monolayer culture (Figure 2B), by colony number (Figure 2C), or by cell number per colony (Figure 2D) in clonogenic assays. This growth arrest was associated with a change in localization of AR protein. In untreated 957E/hTERT-AR cells, AR was located predominately in the cytosol but was in the nucleus in cells exposed to androgen (Figure 4A).
AR Suppresses the Growth of Immortalized PrECs. The 957E/hTERT prostate cell line is an hTERT-immortalized PrEC cell line (* indicates a p-value < 0.05). (A) Expression of AR protein in 957E/hTERT-AR cells was compared to LNCaP and a vector only control (Control). (B) 957E/hTERT-AR cells were suppressed for growth in presence of 1nM R1881 or 1nM DHT after 5 days. The growth suppression was blocked partially by administration of Casodex. (C and D) Clonogenic growth of 957E/hTERT-AR cells in the presence of 1nM R1881. 957E/hTERT-AR cells formed significantly fewer colonies (C) that were smaller (D) compared to 957E/hTERT-Control cells (Control). (E) AR activation in 957E/hTERT-AR cells (AR and AR + R1881) resulted in a G0/G1 arrest. 957E/hTERT-Controls (Control and Control + R1881) were not affected by addition of R1881. (F) AR activation in 957E/hTERT-AR cells resulted in significant decrease in percentage of cells expressing the proliferation marker Ki-67 (black bars) and the basal cell marker ΔNp63 (grey bars). As an additional control, removal of media growth factors (GFs) resulted in decreased Ki67 expression but no change in ΔNp63 expression.
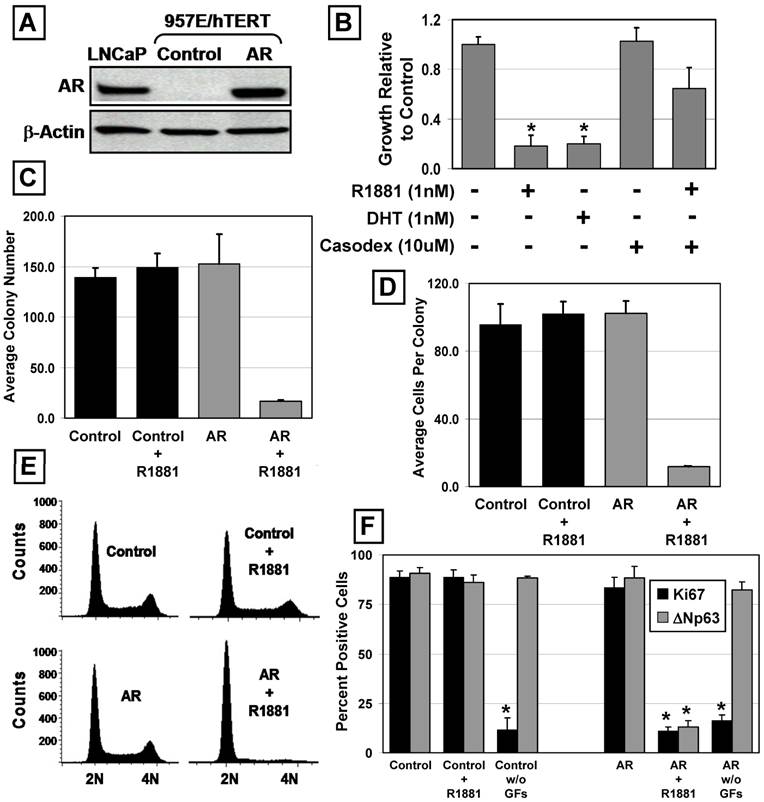
Androgen-induced growth arrest of 957E/hTERT-AR cells requiring AR signaling was supported by two experimental approaches. In the first approach, 957E/hTERT-AR cells were co-exposed to androgen and Casodex and the growth response evaluated. These studies documented that Casodex retarded the growth arrest induced by androgen (Figure 2B). In the second approach, the floxed AR gene was excised using CRE-Lox mediated recombination and clones were isolated and evaluated for AR expression and growth arrest response to androgen (data not shown). These results documented that when AR protein expression was undetectable in a clone, its growth was not inhibited by added androgen.
Flow cytometric analyses of DNA content documented that androgen-exposure of 957E/hTERT-AR cells induce growth arrest in G0/G1 (Figure 2E). Despite this ligand-dependent growth arrest, R1881-treated cells continued to remain viable without increased apoptosis as documented with flow cytometric DNA analysis [i.e. no sub-G0 population detected] (Figure 2E) and time-lapse fluorescence microscopy. Such androgen-induced growth inhibition was associated with a significant (p<0.05) increase in the proportion of cells exiting cycle and arresting in G0 as monitored by lack of nuclear Ki-67 expression and undergoing differentiation as monitored by down regulation of ΔNp63 expression (Figure 2F and Figure 3A) and increased (p<0.05) mRNA expression for PSA, an androgen-regulated gene whose expression is characteristic of differentiated prostatic secretory-luminal cells (Figure 3B). Such androgen induced G0 arrest and differentiation were irreversible as established by the lack of clonogenic growth (Figure 4B) or restoration of ΔNp63 expression (data not shown) upon replacement with media lacking androgen. The fact that AR signaling induced both terminal growth arrest and differentiation of 957E/hTERT-AR cells could be coincidental or causally linked. In support of a causal link is the observation that 957E/hTERT-AR cells could be growth arrested simply by switching them to media lacking the supplemental GFs [24]. Under this GF restriction the cells did not differentiate as monitored by a lack of change in their ΔNp63 expression even though they growth arrested in G0 as monitored by a decrease in nuclear Ki-67 expression (Figure 2F). This GF restriction-induced growth arrest was reversible, since re-addition of GFs stimulated subsequent growth (data not shown). Prostate epithelial cells must differentiate to terminally growth arrest, and androgen stimulates both processes via AR-dependent signaling.
