Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Materials
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(7):664-676. doi:10.7150/ijbs.8232 This issue Cite
Research Paper
Long Non-coding RNA URHC Regulates Cell Proliferation and Apoptosis via ZAK through the ERK/MAPK Signaling Pathway in Hepatocellular Carcinoma
Wei-Hua Xu1*, Jian-Bin Zhang2*, Zheng Dang1*, Xiao Li1*, Ti Zhou1, Jie Liu1, De-Sheng Wang1, Wen-Jie Song1 ![]() , Ke-Feng Dou1
, Ke-Feng Dou1 ![]()
1. Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi, 710032, People's Republic of China
2. Department of Occupational and Environmental Health, Faculty of Military Preventive Medicine, Fourth Military Medical University, Xi'an, Shaanxi, 710032, People's Republic of China
* Wei-Hua Xu, Jian-Bin Zhang, Zheng Dang and Xiao Li are co-first authors and contributed equally to this work.
Received 2013-11-27; Accepted 2014-5-19; Published 2014-6-10
Abstract
Long non-coding RNAs (lncRNAs) have previously been implicated in human disease states, especially cancer. Although the aberrant expression of lncRNAs has been observed in cancer, the biological functions and molecular mechanisms underlying aberrantly expressed lncRNAs in hepatocellular carcinoma (HCC) have not been widely established. In the present study, we investigated a novel lncRNA, termed URHC (up-regulated in hepatocellular carcinoma), and evaluated its role in the progression of HCC. Expression profiling using a lncRNA microarray revealed that URHC was highly expressed in 3 HCC cell lines compared to normal hepatocytes. Quantitative real-time polymerase chain reaction (qRT-PCR) analyses confirmed that URHC expression was increased in hepatoma cells and HCC tissues. Moreover, using qRT-PCR, we confirmed that URHC expression was up-regulated in 30 HCC cases (57.7%) and that its higher expression was correlated with poor overall survival. We further demonstrated that URHC inhibition reduced cell proliferation and promoted apoptosis. We hypothesize that URHC may function by regulating the sterile alpha motif and leucine zipper containing kinase AZK (ZAK) gene, which is located near URHC on the same chromosome. We found that ZAK mRNA levels were down-regulated in HCC tissues and the expression levels of ZAK were negatively correlated with those of URHC in the above HCC tissues. Next, we confirmed that URHC down-regulated ZAK, which is involved in URHC-mediated cell proliferation and apoptosis. Furthermore, ERK/MAPK pathway inactivation partially accounted for URHC-ZAK-induced cell growth and apoptosis. Thus, we concluded that high URHC expression can promote cell proliferation and inhibit apoptosis by repressing ZAK expression through inactivation of the ERK/MAPK pathway. These findings may provide a novel mechanism and therapeutic targets for the treatment of HCC.
Keywords: Long non-coding RNA, URHC, HCC, ZAK, proliferation, apoptosis.
Introduction
Hepatocellular carcinoma (HCC) is the most commonly occurring primary malignancy of the liver in adults. HCC is currently the fifth most frequently diagnosed tumor and the third leading cause of cancer-related deaths, with an estimated 748,300 new liver cancer cases and 695,900 HCC-related deaths worldwide in 2008 [1, 2]. The majority of these cases and deaths occur in developing countries, including China [3, 4]. Despite the therapeutic advances that have been made in HCC, such as surgical resection, liver transplantation, and adjuvant therapy, the overall 5-year survival rate of HCC patients has not significantly improved [3]. Earlier studies have indicated that hepatocarcinogenesis is a complex, multistep process characterized by a large number of abnormal molecules, and several HCC-related molecular signaling pathways have been identified [2, 5-9]. However, the precise molecular mechanisms underlying liver carcinogenesis remain poorly understood. Therefore, it is of paramount importance to identify novel biomarkers and effective therapeutic targets for HCC.
