Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction to drug...
Activity-based vs in silico drug...
Drug repositioning for...
Concluding remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):654-663. doi:10.7150/ijbs.9224 This issue Cite
Review
Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs
Joong Sup Shim1 ![]() , Jun O. Liu2
, Jun O. Liu2 ![]()
1. Faculty of Health Sciences, University of Macau, Av. Padre Tomas Pereira, Taipa, Macau SAR, China.
2. Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, 725 N Wolfe St, Baltimore, MD 21205, USA.
Received 2014-3-26; Accepted 2014-5-6; Published 2014-6-10
Abstract
Drug repositioning (also referred to as drug repurposing), the process of finding new uses of existing drugs, has been gaining popularity in recent years. The availability of several established clinical drug libraries and rapid advances in disease biology, genomics and bioinformatics has accelerated the pace of both activity-based and in silico drug repositioning. Drug repositioning has attracted particular attention from the communities engaged in anticancer drug discovery due to the combination of great demand for new anticancer drugs and the availability of a wide variety of cell- and target-based screening assays. With the successful clinical introduction of a number of non-cancer drugs for cancer treatment, drug repositioning now became a powerful alternative strategy to discover and develop novel anticancer drug candidates from the existing drug space. In this review, recent successful examples of drug repositioning for anticancer drug discovery from non-cancer drugs will be discussed.
Keywords: drug repositioning, drug discovery, cancer, angiogenesis, drug screening, drug library.
Introduction to drug repositioning
The traditional approach to drug discovery involves de novo identification and validation of new molecular entities (NME), which is a time-consuming and costly process. Despite huge investment in drug discovery and development and explosive advancement in biological/informational technologies during past decades, the number of new drugs introduced into the clinic has not increased significantly. For example, while the total R&D expenditure for the drug discovery worldwide increased 10 times from 1975 (US $4 billion) to 2009 ($40 billion), the number of NMEs approved has remained largely flat (26 new drugs approved in 1976 and 27 new drugs approved in 2013) [1, 2]. The average time required for drug development has also increased over time. It has been estimated that the average drug development time from discovery to market launch in US and EU countries was 9.7 years during 1990s, but has increased to 13.9 years from 2000 onwards [3]. Those hurdles in discovering and developing new drugs call for alternative approaches including drug repositioning.
Drug repositioning refers to the identification of new indications from existing drugs and the application of the newly identified drugs to the treatment of diseases other than the drug's intended disease. A well-known example of drug repositioning is the use of sildenafil (Viagra) in erectile dysfunctions. Sildenafil is an inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5) and was originally developed for the treatment of coronary artery disease by Pfizer in 1980s. The side effect of sildenafil, marked induction of penile erections, was serendipitously found during the Phase I clinical trials for the patients with hypertension and angina pectoris [4]. After sildenafil failed in Phase II clinical trials for the treatment of angina, it was redirected to the treatment of erectile dysfunctions. Sildenafil received a US-Food and Drug Administration (FDA) approval and entered the US market in 1998, quickly becoming a blockbuster. Another well-known example of drug repositioning is thalidomide. Thalidomide was originally developed as a sedative by the German pharmaceutical company Grünenthal in 1957. It had been used to alleviate morning sickness in pregnant women. Not long after the drug was introduced, it was found to cause serious birth defects. More than 10,000 children in 46 countries were born with malformation of the limbs and other body extremities due to the use of thalidomide, and around half of them died within a few months after birth [5], leading to its withdrawal from the market. In the ensuing decades, several research groups found that thalidomide possesses anticancer activity. It was found to inhibit angiogenesis in animal models by Robert D'Amato and Judah Folkman [6] and was subsequently shown to have promising therapeutic effect on refractory multiple myeloma and metastatic prostate cancer [7, 8]. In 2006, thalidomide received US-FDA approval for the treatment of multiple myeloma in combination with dexamethasone.
Activity-based vs in silico drug repositioning
Several success stories of drug repositioning brought global attention to the existing drug space for potential off-target effects that may be beneficial to certain diseases such as cancer. Since existing drugs have already been used in humans, they have well-established dose regimen with favorable pharmacokinetics (PK) and pharmacodynamics (PD) properties as well as tolerable side effects, making old drugs useful sources of new anticancer drug discovery. In early 2000s, we launched a new initiative to assemble a library of existing drugs, dubbed the Johns Hopkins Drug Library (JHDL) [9]. JHDL has about 2,200 drugs that have been approved by US-FDA or by its foreign counterparts and about 800 non-approved drug candidates that have entered various phases of human clinical trials. We note that NIH Chemical Genomics Center (NCGC) recently built a collection of existing drugs called NCGC Pharmaceutical Collection (NPC) which contains 2,400 small molecular entities that have been approved for clinical use in US (FDA), EU (EMA), Japan (NHI), and Canada (HC) [10, 11]. In addition to these, many of clinical drug collections are currently commercially available. These clinical drug collections have proven to be useful sources to find new indications of existing drugs.
The term 'activity-based drug repositioning' we shall use in this review refers to the application of actual drugs for screening. In contrast, 'in silico drug repositioning' utilizes public databases and bioinformatics tools to systematically identify interaction networks between drugs and protein targets [12]. This latter approach has become successful since a large amount of information on the structure of proteins and pharmacophores has been accumulated over the past few decades along with the advancement of bioinformatics and computational science. Most pharmaceutical companies have already adopted the in silico models for drug discovery from diverse chemical spaces. In silico drug repositioning is a potentially powerful technology and has some advantages over the activity-based drug repositioning, including increased speed and reduced cost. However, it also has some limitations since it requires high-resolution structural information of targets. It also requires disease/phenotype information or gene expression profiles of drugs when a screen does not involve protein targets. In contrast, activity-based drug repositioning can employ both protein target-based and cell/organism-based screens without requiring structural information of target proteins or database. Thus, activity-based and in silico drug repositioning represent two alternative and complementary approaches to new drug discovery (Table 1). Here, we briefly summarize a few recent discoveries of new anti-angiogenic and anticancer activities of existing drugs through activity-based screening of the JHDL along with the subsequent mechanistic and translational follow-up studies.