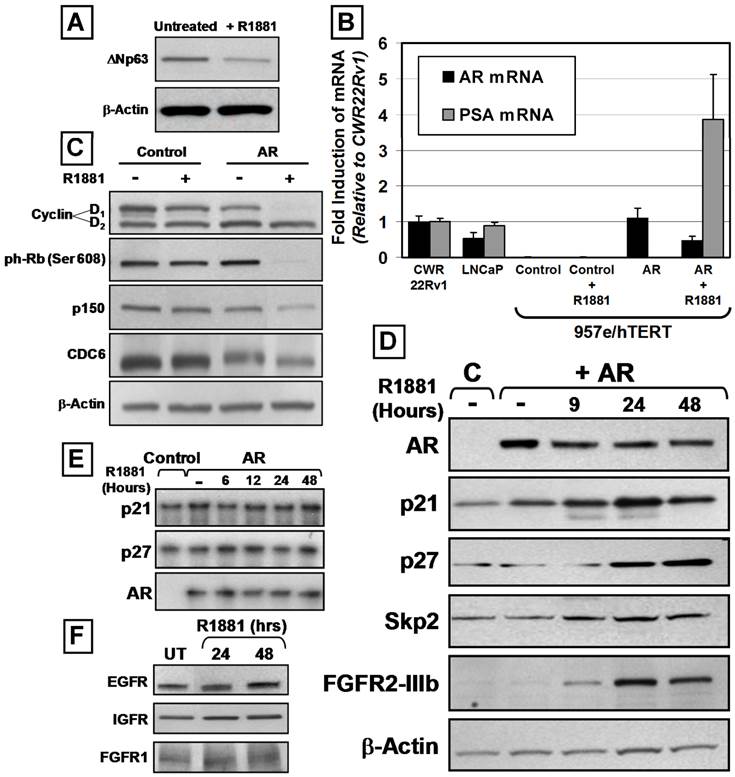
AR Expression Induces Luminal Differentiation and SKP-2-Independent Increases in p21 and p27 Protein Expression. (A) Decreased ΔNp63 expression of 957E/hTERT-AR cells in the presence of 1nM R1881 (+R1881) compared to untreated controls (Untreated). Actin was used as a loading control. (B) PSA mRNA expression increased in androgen-stimulated 957E/hTERT-AR cells. Quantitative real-time PCR was normalized to the CWR22R prostate cancer cell line. Analysis of AR mRNA expression documented undetectable AR in 957E/hTERT-Control cells and increased in AR mRNA stability in presence of R1881. (C) Decreased Cyclin D1, phospho-RB, p150, and CDC6 protein expression in 957E/hTERT-AR cells (AR) after 48 hours in the presence of 1nM of R1881. 957E/hTERT-Controls cells (Control) exhibited no response in presence of R1881. Actin was used as a loading control. (D) Elevated p21, p27, SKP-2 and FGFR2 protein expression in 957E/hTERT-AR (+AR) cells upon exposure to R1881 was compared to 957E/hTERT-Control (C) cells. Expression was assayed 9, 24, and 48 hours after exposure to R1881. Actin was used as a loading control. (E) RNAse protection showed no changes of p21, p27, and AR mRNA expression in androgen-stimulated 957E/hTERT-AR cells. 957E/hTERT-Control cells (Control) served as controls. 957E/hTERT-AR (AR) cells were exposed to 1nM R1881 over 48 hours. (F) Western blotting documented no change in EGFR, IGFR, and FGFR1 expression within 957E/hTERT-AR cells after exposure to 1 nM R1881. Untreated cells (UT) were used for comparison.
AR is Localized to the Nucleus and Results in Irreversible Growth Inhibition without Changes in FGFR2 Isoform Expression. (A) Nuclear translocation of wild-type and mutant A573D AR in response to AR ligand (1.0 nM DHT). (B) Growth suppression of 957E/hTERT-AR cells in presence of R1881 was irreversible. Growth-suppressed 957E/hTERT-AR clones in the presence of ligand were washed and re-fed with complete media no longer containing R1881. No further clonogenic growth was observed after one week. (C) Increased in FGFR2 protein expression in 957E/hTERT-AR cells (Figure 3D) did not result in a conversion to a different isoform. RT-PCR and restriction endonuclease digestion documented that FGFR2 expressed is the IIIb isoform since the PCR-amplified FGFR2 cDNA was only digested by the Ava I (A) restriction endonuclease (37).
Molecular Analyses of Androgen-Dependent AR-Mediated Terminal G0 Growth Arrest
Western blotting of 957E/hTERT-AR cells documented that both Cyclin D1 and phospho-RB became undetectable upon exposure of 957E/hTERT-AR cells to androgen (Figure 3C). Decreased expression of the S-phase specific protein p150 and the DNA licensing factor CDC6 also was observed (Figure 3C). Androgen-dependent AR-mediated terminal G0 growth arrest of 957E/hTERT-AR cells was associated with increased expression of the Cdk inhibitors p21 and p27 proteins (Figure 3D), similar to androgen-mediated terminal growth arrest of PrEC-AR cells. RNAse protection assays documented that the steady state level of p21, p27, and AR mRNA remained unchanged throughout growth arrest induced during the first 48 hour exposure to androgen (Figure 3E) even though there were increases in p21 and p27 protein levels during this period of androgen exposure (Figure 3D). The lack of change in p21 mRNA expression was unanticipated since the p21 gene contains a putative AR-response element (ARE) within its promoter and p21 expression is transcriptionally regulated via AR binding to its proximal promoter in LNCaP cells [35]. In contrast to prostate cancer cells, the data demonstrated that p21 and p27 protein expression during androgen induced AR-mediated G0 growth arrest was regulated by non-transcriptional mechanisms in normal prostate epithelial cells. One possible mechanism for increased protein expression of p21 and p27 in the absence of increased mRNA expression is inhibition of ubiquitin-mediated protein degradation. In particular, Skp2 is an F-box protein and a component of the E3 ubiquitin ligase complex, which has been implicated in regulating cell-cycle progression, and p21 and p27 are substrates for Skp2-mediated degradation [36]. However, Skp2 protein levels increased in conjunction with p21 and p27 during androgen induced G0 growth arrest of 957E/hTERT-AR cells (Figure 3D); thus increased p21 and p27 protein expression occured via a Skp2-independent mechanism.