In recent years, genome-wide transcriptional studies have indicated that only approximately 1% of the human genome produces biologically meaningful RNA transcripts, whereas a much larger proportion of the genome is transcribed into non-coding RNAs (ncRNAs) [10, 11]. Compared with the diversity of RNA transcripts that encode for proteins, functions have only been demonstrated for a small number of ncRNAs. Long non-coding RNAs (lncRNAs), which are currently defined as transcripts greater than 200 nucleotides in length without obvious protein coding functions, have been identified as a new class of functional ncRNAs [12, 13]. In many cases, lncRNAs act as key molecules regulating gene expression, chromatin remodeling, transcription, and post-transcriptional processing [14-17]. In addition, increasing numbers of reports have demonstrated that lncRNAs regulate multiple biological processes, including cell growth, cell cycle progression, differentiation, and apoptosis [18-21]. Furthermore, a large and growing body of literature has indicated the involvement of lncRNAs in a variety of diseases, especially cancer [10, 22-24]. Those lncRNAs that have been associated with cancers are often aberrantly expressed and play functional roles in carcinogenesis, tumor growth, metastasis, and recurrence [16, 23]. Recent studies have demonstrated that dysregulation of several lncRNAs is related to human cancers, such as PCGEM 1 (a prostate tissue-specific, androgen-regulated gene), MALAT-1 (metastasis-associated lung adenocarcinoma transcript 1), and HULC (highly up-regulated in liver cancer) [25-27]. Although altered expression of several lncRNAs has been described in some cancers, the specific role of aberrantly expressed lncRNAs in HCC remains largely unknown.
The aim of the present study was to understand the involvement of lncRNAs in HCC and to investigate differentially expressed lncRNAs in HCC, thereby characterizing their potential roles in tumor growth. Our results indicated that a novel lncRNA, termed URHC (up-regulated in hepatocellular carcinoma), was identified to be one of the most frequently up-regulated lncRNAs in malignant hepatocytes. We found that URHC levels were significantly increased in HCC tissues, suggesting that increases in the expression levels may play significant roles in HCC carcinogenesis. Moreover, we demonstrated that down-regulation of URHC expression inhibited hepatoma cell proliferation and induced apoptosis in vitro. Our results indicated that ZAK was a functional target of URHC and that URHC regulated cell proliferation and apoptosis via down-regulating ZAK. Furthermore, we found that inactivation of the ERK/MAPK pathway functioned as the downstream of URHC-ZAK axis. Thus, these results may provide us new insight into the role of lncRNAs in the development of HCC and indicate the potential application of URHC in the treatment of HCC.
Materials and Methods
Cell lines and samples
The HepG2, SMMC7721 and Huh7 human HCC cell lines and the HL-7702 human immortalized normal hepatocyte cells were maintained in our lab. All of the cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS, Gibco) and incubated at 37℃ in a humidified environment containing 5% CO2. Samples of HCC tissues and corresponding benign liver tissues (3 cm away from the tumor) were collected from patients who had undergone curative surgery for HCC at Xijing Hospital, which is affiliated with the Fourth Military Medical University, between 2005 and 2007. The tissue samples were snap-frozen with liquid nitrogen after surgery and stored at -80℃ prior to RNA extraction. All HCC and normal liver tissue samples were pathologically confirmed. Written informed consent for the biological studies was obtained from each patient involved in the study, and the study was approved by the Ethics Committee of Xijing Hospital.
RNA extraction
Total RNA was extracted from the cells or tissue samples using TRIzol (Life Technologies, Carlsbad, CA, USA) reagent according to the manufacturer's recommended instructions. RNA purity and integrity were analyzed using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Quantified total RNA was further purified using an RNeasy Mini Kit (QIAGEN, GmBH, Germany) and an RNase-free DNase set (QIAGEN).
LncRNA microarray analysis
A lncRNA microarray assay was performed by Biotechnology corporation (Shanghai, China). Briefly, total RNA was amplified and transcribed into fluorescent cRNA using a Low Input Quick Amp Labeling Kit, One-Color (Agilent Technologies). The labeled cRNA was purified using an RNeasy Mini Kit (QIAGEN) and hybridized to the Agilent Human LncRNA Microarray (4×180K, platform: GPL17885, Agilent Technologies). Approximately 38,000 lncRNAs were collected from the authoritative data sources, including NCBI_refseq, NCBI_unigene, NCBI_other, Ensembl, UCSC, LNCRNA-DB, Agilent, ncRNASCAN and published papers. Moreover, the correlations between approximately 41,000 mRNAs and lncRNAs were also analyzed using this array. After a 17-hour hybridization, the arrays were washed in staining dishes with a Gene Expression Wash Buffer Kit (Agilent Technologies). The arrays were scanned using an Agilent Microarray Scanner (Agilent Technologies), and the data were extracted using Feature Extraction Software (Version 10.7, Agilent Technologies). Subsequent raw data were normalized using a Quantile algorithm with Gene Spring Software (Version 11.0, Agilent Technologies).