Activity-based and in silico approaches for drug repositioning.
| Approaches | Pros | Cons |
|---|---|---|
| Activity-based | * No limitation for target-based and cell-based screening assays * Easy to validate screening hits * Lower rate of false positive hits during the screening | * Time and labor consuming * Requires an entire collection of existing drugs * Need to develop a screening assay |
| In silico | * Time and labor efficient * No need an entire collection of existing drugs * No need to develop a screening assay | * Limitation for target-based and cell based screenings (requires structural information of target proteins and drug-induced cell/disease phenotype information) * Higher rate of false positive hits during the screening |
Drug repositioning for anticancer applications
Itraconazole
Itraconazole is a triazole antifungal drug developed in 1980s. Like other azole family of antifungal drugs, it is effective in a variety of systemic fungal infections [13]. The mechanism of antifungal activity of itraconazole has been well established. It is known to inhibit cytochrome P450-dependent lanosterol 14-α-demethylation (14DM) in the ergosterol biosynthesis pathway in fungi [14]. Ergosterol is the main sterol in most yeasts and fungi, being responsible for their membrane integrity and function. It is required for fungal cell proliferation [15]. By inhibiting 14DM, itraconazole and related azole compounds cause the depletion of ergosterol and induce accumulation of 14-α methylsterols that can impair membrane functions, thereby suppressing the fungal growth [14, 16]. Although itraconazole is a well-tolerated drug, it has some side effects including hepatotoxicity (rare but sometimes serious), cardiovascular toxicity and diarrhea (when prepared with cyclodextrin) [17].
Anticancer activity of itraconazole was first reported by Chong et al. in 2007 due to its newly discovered anti-angiogenic activity [18]. In this study, the JHDL was screened for inhibitors of human umbilical vein endothelial cell (HUVEC) proliferation, a proxy for angiogenesis, and itraconazole was identified as one of the most potent hits. In follow-up studies, itraconazole, either alone or in combination with other anticancer drugs, showed strong anticancer activities in preclinical models including non-small cell lung cancer (NSCLC), medulloblastoma, and basal cell carcinoma [19, 20]. Prompted by such encouraging preclinical results, itraconazole has entered several Phase II clinical studies for the treatment of various types of cancer. Most recently, positive clinical results have been reported from advanced lung cancer and prostate cancer trials (both at Johns Hopkins Sidney Kimmel Comprehensive Cancer Center) and from basal cell carcinoma trial (at Stanford University) [21-23]. Itraconazole in combination with pemetrexed showed significant survival benefit in patients with progressive nonsquamous non-small-cell lung cancer compared to the control arm of pemetrexed alone [21]. High dose (600 mg/day) of itraconazole also showed modest anticancer activity in patients with metastatic castration-resistant prostate cancer (CRPC) [22]. In the basal cell carcinoma trial, patients were received oral itraconazole 200 mg twice per day for 1 month or 100 mg twice per day for an average of 2.3 months. In this exploratory trial, itraconazole reduced tumor size by 24% from the patients [23].
Overall, itraconazole was well-tolerated by the patients in all three aforementioned Phase II trials with a few common toxicities including fatigue, nausea and anorexia. Recent study about therapeutic drug monitoring (TDM) of itraconazole suggests that serum concentration of 5 μg/ml (7 μM) is associated with 26% probability of adverse effect [24]. The probability increases progressively with increasing serum concentrations of itraconazole. Classification and regression tree (CART) analysis suggests that serum itraconazole level of 17.1 μg/ml (24.4 μM) is upper limit for TDM [24]. Considering that itraconazole's IC50 values for angiogenesis range from sub-micromolar to single digit micromolar concentrations (0.5 ~ 3 μM), it has a moderate therapeutic window. However, occurrence of some rare but serious side effects such as hepatotoxicity and congestive heart failure should be monitored in the clinical settings, though it is interesting that the higher dose of itraconazole (600 mg daily) did not cause those major side effects in prostate cancer patients as would have been anticipated [17, 22].
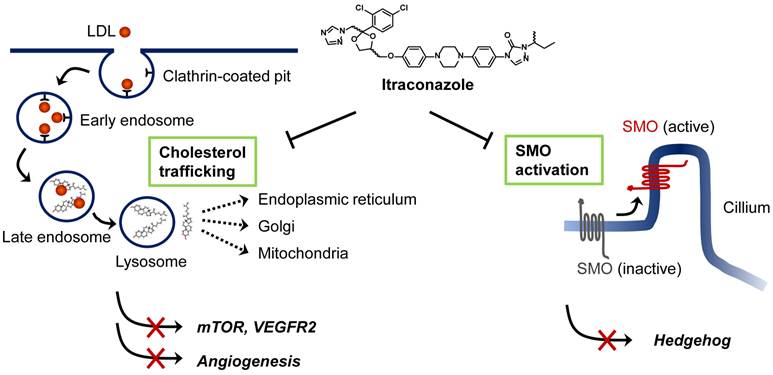
Although itraconazole showed promising anticancer activity in several types of cancer, its precise anticancer mechanism has remained elusive. To date, two anticancer mechanisms of itraconazole have been proposed; inhibition of angiogenesis and inhibition of Hedgehog signaling pathway in certain cancer cells (Figure 1). The studies of Xu et al. [25], and Nacev et al. [26] showed that itraconazole inhibited cholesterol trafficking in human endothelial cells, leading to inhibition of mammalian target of rapamycin (mTOR) and vascular endothelial growth factor receptor type 2 (VEGFR2) signaling pathways that are critical for endothelial cell proliferation and angiogenesis. In a separate study, Kim et al. demonstrated that itraconazole inhibited Hedgehog signaling pathway, thereby suppressing the growths of medulloblastoma and basal cell carcinoma [19]. Ongoing studies are being focused on identifying the molecular target of itraconazole in mammalian cells, which will further our understanding of the precise mode of action of itraconazole for its anticancer activity, facilitating its development as a new anticancer and anti-angiogenic drug.