One possible mechanism for the AR-mediated growth arrest of 957E/hTERT-AR cells could involve androgen-induced AR signaling repression of the expression of growth factor receptors thereby blocking growth factor signaling. EGF, IGF-1, and FGFs are several GFs added to SFD media needed for growth of normal prostate epithelial cells [24]. These ligands signal via their cognate receptors; EGFR or Erb1 for EGF and IGF-type1 receptor (IGF-1R) for IGF, and FGFR2 for FGFs. Western blot analysis documented that androgen-mediated AR signaling in 957E/hTERT-AR cells did not modify measurably the expression of EGFR or IGFR proteins (Figure 3F) and instead elevated the expression of FGFR2 protein (i.e. KGF receptor) (Figure 3D). Such induction of FGFR2 protein did not result from a change in the isotype expressed, since the only form of FGFR2 receptor detected is the IIIB isoform (Figure 4C) [37].
AR Signaling in Human Prostate Stromal Cells and Malignantly-Initiated Prostate Epithelial Cells Did Not Induce Growth Arrest
We have documented previously that pure cultures of normal human prostate stromal cells (termed PrSCs) containing smooth muscle cells and fibroblasts can be established routinely from fresh surgical material using high calcium (i.e.>600nM)-fetal calf serum (FCS) containing media [6]. Non-immortalized PrSC cultures [i.e. PrSC6, [6] and RT006 [38]] were established from patients who underwent radical prostatectomy. The SV-40 immortalized, but not transformed, stromal WPMY-1 cell line also was evaluated [39]. All of these prostate-derived stromal cells expressed detectable endogenous AR protein, which was stabilized by ligand (Figure 5A). In contrast to PrEC cells, none of these independently derived stromal cell cultures was growth inhibited by androgen-induced AR signaling (Figure 5B). The level of endogenous AR protein expression by these stromal cells was ~40 times lower than that of LNCaP cells, even in the presence of ligand (Figure 2A). To test whether elevated AR protein expression could inhibit stromal cell growth, WPMY-1-AR and WPMY-1-Control cells were derived that expressed ~8-fold higher levels of AR protein with or without androgen treatment compared to the similarly treated parental WPMY-1 cells (Figure 5A). The elevated level of AR protein expression failed to inhibit or accelerate cell proliferation in the presence of ligand (Figure 5B). Thus, androgen-induced AR signaling within prostatic stromal cells does not regulate their growth.
AR-Mediated Growth Inhibition is Cell-Context Dependent. (A) AR protein expression in late passage (PrSC6), early passage (RT006), and immortalized (WPMY-1) prostate stromal cell cultures. LNCaP cells were used as a control, and different cell numbers were used to compare expression at the same exposure. R1881 increased AR protein expression after 48 hours. Lenti-GFP and Lenti-GFP/AR were expressed in WPMY-1 cells (GFP and GFP/AR). (B) Treatment of prostate stromal cells with 1nM R1881 did not modulate stromal cell proliferation. (C) BPH-1 cells did not express endogenous AR protein, but expressed the basal marker, ΔNp63. LNCaP cells were used as a control. (D) BPH-1-AR cells did not exhibit increased p21 protein expression in response to 1nM R1881 for 48 hours. LNCaP cells were used as a control. Expression of p27 protein remained high in response to androgen. (E) 1 nM R1881 did not inhibit the growth of BPH-1 cells expressing exogenous AR (BPH-1-AR) compared to vector alone (Control). (F) AR-mediated growth inhibition in the absence of p53 function using the HVP-immortalized PrEC line HPr-1AR.