Quantitative real-time polymerase chain reaction (qRT-PCR)
The complementary DNA (cDNA) was synthesized using an iScript cDNA Synthesis Kit (BIO-RAD, Hercules, CA, USA) in accordance with the manufacturer's recommended instructions. The RNA expression levels were detected using qRT-PCR, which was performed using the 7900 HT Sequence Detection System (ABI, Foster City, CA, USA) and ABI Power SYBR Green PCR Master Mix (ABI). The expression of β-actin was used as a reference to normalize the amount of lncRNA or mRNA in each sample. All of the real-time PCR reactions were performed in triplicate. The relative RNA expression was calculated using the 2-ΔCt method. The primer sequences that were tested in this study were as follows: URHC-forward 5'-TGTTTATGTGAGAGGAGAAAGGAAG-3', URHC-reverse, 5'-CACTAGAGGTCTGCAAATAAAGTGA-3'; ZAK-forward 5'-TCAGACTCCACCTTTGTTTGCA-3', ZAK-reverse, 5'-GTAGGTGCTTGGAACTCTAGTTTTGA-3'; and β-actin-forward, 5'-CTGGAACGGTGAAGGTGACA-3', β-actin-reverse, 5'-CGGCCACATTGTGAACTTTG-3'.
RNA interference and transfection
Specific siRNAs targeting URHC mRNA (URHC-siRNA #1 target sequence: 5'-CCCUUGA CUUGGGAUUAAUTT-3' and URHC-siRNA #2 target sequence: 5'-GGGAUACAUACGUACA UAATT-3'), ZAK mRNA (ZAK-siRNA #1 target sequence: 5'-CAACACGGACAUCAUACGATT-3' and ZAK-siRNA #2 target sequence: 5'-UGUUCAACUCCUAACUGCGTT-3'), and the control siRNAs were synthesized by GenePharma (Shanghai, China). Cells were transfected with 50 nmol/L of either siRNA targeting URHC or ZAK using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's recommended instructions. To monitor transfection efficiency, negative siRNA was used as a control. The cells were harvested 48 h after transfection.
Methyl thiazolyl tetrazolium (MTT) assay
Appropriate cells that had been transfected with URHC-siRNA, ZAK-siRNA, URHC-siRNA plus ZAK-siRNA, or negative control siRNA were seeded in 96-well plates in triplicate at densities of approximately 1000 cells per well and cultured in DMEM containing 10% FBS. Cell viability was analyzed using MTT (Sigma, CA, USA) assay at the indicated time points.
5-ethynyl-2′-deoxyuridine (EdU) incorporation assay
SMMC7721 or HepG2 cells were transfected with URHC-siRNA or control siRNA in 96-well plates. Forty-eight hours after transfection, EdU incorporation assay was carried out using the Cell-Light EdU imaging detecting kit (RiboBio, Guangzhou, China) according to the manufacturer's instructions. EdU is an alternative thymidine analog whose incorporation can be used to label and identify cells undergoing DNA replication [28]. EdU positive cells were calculated with (EdU add-in cells/Hoechst stained cells) ×100%.
Cell cycle assay
After 48 h transfection as earlier described, the cells were collected and washed with phosphate-buffered saline (PBS). The washed cells were resuspended in PBS and fixed in 75% ethanol. Then, the fixed cells were stained with propidium iodide (PI) supplemented with RNaseA (Sigma) for cell cycle analysis with a FACScan flow cytometer (BD Biosciences, CA, USA). Data were collected and analyzed with the ModFit software (BD Biosciences).