Proposed mechanisms of anticancer activity of itraconazole. Extracellular cholesterols are transported into cells in the form of low-density lipoprotein (LDL) through binding to LDL receptor. Cholesterol esters are then hydrolyzed in the late endosome/lysosomes and transported to various cellular destinations through cholesterol trafficking system. Itraconazole is known to block the cholesterol release from the late endosome/lysosomes causing hyper-accumulation of cholesterols in the organelle (so-called Niemann-Pick C phenotype). This leads to the inhibition of mTOR activity and VEGFR2 glycosylation in endothelial cells. Itraconazole is also known to inhibit Smoothened (SMO) activation in Hedgehog signaling by a mechanism distinct from that of cyclopamine and other known SMO antagonists. Itraconazole suppresses Sonic hedgehog (SHH)-induced accumulation of SMO in the primary cilium.
Nelfinavir
Nelfinavir is a competitive inhibitor of human immunodeficiency virus (HIV) aspartyl protease and is being used in combination with other antiretroviral drugs to treat patients with HIV infection [27]. It received the US-FDA approval in 1997 for an oral dose regimen of 750 mg three times daily. It was later modified to a regimen of 1250 mg twice daily as recommended by US-FDA. Both regimens were proven to be equally effective [28]. The average peak plasma level of nelfinavir is around 8 μM and the bioavailability is known to be increased when taken with food [29]. Nelfinavir is a well-tolerated drug with some common side effects such as insulin resistance, hyperglycemia and lipodystrophy. From early 2000s, researchers have found potential anticancer activity of nelfinavir. It was reported to inhibit the growths of Kaposi's sarcoma [30], multiple myeloma [31], NSCLC [32, 33], prostate cancer [34], and breast cancer [35, 36]. Nelfinavir exhibited a broad-spectrum anticancer activity in vivo, being efficacious in several preclinical cancer models.
There has been an increasing interest in underlying mechanism of anticancer activity of nelfinavir. A common side effect of nelfinavir was insulin resistance, which was later found to be through inhibition of phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway. AKT was recognized as an important mediator for cancer cell survival. In addition, activation of AKT signaling promotes resistance to chemo- and radiation therapy. Brunner et al. recently conducted a Phase I clinical trial of nelfinavir and chemoradiation for locally advanced pancreatic cancer [37]. In this trial, nelfinavir showed potent radiosensitizing and antitumor activities without adding toxicity in patients with pancreatic cancer. Although nelfinavir is known to inhibit AKT signaling pathway, it does not directly inhibit the kinase activity of AKT. Gupta et al. showed that nelfinavir down-regulates AKT phosphorylation by inhibiting 20S proteasome activity [38]. Hamel et al. also showed that nelfinavir inhibited chymotrypsin- and trypsin-like activities of the rat proteasome preparation in vitro [39]. However, whether proteasome is the relevant target of nelfinavir for its anticancer and anti-AKT activity has been debated since several conflicting results have been reported. Nelfinavir was shown to inhibit cyclin-dependent kinase 2 (CDK2) activity by enhancing proteasome-dependent degradation of Cdc25A phosphatase [40]. In addition, known proteasome inhibitors including MG132 and bortezomib did not recapitulate effects of nelfinavir in breast cancer cells, but, instead, they rescued the AKT inhibition by nelfinavir [36].
Recently, Srirangam et al. [41] and Shim et al. [36] demonstrated that nelfinavir is a novel inhibitor of heat shock protein-90 (HSP90). Srirangam et al. showed that ritonavir, a structurally related HIV protease inhibitor to nelfinavir, bound to HSP90 and inhibited interaction between HSP90 and AKT. Shim et al. conducted a pharmacological profiling of seven genotypically different breast cancer cell lines using JHDL and found that nelfinavir selectively inhibited the proliferation of human epidermal growth factor receptor 2 (HER2)-positive breast cancer cells over HER2-negative ones. In HER2-positive breast cancer cells, nelfinavir caused degradation of HER2 and AKT by inhibiting their association with HSP90. In addition to HER2 and AKT, the study further showed that nelfinavir decreased the levels of other known HSP90 client proteins including CDK4 and CDK6 [36]. These studies explained in part how nelfinavir inhibited AKT signaling in cancer cells.
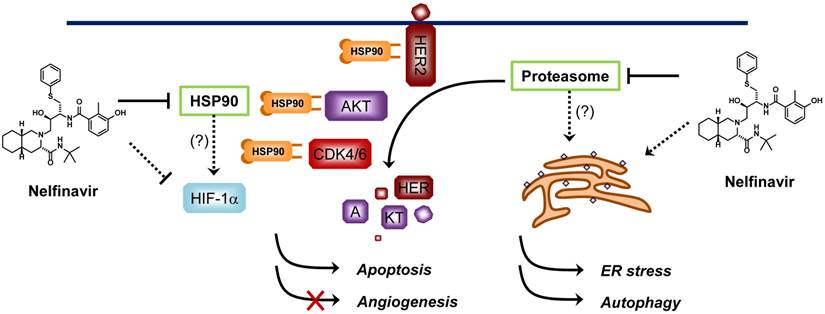
Other proposed anticancer mechanisms of nelfinavir include induction of endoplasmic reticulum (ER) stress and autophagy in cancer cells and inhibition of angiogenesis through down-regulation of hypoxia-inducible factor-1α (HIF-1α) [32, 42, 43]. Since nelfinavir can potentially interact with multiple proteins in cells, its anticancer activity might be a consequence of simultaneous inhibition of multiple pathways essential for cancer cell proliferation and survival (Figure 2). Nelfinavir is now under more than 20 Phase I/II clinical trials for cancer (http://clinicaltrials.gov/). Although the anticancer mechanism of nelfinavir remains to be completely elucidated, promising anticancer activities have been reported from the clinical studies [44-46].