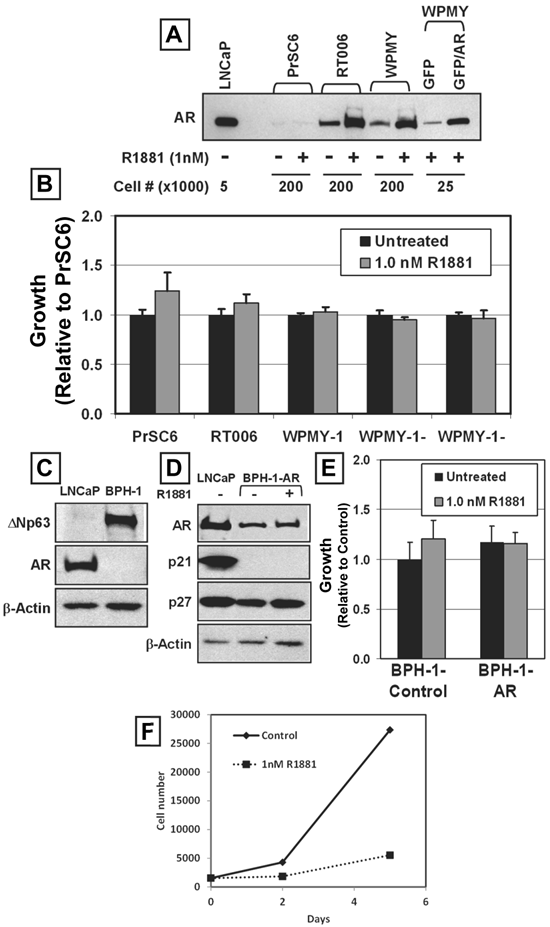
The immortalized BPH-1 human prostate epithelial cell line was established by immortalizing human prostate epithelial cells from a BPH surgical specimen using SV40 early virus region, which includes both large- and small-T antigens [25]. Like the 957E/hTERT cells, BPH-1 cells expressed the basal cell marker ΔNp63, did not express AR or PSA [25] (Figure 5C), did not express p21 but do expressed p27 protein (Figure 5D). In contrast to 957E/hTERT immortalized cells, BPH-1 cells are aneuploid with numerous structural abnormalities [25]. These cells are tumorigenic, but only when co-inoculated into nude mice with human prostate cancer associated fibroblasts or rodent Urogenital Sinus Mesenchyme [40]. Therefore BPH-1 cells are malignantly “initiated” but not fully tumorigenic when inoculated alone. BPH-1-AR and BPH-1-Control cultures were established to test whether AR expression and ligand activation suppressed the growth of these cells (Figure 5D). In contrast to normal or hTERT-immortalized AR-expressing prostate epithelial cells, treatment of AR-expressing BPH-1 cells with androgen did not induce growth arrest (Figure 5E). The lack of androgen-induced growth arrest in AR expressing BPH-1 cells was associated with no change in p27 protein expression and no detectable expression of p21 protein (Figure 5D).
To account for the role of p53 in mediating AR growth suppression, we used the HPV-immortalized PrEC line HPr-1AR. In this cell line, HPV E6 protein binds and neutralizes p53, much like SV40 large T-antigen. Activation of AR in HPr-1AR cells also led to inhibition of cell growth and demonstrated AR-mediated growth inhibition even in the absence of p53 function (Figure 5F). These results, when combined with those for stromal cells, documented that the AR-dependent terminal G0 growth arrest in the normal prostatic epithelial PrEC-AR and 957E/hTERT-AR cells is a cell-context dependent, is p53-independent, and not a ubiquitous cellular response.
Knockdown of RB, p21, or p27 does not Prevent Androgen-Induced Growth Arrest of Human Prostate Epithelial Cells
Inhibition of RB protein phosphorylation within growth arrested 957E/hTERT-AR cells (Figure 3C), and the well-documented requirement of RB protein phosphorylation for normal cell cycle progression [41] raised the question of whether RB protein is critically required for androgen-induced growth arrest of these prostate epithelial cells. Treatment with appropriately designed shRNA resulted in knockdown of RB protein expression to undetectable levels (Figure 6A). Such targeted knockdown of RB protein did not, however, overcome the AR-induced G0 growth arrest of the 957E/hTERT-AR cells, which documented that AR-mediated growth suppression does not require RB expression (Figure 6B).
AR-Mediated Growth Inhibition does not Require RB, p21 or p27 but does Require DNA Binding. (A) 957E/hTERT-AR cells expressed stable shRNAs to RB. Western blotting documented knockdown of RB protein in 957E/hTERT-Control cells (Control) and 957E/hTERT-AR cells (AR). Actin was used as a loading control. (B) Knockdown of RB failed to override AR-mediate growth inhibition of 957E/hTERT-AR cells (* indicates a p-value < 0.05). (C) Western blotting documented single or double knockdown of p21 and p27 in the same 957E/hTERT-AR cell population; treatment with R1881 failed to produce an elevation of p21 and p27 expression. (D) Clonogenic growth of 957E/hTERT-AR cells in the presence of R1881 documented that individual and double p21 and p27 knockdown failed to prevent growth inhibition by AR (* indicates p-value < 0.05). (E) Western blotting of 957E/hTERT-AR(A573D) cells after treatment with 1nM R1881 for 48 hours. Untreated cells were used as a control (UT). (F) Stable expression of a DNA-binding mutant of AR (A573D) failed to inhibit the clonogenic growth of 957E/hTERT cells in the presence of ligand (+R1881) compared to untreated controls (Untreated).
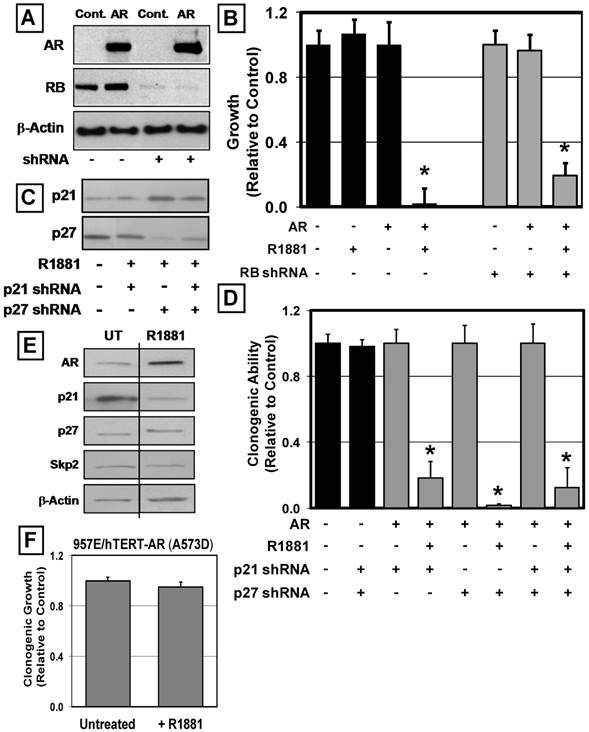
A similar shRNA approach was used to evaluate a causal role for p21 and p27 protein increases in androgen-induced G0 growth arrest of 957E/hTERT-AR cells using constructs to specifically target p21 or p27 protein expression [31]. Similar to the observation with the targeted knockdown of RB, targeted knockdown of p21 or p27 proteins (Figure 6C) did not override androgen-induced, AR-mediated growth suppression (Figure 6D). Moreover, double knockdown of both p21 and p27 within 957E/hTERT-AR cells failed to override AR-mediated growth suppression (Figure 6C and D). These data document that the AR-mediated growth inhibition of normal prostate epithelial cells does not require either RB or the CDK inhibitors p21 or p27.