Western blot analysis
Western blotting was carried out as previously described [29]. Briefly, total proteins were prepared from the samples by complete cell lysis (Beyotime, Shanghai, China) with protease and phosphatase inhibitors. Identical quantities of proteins were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred onto polyvinylidene difluoride membranes. After incubation with antibodies specific for Bcl-2 (Cell Signaling Technology, Beverly, MA, USA), Bax (Cell Signaling Technology), cleaved caspase 3 (Cell Signaling Technology), caspase 3 (Cell Signaling Technology), ZAK (Abcam, MA, USA), ERK (Cell Signaling Technology), p-ERK (Cell Signaling Technology), JNK (Cell Signaling Technology), p-JNK (Cell Signaling Technology), p38 (Cell Signaling Technology), p-p38 (Cell Signaling Technology) and β-actin (Sigma), the blots were incubated with goat anti-rabbit or anti-mouse secondary antibodies (Cell Signaling Technology). The bands were then scanned using a ChemiDocXRS+ Imaging System (BIO-RAD) and quantified using Quantity One v4.6.2 software (BIO-RAD).
Inhibition of ERK phosphorylation
SMMC7721 cells that had been transfected with URHC-siRNA or negative control siRNA were treated with an ERK inhibitor (PD98059, 10 μM, Cell Signaling Technology) in DMEM that had been supplemented with 10% FBS. Subsequent MTT, cell cycle and western blot assays were performed 48 h after inhibition.
Statistical analyses
All of the data are summarized and presented as the means ± standard deviation (SD) from at least 3 separate experiments. The differences between independent groups were analyzed using ANOVA followed by Student's t-test. The correlations between URHC and clinicopathological characteristics were analyzed using Pearson's chi-squared test or Fisher's exact test. The expression levels of ZAK in clinical HCC tissues and their adjacent non-tumorous tissues were compared using a Wilcoxon signed-rank test. The relationship between expression levels of URHC and ZAK was analyzed by Pearson's correlation. The overall survival probability was evaluated using the Kaplan-Meier method, and the differences between 2 groups were estimated using the log-rank test. All statistical analyses were performed using SPSS 17.0 software (Chicago, IL, USA) and GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). A P < 0.05 was considered to be statistically significant.
Results
URHC expression is up-regulated in HCC cell lines and tissues
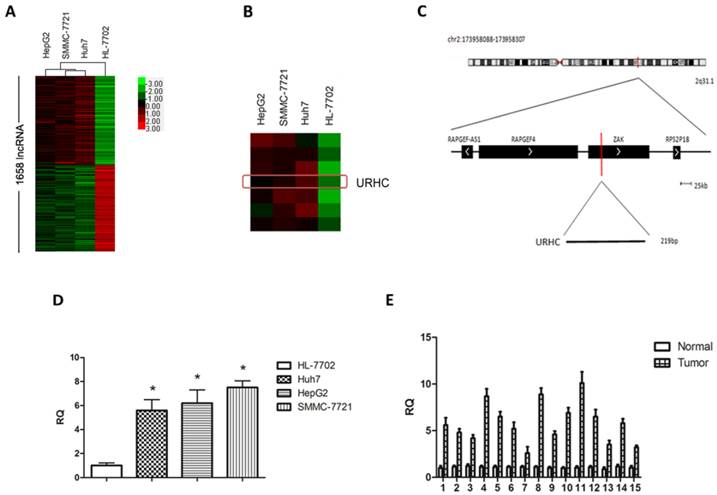
To determine changes of lncRNAs in malignant hepatocytes, we performed a hybridization-based microarray analysis for lncRNA expression (The microarray data discussed in this paper have been deposited in NCBI Gene Expression Omnibus (GEO) and are accessible through GEO accession number GSE56384, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE56384). Compared with normal hepatocytes, 1658 lncRNAs were aberrantly expressed in 3 hepatoma cell lines simultaneously and were regarded as differentially expressed lncRNAs (Fold change >2, Additional file 1: Supplementary Table 1), accounting for 3.9% of all lncRNAs. Hierarchical clustering was used to analyze the differential lncRNA expression profiles in HCC cell lines (Fig. 1A). GO annotation and KEGG pathway analyses indicated that aberrantly expressed lncRNAs affected many genes that may be associated with tumor biological processes, such as cell proliferation, cell cycle progression, apoptosis, migration and/or invasion (http://david.abcc.ncifcrf.gov/, Additional file 1: Supplementary Table 2). Next, to understand the behavior of these differentially expressed lncRNAs in HCC, we screened several HCC-related lncRNAs from the aspect of expression levels, the length of lncRNAs, GO annotation and KEGG pathway analysis, and finally focused on 8 overexpressed lncRNAs in the microarray analysis with fold change >2. We then determined the expression levels of the above lncRNAs in an independent 15 HCC tissues (Additional file 2: Supplementary Figure S1), and named one of the most up-regulated lncRNAs in malignant hepatocytes and designated it as URHC (up-regulated in hepatocellular carcinoma) (Fold change=3.335, Fig. 1B). Information from the Genbank revealed that URHC is located on the forward strand of human chromosome 2: 173958088-173958307 and that its transcript length is 219 bp (Fig. 1C). To further validate this result, we verified the expression of URHC in HCC cell lines using real-time PCR analyses (Fig. 1D). The PCR findings were similar to the expression levels observed using the lncRNA microarray. We further confirmed the expression pattern of URHC in 15 HCC samples and found that it was significantly up-regulated by 2-fold or more (2-to 10-fold) compared to the matching benign liver tissues (Fig. 1E). These data indicate that URHC expression levels are frequently increased in human HCC and may contribute to the pathogenesis of HCC.