Proposed mechanisms of anticancer activity of nelfinavir. Nelfinavir is known to have a strong anticancer activity through multiple pathways including induction of ER stress, apoptosis and autophagy, and inhibition of AKT pathway and hypoxia-inducible factor 1α (HIF-1α)-dependent angiogenesis. Nelfinavir was shown to inhibit the chymotrypsin- and trypsin-like activities of 20S human proteasome. However, whether anti-proteasome effect is the primary mechanism of nelfinavir for anticancer activity remains elusive since nelfinavir causes proteasome-dependent degradation of several proteins. HSP90 is another proposed molecular target of nelfinavir, of which the inhibition leads to a decrease in the levels of its client proteins including HER2, AKT and CDKs through proteasome-dependent degradation.
Digoxin
Digoxin is a cardiac glycoside isolated from foxglove [47]. It has a long history of use in the treatment of various heart conditions including heart failure and arrhythmia. Digoxin is known as a potent inhibitor of Na+/K+-ATPase pump in cell membrane [48]. Na+/K+-ATPase regulates sodium ion gradient across the cell membrane to efflux intracellular Ca2+ ions. Inhibition of Na+/K+-ATPase by digoxin causes an increase in intracellular Ca2+ concentration in myocardiocytes and pacemaker cells, thereby lengthening cardiac action potential [49].
From early 1980s, a few cohort studies with a small group of breast cancer patients have shown that the use of digoxin decreased the breast cancer recurrence and aggressiveness [50, 51]. These observations suggested a potential anticancer activity of digoxin against breast cancer. It was believed that digoxin, as a phytoestrogen, could interfere with estrogen receptor (ER) signaling in cancer cells, thereby suppressing the growth of breast cancer [52, 53]. Two decades later, however, conflicting results were reported. Haux et al. showed that the population who were taking digitoxin, another cardiac glycoside, had a higher incidence of cancer compared to the control population [54]. In addition, Ahern et al. [55] and Biggar et al. [56] reported that the use of digoxin significantly increased the breast cancer incidence among women in Denmark. Among the digoxin users, there was the higher risk for developing ER-positive breast cancers than ER-negative breast cancers [56]. These data suggested that digoxin, in certain conditions, might act as an estrogen-like molecule rather than an anti-estrogen in women, thus increasing ER-positive breast cancer risk.
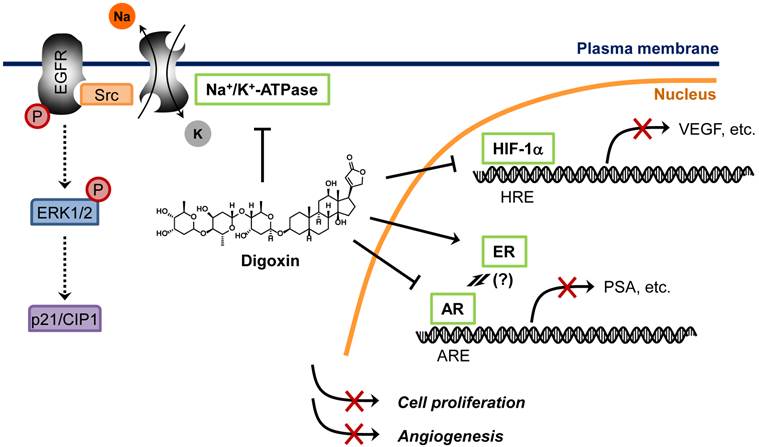
In a recently study, Platz et al. conducted two-stage multidisciplinary studies to identify possible anti-prostate cancer drugs from JHDL [57]. The authors screened JHDL and identified digoxin as one of the most potent inhibitors of prostate cancer cell proliferation. A subsequent large-scale cohort study with long-term follow-up demonstrated that digoxin significantly reduced the incidence of prostate cancer by 25% among men [57]. Moreover, men who had used digoxin for longer than 10 years showed 46% lower incidence of prostate cancer, suggesting a potential anti-prostate cancer activity of digoxin. This encouraging observation led to the recent Phase 2 clinical trial for recurrent prostate cancer. How digoxin showed opposite effects between breast cancer and prostate cancer remains unclear. The fact that digoxin increased the risk of only estrogen sensitive cancers including breast and uterus cancers, but not ovary or cervix cancer, suggests that the tumor promoting mechanism is mediated through its estrogenic effect [58]. Paradoxically, estrogens suppress androgen levels and inhibit prostate cancer growth [59]. Hedelin et al. reported that intake of dietary phytoestrogens significantly reduced prostate cancer risk among the population in Sweden [60]. Moreover, digoxin and other cardiac glycosides decreased secretion of prostate specific antigen (PSA) in androgen receptor (AR)-dependent prostate cancer cells [61]. These observations strongly suggest that estrogenic effect of digoxin is beneficial for the treatment of androgen-dependent cancers, such as prostate cancer. In addition to the estrogenic effect, other anticancer mechanisms of digoxin have been also proposed, such as inhibition of Na+/K+-ATPase and HIF-1α synthesis [62, 63]. Proposed anticancer mechanisms of digoxin are summarized in Figure 3.
Proposed mechanisms of anticancer activity of digoxin. Digoxin is a phytoestrogen which inhibits AR signaling pathway by preventing AR binding to AR-responsive element (ARE), leading to decrease in AR target genes such as PSA in prostate cancer cells. Digoxin is also known to inhibit HIF-1α synthesis, thereby reducing HIF-1α binding to its cognate element, hypoxia-responsive element (HRE), and suppressing the expression of HIF-1α target genes such as VEGF in cancer cells. Binding of cardiac glycosides to Na+/K+-ATPase is known to activate Src, epidermal growth factor receptor (EGFR) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation, which leads to an accumulation of p21/CIP1 and induction of cell cycle arrest in cancer cells.