DNA Binding of AR is required for Growth Suppression of Prostate Epithelial Cells
Given the observation that p21 and p27 mRNA levels remain unchanged despite detectable changes in their protein expression (Figure 3E), this raised the issue of whether AR suppression of growth of normal human prostate epithelial cells occurs via a transcriptionally-independent mechanism. To evaluate such a possible mechanism, a mutant AR lacking DNA binding was tested for its ability to inhibit prostate epithelial cell growth. For these studies, the A573D mutant AR was used [28]. This A-D mutation was detected in a patient with complete androgen insensitivity syndrome and completely blocks DNA binding without affecting ligand binding or cytoplasmic-to-nuclear translocation upon ligand binding [28]. Using the same GFP-expressing lentiviral construction, 957E/hTERT-AR(A573D) cells were derived. As expected, the mutant AR in 957E/hTERT-AR(A573D) cells is driven to the nucleus upon exposure to androgen (Figure 4A). In contrast to cells expressing wild type AR, the 957E/hTERT-AR(A573D) cells expressing mutant AR exhibit no change in clonogenic growth over a nine-day period in the presence of R1881 (Figure 6F). Moreover, unlike the enhanced expression of p21, p27, and Skp2 proteins occurring following androgen induced wild type AR signaling as part of the 957E/hTERT-AR growth arrest (Figure 3D), there is a decrease in p21 protein expression with no change in p27 or Skp2 upon administering androgen when these cells express the DNA-binding mutant AR protein (Figure 6E). These combined results demonstrate that androgen-activated AR must bind DNA in order to induce G0 growth arrest of prostate epithelial cells.
AR-Mediated Growth Suppression of Prostate Epithelial Cells is Dependent upon Down-Regulation of c-Myc Expression
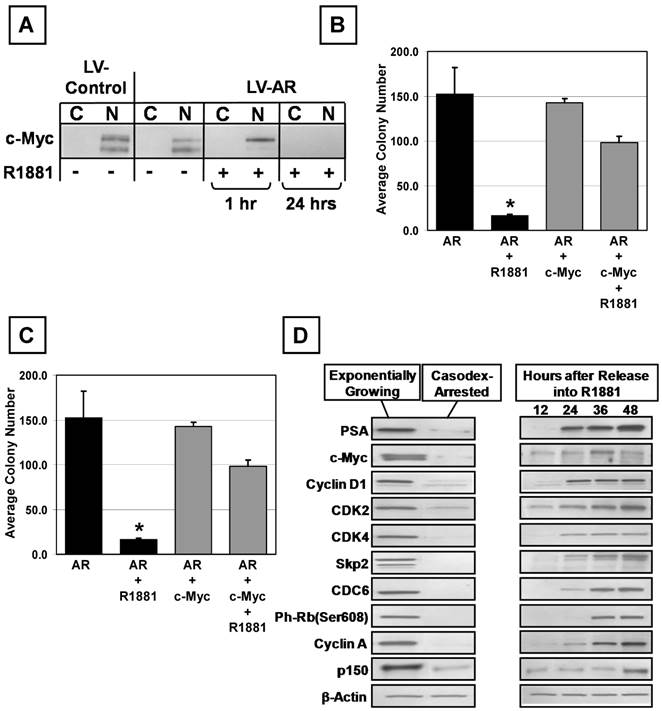
Previous studies documented that AR-induced growth suppression of prostate epithelial cells is associated with decreased expression of c-Myc [17], which was confirmed using 957E/hTERT-AR cells. Western blotting of these cells demonstrated that in the presence of ligand, c-Myc mRNA and protein expression decreases; and resulted in a total loss of detectable c-Myc protein from the nucleus and cytosol within 24 hr of androgen treatment (Figure 7A). 957E/hTERT-AR cells were transduced with an AR independent constitutive c-Myc expression vector to determine whether the decrease in c-Myc expression is an indirect result of AR-mediated growth inhibition or blocks proliferation of prostate epithelial cells. c-Myc mRNA and protein were not decreased when androgen was added to media (Figure 7A). These results document that androgen-stimulated AR signaling induced down-regulation of c-Myc expression in 957E/hTERT-AR cells did not involve a post transcriptional process that either decreased c-Myc mRNA stability or prevented c-Myc mRNA translation. These results are consistent with the decrease in c-Myc mRNA in the presence of AR-signaling in non-transduced parental 957E/hTERT-AR cells via transcriptional inhibition. This transcriptional effect would explain why androgen is not able to inhibit growth of 957E/hTERT cells that express a transcriptionally inactive mutant AR unable to bind to DNA (Figure 6F). Stable constitutive expression of c-Myc in the transduced 957E/hTERT cells expressing wild-type AR abrogated the cell proliferation block induced by ligand-activated AR in the non c-Myc transduced wild-type AR expressing parent cells, which was demonstrated by continued increase in clonogenic colony number (Figure 7B) and colony size (Figure 7C) over time even in the presence of ligand. These data demonstrated that androgen induced AR-dependent down-regulation of c-Myc expression is required for the growth arrest of normal prostate epithelial cells.