URHC expression in HCC cell lines and tissues. (A) Differentially expressed lncRNAs in human malignant and normal liver cells were analyzed as described in the Materials and Methods section using a lncRNA microassay. Red represents higher expression levels and green represents lower expression levels. (B) Hierarchical clustering of URHC expression levels in HCC cell lines compared with the normal control. The red rectangle indicates the hybridization signal of URHC. (C) The URHC information was described in the Genbank. (D) Analyses of URHC expression levels were performed in 3 HCC cell lines using real-time PCR analyses. (E) URHC expression levels were determined in HCC tissues and matched normal tissues (n=15). The 15 cases of HCC tissues and their matched normal tissues were included for further experiments and statistical analysis. β-actin was used as the internal control. The results show the means ± SD from 3 separate experiments. *, P < 0.05.
Association between URHC expression and clinicopathological factors
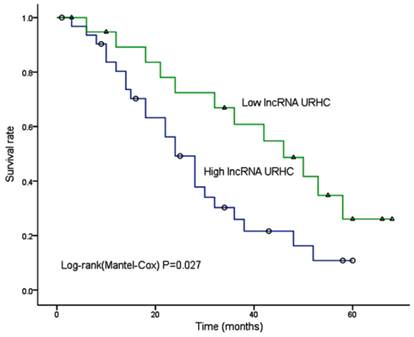
We further sought to determine whether the URHC expression pattern in HCC was associated with specific clinicopathological factors by analyzing samples from 52 HCC patients who had received liver resection therapy. We examined URHC expression levels using qRT-PCR, and fold changes of >2 were designated as up-regulated. We found that URHC was up-regulated in 57.7% (30/52) of HCC tissues compared with adjacent benign tissues, whereas URHC was either down-regulated or was not obviously altered in 42.3% (22/52) of HCC tissues (Table 1). Statistical analyses revealed that high URHC expression levels were significantly associated with both tumor size (χ2=8.868, P=0.003) and tumor number (χ2=6.857, P=0.009, Table 1). However, we did not find any correlation between URHC expression levels and other clinicopathological features, including gender, age, AFP level, tumor metastasis, PVTT (portal vein tumor thrombi), histological differentiation and AJCC (American Joint Committee on Cancer) stage. Next, we examined whether URHC expression levels were correlated with HCC patient outcomes following hepatectomy. A Kaplan-Meier survival analysis revealed that high URHC expression was correlated with shorter overall survival after surgery (χ2=4.863, P=0.027, Fig. 2). These results indicate that URHC may be involved in HCC tumor growth and may predict overall survival in HCC patients.
Correlation between URHC expression and clinicopathologic factors in human HCC.