Digoxin is known to have a narrow therapeutic index (2 to 3), suggesting that doubling or tripling its recommended dose may cause toxicity [64]. The therapeutic serum level of digoxin for heart rate control is about 2 ng/ml (2.6 nM). However, IC50 of digoxin for prostate cancer cell proliferation was about 5-10 times the therapeutic serum level, suggesting a discrepancy between in vitro and in vivo anti-prostate cancer activity of digoxin [57]. Although the mechanism by which digoxin exerts anticancer activity in vivo with its therapeutic serum level remains unclear, it is intriguing to postulate that digoxin may accumulate in prostate tissue or that it may indirectly inhibit prostate cancer growth through other mechanisms such as inhibition of angiogenesis during the long-term, low-dose treatment. Nonetheless, it is clear that digoxin has a beneficial effect on patients with certain types of cancer and is currently undergoing several clinical trials for the treatment of cancer as a monotherapy or in combination with other chemotherapy drugs (http://clinicaltrials.gov/).
Nitroxoline
Nitroxoline is an old antibiotic which has been widely used in European, Asian and African countries from 1960s. It is particularly effective for the treatment of urinary tract infections (UTI) due to the drug's unique PK property. When administered orally, nitroxoline is rapidly absorbed into the plasma and is subsequently excreted into urine [65]. It has a long retention time in urine, thus making it ideal for UTI treatment. Nitroxoline is known to be able to chelate divalent metal ions such as Mg2+ and Mn2+, which is appreciated as a possible mechanism for its antibacterial activity [66].
Shim et al. first reported anticancer activity of nitroxoline in 2010 [67]. The authors conducted two distinct screens, a target-based (methionine aminopeptidase-2 or MetAP2 as a target) and cell-based (HUVEC) screens to identify novel anti-angiogenic agents from a diverse chemical compound library and JHDL, respectively. Nitroxoline was found to be a common hit from both screens [67]. As it was identified from the MetAP2 inhibitor screen, it is not surprising that nitroxoline potently inhibited MetAP2 activity in vitro (IC50 = 54 nM) and in endothelial cells. It is well established that inhibition of MetAP2 activity in endothelial cells causes an increase in p53 level and an activation of retinoblastoma protein (pRb) by decreasing its phosphorylation, leading to the inhibition of endothelial cell proliferation [68]. Similar to a known MetAP2 inhibitor TNP-470, nitroxoline increases the level of p53 and induces hypo-phosphorylation of pRb in HUVEC. In addition, nitroxoline also causes an increase in acetylation of p53 (K382), α-tubulin and histone H3, hallmarks of inhibition of human sirtuins 1 and 2. Subsequent in vitro and in vivo studies showed that nitroxoline inhibited angiogenesis and the growth of cancer xenograft in mouse models. Given that nitroxoline has a long retention time in urine, it was postulated that the drug might be particularly effective in urological cancers such as bladder cancer. Nitroxoline was tested in an orthotopic bladder cancer model in mice and was administered orally for two weeks to assess its anticancer activity. Cancers from control group grew continuously, whereas the cancer growth was significantly delayed in nitroxoline treated group, suggesting a potential anticancer activity of nitroxoline against bladder cancer in vivo. From the translational perspective, the concentration of nitroxoline required for inhibition of endothelial cell proliferation (IC50 = 1.9 μM) was well below the maximal clinically achievable concentration (Cmax > 10 μM) in both human plasma and urine. Taking into account that antibacterial activity of nitroxoline was shown at greater than 10 μM and that daily nitroxoline dosage of 400-750 mg (for adult) was sufficient to show antibacterial activity in human, the current nitroxoline dosage regimen for UTI treatment is likely to be sufficient for blocking angiogenesis in vivo.
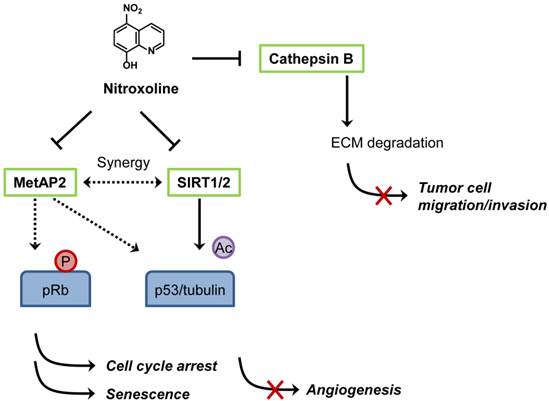
Other recent studies also supported the anticancer activity of nitroxoline. Mirkovic et al. showed that nitroxoline inhibited cathepsin B activity and suppressed breast cancer cell invasion [69]. Cathepsin B plays a role in degradation of extracellular matrix (ECM) and is implicated in tumor cell migration, invasion and metastasis. However, the Ki values of nitroxoline for endopeptidase activity of cathepsin B were 154.4 μM (for dissociation of EI complex) and 39.5 μM (for dissociation of ESI complex), calling into question the relevance of this effect of nitroxoline to its anti-angiogenic activity. However, it cannot be ruled out that the anti-cathepsin B effect of nitroxoline contributes to its anticancer activity in vivo by suppressing tumor cell migration and invasion. The proposed mechanisms of anticancer activity of nitroxoline are summarized in Figure 4. In a separate study, Jiang et al. recently reported that nitroxoline showed strong anticancer activity against lymphoma, leukemia, pancreatic cancer and ovarian cancer cells [70]. Nitroxoline has been used in many European countries as UTI drug for over 50 years and no apparent human toxicity has been reported, making the drug an excellent candidate for anticancer drug repositioning. With the unique PK property and current dosage regimen, human clinical studies of nitroxoline for the treatment of cancer, especially, bladder cancer are warranted.