MYC Expression Overrides AR-Mediated Growth Inhibition of Prostate Epithelial Cells. (A) Undetectable c-MYC protein expression in the cytosol (C) or nucleus (N) of 957E/hTERT-LV-AR cells after 24 hours in the presence of 1nM R1881. The PrEC-hTERT-LV-Control cells are used as a control. (B and C) 957E/hTERT-AR cells were infected stably with MYC retrovirus and tested for clonogenic ability in the presence of R1881. 957E/hTERT-AR cells expressing exogenous MYC are more clonogenic in the presence of R1881; MYC expression resulted in a greater number of cells per colony. (D) Treatment of the LNCaP cells with 10 μM Casodex resulted in decreased MYC expression.
AR Promotes the Expression of c-Myc in Prostate Cancer Cells
Paracrine signaling converts to an autocrine mechanism of AR-stimulated growth during prostate carcinogenesis [6, 22]. Elevated c-Myc expression occurs very early during prostatic carcinogenesis and is detectable in cells that co-express AR [42]. Elevation of c-Myc expression in AR expressing prostate cancer cells is paradoxical to the situation in normal prostate epithelial cells in which c-Myc expression is undetectable in AR positive luminal cells [42]. Activated AR's ability to down regulate c-Myc expression in normal prostate epithelial cells is lost as AR is converted from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer. Casodex was used to block AR signaling in LNCaP cells and expression of c-Myc analyzed. When LNCaP cells growing on standard tissue culture plastic were exposed to 10 μM Casodex, they rapidly became loosely attached to the flask, acquired a dendritic morphology, and began dying within ~ 3-4 days. In contrast, if cells were grown on poly-D-lysine coated tissue culture flasks and treated, cells were growth arrested but remained attached, did not undergo morphological changes, and were viable for more than two weeks. LNCaP cells were maintained with and without 10 μM Casodex for one week in poly-D-lysine coated culture flasks. Ki-67 staining, MTT growth assays, and Western blotting documented that when LNCaP cells are in media without Casodex, the cells grew exponentially (i.e. 90+/- 5 % Ki-67 positive) with doubling time 40+/- 4 hr and had a high level protein expression of c-Myc, CDC6, Cyclin D1, phospho-RB, Cyclin A, CDK2&4, p150, and Skp2 (Figure 7D). When AR signaling was blocked by Casodex, cells on poly-D-lysine growth-arrested and did not increase in cell number after day 3 and less than 4% of the cells were Ki-67 positive by day 7. AR blockade induced growth arrest of LNCaP cells that was associated with undetectable expression of c-Myc and other cell cycle proteins and PSA (Figure 7D). Dose-response assays documented that these Casodex-synchronized cells were stimulated maximally back into cell cycle by addition of 1nM R1881 to the Casodex containing media (data not shown). Re-entry into the cell is associated with induction of c-Myc expression, which is detectable at the protein level ~12 hours earlier than the other cell cycle markers and PSA (Figure 7D). These results document that c-Myc expression is up-regulated rapidly by AR signaling in prostate cancer cells in contrast to normal prostate epithelial cells where AR signaling down-regulates c-Myc expression.
Discussion
Previous studies have demonstrated that androgen-induced AR signaling acquires a gain of oncogenic function to promote cell division in prostate cancer cells. Thus, the majority of prostate cancers express AR that provides the rationale for why androgen deprivation therapy is standard therapy for metastatic prostate cancer since it inhibits proliferation and activates apoptotic death of metastatic cells. Moreover, AR remains a central target even for castration-resistant metastatic prostate cancers [43]. AR gene mutation and/or amplification results in elevated AR protein expression in the majority of metastatic prostate cancer tissue sites obtained from autopsies of patients with castration-resistant metastatic disease [44]. Consistent with these clinical observations, AR gene mutation, amplification and protein over expression are observed in the majority of prostate cancer cell lines derived from castration resistant hosts [45]. These castration-resistant prostate cancer cell lines do not undergo apoptosis when androgens are depleted or androgens antagonists are used; however, they stop proliferating and activate cell death if the AR protein level is reduced below a critical level both in vitro [45, 46] and in vivo [47]. Recent data from our group demonstrated that AR functions to drive prostate cancer cell proliferation and AR signaling in host stroma is not required [22]. These observations document that castration-resistant prostate cancer cells are addicted to oncogenic AR signaling for malignant growth [48].
The present studies demonstrated that androgen-stimulated AR signaling induced G0 growth arrest of normal human prostate epithelial cells and directed their differentiation into ΔNp63 negative, PSA-expressing secretory luminal cells. This mechanism limits a potentially endless feed-forward proliferative paracrine loop to prevent continuous prostatic epithelial overgrowth in the presence of androgen induced high levels of stromal andromedins. Additional reports demonstrated that AR induces growth suppression and terminal differentiation. For example, AR is recognized as a growth suppressor for a variety of other normal human epithelial cell types, such as thyroid and adrenocortical epithelial cells, where ligand-dependent endogenous AR signaling suppressed growth [49]. Cunha et al. demonstrated that when AR wild type embryonic Urogenital Sinus (UGS) mesenchymal tissue was recombined with AR-null UGS epithelium, prostatic tissue developed in which luminal cells lacked differentiation, displayed a cuboidal, not columnar, phenotype and lacked the expression of prostatic secretory proteins [5]. Xin et al. demonstrated that expression of AR suppressed the growth of murine prostate epithelial cells using similar tissue recombination methods [50]. Prins et al. reported that AR down-regulation in normal rat prostate epithelial cells as a consequence of transient estrogen treatment during the period of prostatic organogenesis resulted in hyperplastic growth and lack of differentiation within the epithelial compartment [51]. A similar observation was documented using a transgenic mouse model where targeted CRE-expression induced a prostatic epithelial-specific deletion of AR, which resulted in poorly differentiated and hyper-proliferative epithelial structures [18].