| Characteristics | Number of patients | LncRNA URHC | χ2 | * P value | |
|---|---|---|---|---|---|
| High | Low | ||||
| All cases | 52 | 30(57.7%) | 22(42.3%) | ||
| Gender | - | 0.685 | |||
| Male | 45 | 25 | 20 | ||
| Female | 7 | 5 | 2 | ||
| Age (yr) | 2.340 | 0.126 | |||
| ≥50 | 22 | 10 | 12 | ||
| <50 | 30 | 20 | 10 | ||
| Tumor size (cm) | 8.868 | 0.003** | |||
| ≤5 | 23 | 8 | 15 | ||
| >5 | 29 | 22 | 7 | ||
| PVTT | 0.552 | 0.458 | |||
| Absent | 30 | 16 | 14 | ||
| Present | 22 | 14 | 8 | ||
| Tumor number | 6.857 | 0.009** | |||
| Single | 20 | 7 | 13 | ||
| Multiple | 32 | 23 | 9 | ||
| AFP level(ng/ml) | 0.227 | 0.634 | |||
| ≤400 | 28 | 17 | 11 | ||
| >400 | 24 | 13 | 11 | ||
| Lymph node metastasis | 0.023 | 0.879 | |||
| Yes | 23 | 13 | 10 | ||
| No | 29 | 17 | 12 | ||
| Histological differentiation | 0.004 | 0.947 | |||
| High+ moderate | 31 | 18 | 13 | ||
| Low | 21 | 12 | 9 | ||
| Distal metastasis | - | 0.567 | |||
| Yes | 3 | 1 | 2 | ||
| No | 49 | 29 | 20 | ||
| AJCC stage | 0.366 | 0.545 | |||
| I + II | 19 | 12 | 7 | ||
| III + IV | 33 | 18 | 15 | ||
* Statistical analyses were performed using the chi-squared test or Fisher's exact test. **, P < 0.05.
Kaplan-Meier analysis of overall survival in 52 HCC patients based on URHC expression levels.
URHC regulates cell proliferation and apoptosis
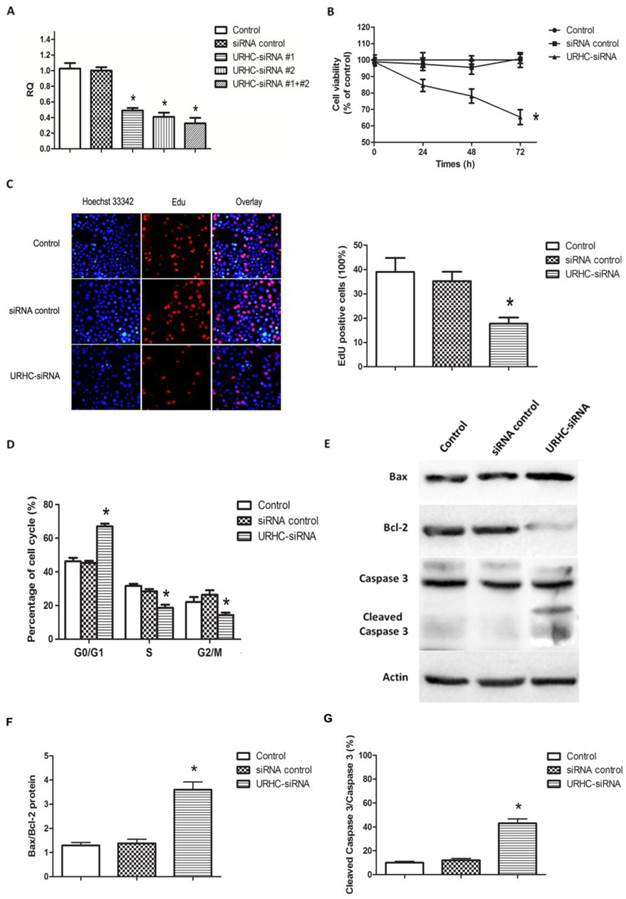
To further assess the biological effects of URHC in HCC, we inhibited URHC expression by transfecting URHC-siRNA into SMMC7721 cells, using control siRNA as the negative control. After 48 h of RNA interference, real-time PCR analyses revealed that URHC expression was efficiently inhibited in SMMC7721 cells co-transfected with two specific siRNAs relative to the controls (Fig. 3A). Next, the cells co-transfected with two siRNAs were used in the subsequent functional experiments. Compared with the control cells, the MTT results revealed that silencing URHC resulted in a significant decrease of cell viability at 24, 48, and 72 h, respectively (Fig. 3B). Further EdU incorporation assays showed EdU positive cells reduced significantly after URHC knockdown, indicating that URHC depletion suppressed the entry of cells into S phase (reflecting DNA synthesization) (Fig. 3C). Moreover, the results of cell cycle assay indicated that repressing URHC expression mainly led to a G0/G1 accumulation at 66.74±3.65% compared with 46.13±4.14%, 45.33%±2.31% of the controls, but a decrease of S phase at 18.67±3.06%, compared with 31.67±2.08%, 28.33±2.52% of the controls, which further support our EdU results (Fig. 3D). In addition, an analysis of apoptosis demonstrated that URHC knockdown promoted apoptosis. We examined whether URHC down-regulation could alter the balance between the proapoptotic protein Bax and the antiapoptotic protein Bcl-2. Western blot analysis showed that SMMC7721 cells in which URHC had