Proposed mechanisms of anticancer activity of nitroxoline. Nitroxoline was shown to inhibit both MetAP2 and sirtuins (SIRT1 and 2) in human endothelial cells. Inhibition of MetAP2 by nitroxoline induced hypo-phosphorylation of retinoblastoma protein (pRb) and increased the level of p53. Inhibition of SIRT1 and 2 caused an increase in acetylation of p53 (K382) and α-tubulin (in the presence of histone deacetylase inhibitor), leading to an induction of endothelial cell senescence. A synergy in increasing the acetylation level of p53 (K382) and inducing senescence was observed when MetAP2 and SIRT1 were inhibited simultaneously, representing a mechanism of nitroxoline for its anti-angiogenic activity. Nitroxoline was also shown to bind and inhibit cathepsin B, an enzyme responsible for extracellular matrix (ECM) protein degradation in cancer cells, thereby blocking cell migration and invasion.
Concluding remarks
In this review, we provided an overview of drug repositioning for anticancer applications with a particular emphasis on activity-based drug repositioning of non-cancer drugs. Several successful case studies including those exemplified in this review are summarized in Table 2. Many of these drugs are under Phase II studies for cancer therapy. Although drug repositioning should significantly reduce the time and cost associated with drug development processes, benefits are limited to a certain process between preclinical to Phase II study. Many challenges still exist after Phase II trials. Phase III studies involves much larger number of patients compared to Phase I and II studies. Due to the size and relatively long duration, Phase III studies are the most expensive and time-consuming trials and these hurdles in Phase III studies have not changed over the years. Another challenge that should be considered for drug repositioning has to do with intellectual property (IP) protection of the repositioned drugs, especially for those drugs that are off patents. Off-patent drugs can be protected in part by method-of-use (MOU) patents which contain one or more claims directed to a method of use. MOU patents are much weaker than the composition-of-matter (COM) patents in terms of the exclusionary right. Nitroxoline, for example, used to be off patent, but is currently under MOU patent protection for anticancer applications since it was found to have anticancer properties [71].
Examples of activity-based drug repositioning for anticancer applications.
| Drugs | Original uses | Proposed anticancer mechanisms | Developmental status |
|---|---|---|---|
| Itraconazole | * Treatment for fungal infections | * Inhibiting endothelial cell cholesterol trafficking and angiogenesis [18, 25] * Inhibiting Hedgehog pathway [19] | Phase I and II |
| Nelfinavir | * Treatment for HIV infections | * Inhibiting 20S proteasome and AKT signaling [38, 39] * Inhibiting HSP90 and HER2 signaling [36, 41] * Inducing ER stress and autophagy, and inhibiting angiogenesis [32, 42, 43] | Phase I and II |
| Digoxin | * Treatment for cardiac diseases | * Inhibiting Na+/K+-ATPase [63] * Acting as a phytoestrogen and inhibiting androgen receptor signaling [59, 61] * Inhibiting HIF-1α synthesis [62] | Phase I and II |
| Nitroxoline | * Treatment for urinary tract infections | * Inhibiting human MetAP2 and sirtuins in endothelial cells [67] * Inducing premature senescence and inhibiting angiogenesis [67] * Inhibiting cathepsin B [69] | Preclinical trials |
| Riluzole | * Treatment for Amyotropic lateral sclerosis | * Inhibiting the release of glutamate [72] * Inhibiting cell proliferation of metabotropic glutamate receptor 1 (GRM1)-expressing human melanoma cells [72] | Phase I and II |
| Mycophenolic acid | * Immunosuppressant | * Inhibiting type-1 inosine monophosphate dehydrogenase (IMPDH-1) and angiogenesis [73] * Inhibiting c-Myc signaling network in endothelium [74] | Phase I |
| Disulfiram | * Treatment for chronic alcoholism | * Inhibiting proteasome when complexed with metals [75] * Inhibiting DNA methyltransferase 1 (DNMT1) [76] | Phase II and III |
Currently, an estimated number of 4,000 of active pharmaceutical ingredients (API) have been approved for human use in the world [10]. Approved drugs keep accumulating over the years; on average 20 to 30 NMEs each year have been approved by US-FDA [2] further expanding the space for drug repositioning. Since more diverse and selective cancer drug targets are being discovered and developed, the approved drug collections will be particularly useful to quickly identify clinically advanced anticancer drugs against those targets. A major problem of conventional cancer chemotherapy drugs (mainly DNA damaging agents) is notorious side effects that significantly reduce the quality of life of patients. As most of non-cancer drugs have little or tolerable side effects in human, repositioning of non-cancer drugs for anticancer therapy as exemplified in this review will be an excellent strategy for future anticancer drug development.