The results of the present studies have significant clinical implications for the use of androgen deprivation therapy for prevention verses treatment of prostate cancer. The present studies raise the issue that AR signaling is perverted from a growth suppressor to a growth promoting function in clinical prostate cancers, but do not clarify at what point in the carcinogenic process this occurs. Clinical data emphasize the critical importance of resolving this issue. For example, male dogs are often neutered at a young age and such long term androgen deprivation is associated with increased incidence of symptomatic prostate cancer and death [52]. A 5 year clinical prevention human trial showed that Finasteride-induced androgen deprivation was associated with a decrease in moderate grade localized prostate cancers, but an increased frequency of high grade localized cancers [53]. These data suggest that there is a time frame when AR signaling is still inhibiting the proliferation of prostate cells undergoing malignant transformation and thus androgen deprivation during this time is harmful. There are experiment data demonstrating this point. For example, androgen deprivation during the early development of prostate cancer induces the development of metastatic disease using the transgenic TRAMP mouse model [54]. In contrast, once human prostate cancers complete full transformation to a lethal phenotype, numerous clinical trials demonstrate that AR signaling stimulates the cancers and thus androgen deprivation therapy is useful [55]. Data presented here implicates differential regulation of cMyc expression as the critical determinant in conversion by AR signaling to growth suppressive to growth promoting. These data justify future studies to elucidate critical changes in the regulation of cMyc expression during prostate carcinogenesis.
Abbreviations
Androgen Receptor (AR), Androgen response element (ARE), Dihydrotestosterone (DHT), Fetal bovine serum (FBS), Fluorescence activated cell sorting (FACS), Green Fluorescence protein (GFP), human telomerase (hTERT), Prostate epithelial cell culture(PrEC), Prostate-Specific Antigen (PSA).
Acknowledgements
We wish to acknowledge the expert assistance of Leslie Meszler and Lillian Dasko-Vincent of the Johns Hopkins Cell Imaging Core Facility, and Lee Blosser and Ada Tam of the Johns Hopkins School of Medicine Flow Sorting Facility for their assistance. We thank Jessica Hicks for the AR, p63, and Ki-67 immunostaining. We thank Dr. CK Choo for the HPr-1AR cell line, and Dr. Simon Hayward for the BPH-1 cell line.
Funding: NIH Grant R01DK52645 and the Maryland Stem Cell Research Fund MSCRFII-0428-00 have generously supported this research. Donald Vander Griend was supported by a Urology Training Grant, NIH T32DK07552 and a DOD Post-Doctoral Training Award, PC060843. He is faculty of the Section of Urology, The Department of Surgery, The University of Chicago.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kyprianou N, Isaacs JT. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 1988;122:552-62
2. Isaacs JT. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model.: U.S. Dept. of Health and Human Services, NIH Publication #87-2881; 1987.
3. Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 2007;25:2760-9
4. Vander Griend DJ, Karthaus WK, Dalrymple S, Meeker AK, De Marzo AM, Isaacs JT. The Role of CD133 in Normal Human Prostate Stem Cells and Malignant Cancer Initiating Cells. Cancer Res. 2008;68:9703-11
5. Cunha GR, Chung LW, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: role of mesenchymal-epithelial interactions. Recent Prog Horm Res. 1983;39:559-98
6. Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61:5038-44
7. Steg PG, Ferrari R, Ford I, Greenlaw N, Tardif JC, Tendera M. et al. Heart rate and use of beta-blockers in stable outpatients with coronary artery disease. PLoS One. 2012;7:e36284. doi:10.1371/journal.pone.0036284
8. Gaisa NT, Graham TA, McDonald SA, Poulsom R, Heidenreich A, Jakse G. et al. Clonal architecture of human prostatic epithelium in benign and malignant conditions. J Pathol. 2011;225:172-80
9. De Marzo AM, Nelson WG, Meeker AK, Coffey DS. Stem cell features of benign and malignant prostate epithelial cells. J Urol. 1998;160:2381-92
10. Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Mol Cancer Res. 2002;1:113-21
11. Wikstrom P, Ohlson N, Stattin P, Bergh A. Nuclear androgen receptors recur in the epithelial and stromal compartments of malignant and non-malignant human prostate tissue several months after castration therapy. Prostate. 2007;67:1277-84
12. Ohlson N, Bergh A, Stattin P, Wikstrom P. Castration-induced epithelial cell death in human prostate tissue is related to locally reduced IGF-1 levels. Prostate. 2007;67:32-40
13. Isaacs JT, Furuya Y, Berges R. The role of androgen in the regulation of programmed cell death/apoptosis in normal and malignant prostatic tissue. Semin Cancer Biol. 1994;5:391-400
14. De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol. 1998;153:911-9
15. Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S. et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res. 2004;64:8867-75 doi:10.1158/0008-5472.CAN-04-2938
16. Ling MT, Chan KW, Choo CK. Androgen induces differentiation of a human papillomavirus 16 E6/E7 immortalized prostate epithelial cell line. J Endocrinol. 2001;170:287-96
17. Whitacre DC, Chauhan S, Davis T, Gordon D, Cress AE, Miesfeld RL. Androgen induction of in vitro prostate cell differentiation. Cell Growth Differ. 2002;13:1-11
18. Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C. et al. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci U S A. 2007;104:12679-84