Acknowledgements
This review was supported by the Start-up Research Grant-SRG2013-00045-FHS (to J.S.S) from the University of Macau. The work described in this review carried out in the Liu lab was supported by the Johns Hopkins Malaria Research Institute, Department of Pharmacology and Molecular Sciences, the Johns Hopkins Fund for Medical Discovery, the Kech Foundation, the Patrick C. Walsh Prostate Cancer Fund, the Prostate Cancer Foundation, Flight Attendant Medical Research Institute Fund, the Commonwealth Foundation, and the National Cancer Institute.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Rick W, Phil L. Report to the President on Propelling Innovation in Drug Discovery, Development, and Evaluation. The President's Council of Advisors on Science and Technology (PCAST). 2012
2. Mullard A. 2013 FDA drug approvals. Nat Rev Drug Discov. 2014;13:85-9
3. Pammolli F, Magazzini L, Riccaboni M. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2011;10:428-38
4. Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM. et al. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8:47-52
5. McBride WG. Thalidomide embryopathy. Teratology. 1977;16:79-82
6. D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082-5
7. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P. et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565-71
8. Ning YM, Gulley JL, Arlen PM, Woo S, Steinberg SM, Wright JJ. et al. Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:2070-6
9. Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ Jr. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415-6
10. Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A. et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16
11. NPC. NCGC Pharmaceutical Collection. Available: http://tripodnihgov/npc/. 2011
12. Dubus E, Ijjaali I, Barberan O, Petitet F. Drug repositioning using in silico compound profiling. Future Med Chem. 2009;1:1723-36
13. Warnock DW. Itraconazole and fluconazole: new drugs for deep fungal infection. J Antimicrob Chemother. 1989;24:275-7
14. Vanden Bossche H, Marichal P, Gorrens J, Geerts H, Janssen PA. Mode of action studies. Basis for the search of new antifungal drugs. Ann N Y Acad Sci. 1988;544:191-207
15. Barrett-Bee K, Dixon G. Ergosterol biosynthesis inhibition: a target for antifungal agents. Acta Biochim Pol. 1995;42:465-79
16. Vanden Bossche H, Marichal P, Le Jeune L, Coene MC, Gorrens J, Cools W. Effects of itraconazole on cytochrome P-450-dependent sterol 14 alpha-demethylation and reduction of 3-ketosteroids in Cryptococcus neoformans. Antimicrob Agents Chemother. 1993;37:2101-5
17. Restrepo A, Robledo J, Gomez I, Tabares AM, Gutierrez R. Itraconazole therapy in lymphangitic and cutaneous sporotrichosis. Arch Dermatol. 1986;122:413-7
18. Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ Jr, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2:263-70
19. Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV. et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388-99
20. Aftab BT, Dobromilskaya I, Liu JO, Rudin CM. Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res. 2011;71:6764-72
21. Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R. et al. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J Thorac Oncol. 2013;8:619-23
22. Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC. et al. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist. 2013;18:163-73
23. Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K. et al. Open-Label, Exploratory Phase II Trial of Oral Itraconazole for the Treatment of Basal Cell Carcinoma. J Clin Oncol. 2014
24. Lestner JM, Roberts SA, Moore CB, Howard SJ, Denning DW, Hope WW. Toxicodynamics of itraconazole: implications for therapeutic drug monitoring. Clin Infect Dis. 2009;49:928-30
25. Xu J, Dang Y, Ren YR, Liu JO. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci U S A. 2010;107:4764-9
26. Nacev BA, Grassi P, Dell A, Haslam SM, Liu JO. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J Biol Chem. 2011;286:44045-56
27. Moyle GJ, Youle M, Higgs C, Monaghan J, Prince W, Chapman S. et al. Safety, pharmacokinetics, and antiretroviral activity of the potent, specific human immunodeficiency virus protease inhibitor nelfinavir: results of a phase I/II trial and extended follow-up in patients infected with human immunodeficiency virus. J Clin Pharmacol. 1998;38:736-43
28. Marzolini C, Buclin T, Decosterd LA, Biollaz J, Telenti A. Nelfinavir plasma levels under twice-daily and three-times-daily regimens: high interpatient and low intrapatient variability. Ther Drug Monit. 2001;23:394-8
29. Bardsley-Elliot A, Plosker GL. Nelfinavir: an update on its use in HIV infection. Drugs. 2000;59:581-620
30. Sgadari C, Monini P, Barillari G, Ensoli B. Use of HIV protease inhibitors to block Kaposi's sarcoma and tumour growth. Lancet Oncol. 2003;4:537-47
31. Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol Cancer Ther. 2004;3:473-9
32. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS. et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13:5183-94
33. Yang Y, Ikezoe T, Nishioka C, Bandobashi K, Takeuchi T, Adachi Y. et al. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br J Cancer. 2006;95:1653-62
34. Yang Y, Ikezoe T, Takeuchi T, Adachi Y, Ohtsuki Y, Takeuchi S. et al. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005;96:425-33
35. Bruning A, Friese K, Burges A, Mylonas I. Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Res. 2010;12:R45
36. Shim JS, Rao R, Beebe K, Neckers L, Han I, Nahta R. et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst. 2012;104:1576-90
37. Brunner TB, Geiger M, Grabenbauer GG, Lang-Welzenbach M, Mantoni TS, Cavallaro A. et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol. 2008;26:2699-706
38. Gupta AK, Li B, Cerniglia GJ, Ahmed MS, Hahn SM, Maity A. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia. 2007;9:271-8
39. Hamel FG, Fawcett J, Tsui BT, Bennett RG, Duckworth WC. Effect of nelfinavir on insulin metabolism, proteasome activity and protein degradation in HepG2 cells. Diabetes Obes Metab. 2006;8:661-8
40. Jiang W, Mikochik PJ, Ra JH, Lei H, Flaherty KT, Winkler JD. et al. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007;67:1221-7
41. Srirangam A, Mitra R, Wang M, Gorski JC, Badve S, Baldridge L. et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin Cancer Res. 2006;12:1883-96
42. Pore N, Gupta AK, Cerniglia GJ, Maity A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia. 2006;8:889-95
43. Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM. et al. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006;66:9252-9
44. Pan J, Mott M, Xi B, Hepner E, Guan M, Fousek K. et al. Phase I study of nelfinavir in liposarcoma. Cancer Chemother Pharmacol. 2012;70:791-9
45. Rengan R, Mick R, Pryma D, Rosen MA, Lin LL, Maity AM. et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response. J Thorac Oncol. 2012;7:709-15
46. Buijsen J, Lammering G, Jansen RL, Beets GL, Wals J, Sosef M. et al. Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother Oncol. 2013;107:184-8
47. Hollman A. Drugs for atrial fibrillation. Digoxin comes from Digitalis lanata. BMJ. 1996;312:912
48. Rossi B, Ponzio G, Lazdunski M. Identification of the segment of the catalytic subunit of (Na+,K+)ATPase containing the digitalis binding site. EMBO J. 1982;1:859-61
49. Belardinelli L, Harder D, Sperelakis N, Rubio R, Berne RM. Cardiac glycoside stimulation of inward Ca++ current in vascular smooth muscle of canine coronary artery. J Pharmacol Exp Ther. 1979;209:62-6
50. Stenkvist B, Bengtsson E, Dahlqvist B, Eriksson O, Jarkrans T, Nordin B. Cardiac glycosides and breast cancer, revisited. N Engl J Med. 1982;306:484
51. Stenkvist B, Bengtsson E, Eriksson O, Holmquist J, Nordin B, Westman-Naeser S. Cardiac glycosides and breast cancer. Lancet. 1979;1:563
52. Rifka SM, Pita JC Jr, Loriaux DL. Mechanism of interaction of digitalis with estradiol binding sites in rat uteri. Endocrinology. 1976;99:1091-6
53. Rifka SM, Pita JC, Vigersky RA, Wilson YA, Loriaux DL. Interaction of digitalis and spironolactone with human sex steroid receptors. J Clin Endocrinol Metab. 1978;46:338-44
54. Haux J, Klepp O, Spigset O, Tretli S. Digitoxin medication and cancer; case control and internal dose-response studies. BMC Cancer. 2001;1:11
55. Ahern TP, Lash TL, Sorensen HT, Pedersen L. Digoxin treatment is associated with an increased incidence of breast cancer: a population-based case-control study. Breast Cancer Res. 2008;10:R102
56. Biggar RJ, Wohlfahrt J, Oudin A, Hjuler T, Melbye M. Digoxin use and the risk of breast cancer in women. J Clin Oncol. 2011;29:2165-70
57. Platz EA, Yegnasubramanian S, Liu JO, Chong CR, Shim JS, Kenfield SA. et al. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011;1:68-77
58. Biggar RJ, Wohlfahrt J, Melbye M. Digoxin use and the risk of cancers of the corpus uteri, ovary and cervix. Int J Cancer. 2012;131:716-21
59. Biggar RJ. Molecular pathways: digoxin use and estrogen-sensitive cancers--risks and possible therapeutic implications. Clin Cancer Res. 2012;18:2133-7
60. Hedelin M, Klint A, Chang ET, Bellocco R, Johansson JE, Andersson SO. et al. Dietary phytoestrogen, serum enterolactone and risk of prostate cancer: the cancer prostate Sweden study (Sweden). Cancer Causes Control. 2006;17:169-80
61. Juang HH, Lin YF, Chang PL, Tsui KH. Cardiac glycosides decrease prostate specific antigen expression by down-regulation of prostate derived Ets factor. J Urol. 2010;184:2158-64
62. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR. et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579-86
63. Kometiani P, Liu L, Askari A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol Pharmacol. 2005;67:929-36
64. Becker DE. Drug therapy in dental practice: general principles. Part 2 - pharmacodynamic considerations. Anesth Prog. 2007;54:19-23 quiz 4-5
65. Mrhar A, Kopitar Z, Kozjek F, Presl V, Karba R. Clinical pharmacokinetics of nitroxoline. Int J Clin Pharmacol Biopharm. 1979;17:476-81
66. Pelletier C, Prognon P, Bourlioux P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob Agents Chemother. 1995;39:707-13
67. Shim JS, Matsui Y, Bhat S, Nacev BA, Xu J, Bhang HE. et al. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J Natl Cancer Inst. 2010;102:1855-73
68. Zhang Y, Griffith EC, Sage J, Jacks T, Liu JO. Cell cycle inhibition by the anti-angiogenic agent TNP-470 is mediated by p53 and p21WAF1/CIP1. Proc Natl Acad Sci U S A. 2000;97:6427-32
69. Mirkovic B, Renko M, Turk S, Sosic I, Jevnikar Z, Obermajer N. et al. Novel mechanism of cathepsin B inhibition by antibiotic nitroxoline and related compounds. ChemMedChem. 2011;6:1351-6
70. Jiang H, Taggart JE, Zhang X, Benbrook DM, Lind SE, Ding WQ. Nitroxoline (8-hydroxy-5-nitroquinoline) is more a potent anti-cancer agent than clioquinol (5-chloro-7-iodo-8-quinoline). Cancer Lett. 2011;312:11-7
71. Liu JO, Shim JS, Chong CR, Bhat S. Quinoline compounds as inhibitors of angiogenesis, human methionine aminopeptidase, and sirt1, and methods of treating disorders. Patent. 2010 WO2010042163 A2
72. Namkoong J, Shin SS, Lee HJ, Marin YE, Wall BA, Goydos JS. et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67:2298-305
73. Chong CR, Qian DZ, Pan F, Wei Y, Pili R, Sullivan DJ Jr. et al. Identification of type 1 inosine monophosphate dehydrogenase as an antiangiogenic drug target. J Med Chem. 2006;49:2677-80
74. Domhan S, Muschal S, Schwager C, Morath C, Wirkner U, Ansorge W. et al. Molecular mechanisms of the antiangiogenic and antitumor effects of mycophenolic acid. Mol Cancer Ther. 2008;7:1656-68
75. Cvek B, Dvorak Z. Targeting of nuclear factor-kappaB and proteasome by dithiocarbamate complexes with metals. Curr Pharm Des. 2007;13:3155-67
76. Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J. et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate. 2011;71:333-43
Author contact
![]() Corresponding author: Joong Sup Shim, Ph.D. Faculty of Health Sciences, University of Macau, Av. Padre Tomas Pereira, Taipa, Macau SAR, China. Tel: +853-8397-8445 Email: jsshimmo; or Jun O. Liu, Ph.D, Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, 725 N Wolfe St, Baltimore, MD 21205. Tel: +1-410-955-4619 Email: joliuedu.
Corresponding author: Joong Sup Shim, Ph.D. Faculty of Health Sciences, University of Macau, Av. Padre Tomas Pereira, Taipa, Macau SAR, China. Tel: +853-8397-8445 Email: jsshimmo; or Jun O. Liu, Ph.D, Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, 725 N Wolfe St, Baltimore, MD 21205. Tel: +1-410-955-4619 Email: joliuedu.