19. Simanainen U, Allan CM, Lim P, McPherson S, Jimenez M, Zajac JD. et al. Disruption of prostate epithelial androgen receptor impedes prostate lobe-specific growth and function. Endocrinology. 2007;148:2264-72
20. Simanainen U, McNamara K, Gao YR, Handelsman DJ. Androgen sensitivity of prostate epithelium is enhanced by postnatal androgen receptor inactivation. Am J Physiol Endocrinol Metab. 2009;296:E1335-43
21. Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles' heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972-82
22. Vander Griend DJ, D'Antonio J, Gurel B, Antony L, Demarzo AM, Isaacs JT. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate. 2010;70:90-9 doi:10.1002/pros.21043
23. Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT. Low-calcium serum-free defined medium selects for growth of normal prostatic epithelial stem cells. Cancer Res. 2006;66:8598-607 doi:10.1158/0008-5472.CAN-06-1228
24. Uzgare AR, Xu Y, Isaacs JT. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. J Cell Biochem. 2004;91:196-205
25. Hayward SW, Dahiya R, Cunha GR, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell Dev Biol Anim. 1995;31:14-24
26. Litvinov IV, Vander Griend DJ, Antony L, Dalrymple SL, Becker R, Cheng L. et al. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329-38
27. Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM. et al. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445-57
28. Farla P, Hersmus R, Trapman J, Houtsmuller AB. Antiandrogens prevent stable DNA-binding of the androgen receptor. J Cell Sci. 2005;118:4187-98
29. Yu X, Zhan X, D'Costa J, Tanavde VM, Ye Z, Peng T. et al. Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol Ther. 2003;7:827-38
30. Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464-74
31. Halfter H, Friedrich M, Resch A, Kullmann M, Stogbauer F, Ringelstein EB. et al. Oncostatin M induces growth arrest by inhibition of Skp2, Cks1, and cyclin A expression and induced p21 expression. Cancer Res. 2006;66:6530-9
32. Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008-13
33. De Marzo AM, DeWeese TL, Platz EA, Meeker AK, Nakayama M, Epstein JI. et al. Pathological and molecular mechanisms of prostate carcinogenesis: implications for diagnosis, detection, prevention, and treatment. J Cell Biochem. 2004;91:459-77
34. Yasunaga Y, Nakamura K, Ewing CM, Isaacs WB, Hukku B, Rhim JS. A novel human cell culture model for the study of familial prostate cancer. Cancer Res. 2001;61:5969-73
35. Lu S, Jenster G, Epner DE. Androgen induction of cyclin-dependent kinase inhibitor p21 gene: role of androgen receptor and transcription factor Sp1 complex. Mol Endocrinol. 2000;14:753-60
36. Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323-33
37. Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene. 1997;15:3059-65
38. Koochekpour S, Maresh GA, Katner A, Parker-Johnson K, Lee TJ, Hebert FE. et al. Establishment and characterization of a primary androgen-responsive African-American prostate cancer cell line, E006AA. Prostate. 2004;60:141-52
39. Webber MM, Trakul N, Thraves PS, Bello-DeOcampo D, Chu WW, Storto PD. et al. A human prostatic stromal myofibroblast cell line WPMY-1: a model for stromal-epithelial interactions in prostatic neoplasia. Carcinogenesis. 1999;20:1185-92
40. Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD. et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135-42
41. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671-82
42. Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C. et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156-67
43. Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nat Med. 2004;10:26-7 doi:10.1038/nm0104-26
44. van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL. et al. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205-25 doi:10.1002/pros.10290
45. Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855-63 doi:10.1210/me.2007-0223
46. Yang Q, Fung KM, Day WV, Kropp BP, Lin HK. Androgen receptor signaling is required for androgen-sensitive human prostate cancer cell proliferation and survival. Cancer Cell Int. 2005;5:8
47. Snoek R, Cheng H, Margiotti K, Wafa LA, Wong CA, Wong EC. et al. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39-47
48. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077-80 discussion 80. doi:10.1158/0008-5472.CAN-07-3293
49. Rossi R, Zatelli MC, Valentini A, Cavazzini P, Fallo F, del Senno L. et al. Evidence for androgen receptor gene expression and growth inhibitory effect of dihydrotestosterone on human adrenocortical cells. J Endocrinol. 1998;159:373-80
50. Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A. 2006;103:7789-94
51. Woodham C, Birch L, Prins GS. Neonatal estrogen down-regulates prostatic androgen receptor through a proteosome-mediated protein degradation pathway. Endocrinology. 2003;144:4841-50
52. Bryan JN, Keeler MR, Henry CJ, Bryan ME, Hahn AW, Caldwell CW. A population study of neutering status as a risk factor for canine prostate cancer. Prostate. 2007;67:1174-81
53. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG. et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215-24
54. Huss WJ, Gray DR, Tavakoli K, Marmillion ME, Durham LE, Johnson MA. et al. Origin of androgen-insensitive poorly differentiated tumors in the transgenic adenocarcinoma of mouse prostate model. Neoplasia. 2007;9:938-50
55. Ryan CJ, Small EJ. Early versus delayed androgen deprivation for prostate cancer: new fuel for an old debate. J Clin Oncol. 2005;23:8225-31
Author contact
![]() Corresponding author: John T. Isaacs, Ph.D., The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, 1650 Orleans St., Baltimore, MD 21231, 410-614-6321, isaacjoedu.
Corresponding author: John T. Isaacs, Ph.D., The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, 1650 Orleans St., Baltimore, MD 21231, 410-614-6321, isaacjoedu.