been down-regulated expressed higher levels of Bax and displayed an approximately 3-fold increase in Bax/Bcl-2 protein levels compared with the control cells (Fig. 3E and F). We further examined the expression of caspase 3 and its active form of cleaved caspase 3, which indicate the direct proteolytic activation of the effector caspase to induce apoptotic cell death during apoptosis [30]. Compared with the negative controls, significant up-regulation of cleaved caspase 3 was observed in cells that had been transfected with URHC-siRNA, and the ratio of cleaved caspase 3/caspase 3 was remarkably increased (Fig. 3E and G). Importantly, the similar results of URHC-mediated cell proliferation and apoptosis were also observed in HepG2 cells that had been transfected with URHC-siRNA (Additional file 2: Supplementary Figure S2). Collectively, our data suggest that URHC expression may play a pivotal role in the proliferation and apoptosis of hepatoma cells in vitro.
Effects of URHC on hepatoma cell proliferation and apoptosis. (A) Decreased expression of URHC was detected using qRT-PCR 48 h post-transfection. (B) SMMC7721 cells were transfected with either 50 nmol/L of URHC-siRNA or the siRNA control, and cell viability was measured using the MTT assay at 24, 48, and 72 h after transfection. (C) EdU and Hoechst staining were performed as described in the Materials and Methods section, and cell proliferation was examined by EdU incorporation assay at 48 h after transfection. (D) SMMC7721 cells were collected and stained with PI and the cell cycle distribution was analyzed with flow cytometer. (E) SMMC7721 cells were transfected as described in (B), Bax, Bcl-2, caspase 3 and cleaved caspase 3 expression were examined using western blot analyses and the ratios of Bax/Bcl-2 (F) and cleaved caspase 3/caspase 3 (G) were determined. β-actin was used as a reference. The results show the means ± SD from at least 3 separate experiments. *, P < 0.05.
ZAK is a potential functional target of URHC, and URHC regulates ZAK expression
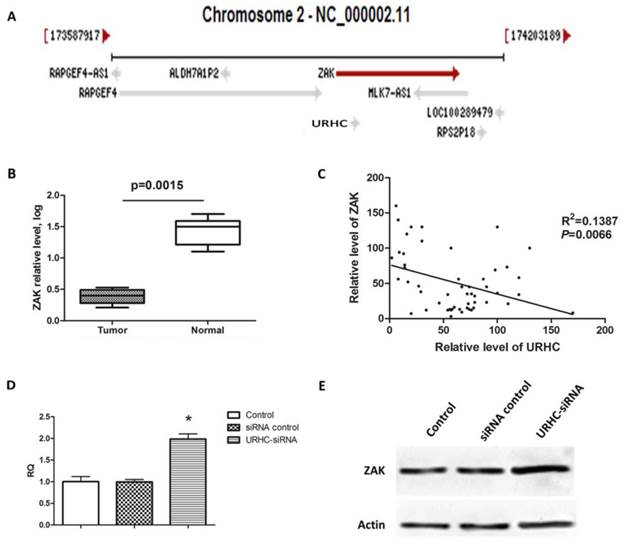
LncRNAs have been reported to act in cis (on neighboring genes) to regulate the expression of genes or in trans (on distantly located genes) to target distant transcriptional activators or repressors via a variety of mechanisms [10]. Therefore, we searched the NCBI database (http://www.ncbi.nlm.nih.gov/gene) and found that ZAK was located near URHC on chromosome 2 (Fig. 4A). ZAK may function as a tumor-suppressor gene, and its role in tumor biology has been well characterized [31]. Next, we determined whether ZAK acted as a potential target for URHC. We assessed the expression of ZAK in 52 human HCC tissues and found that ZAK levels were significantly down-regulated compared with the normal controls (Fig. 4B). We then explored the correlation between URHC and ZAK mRNA expression in the above-mentioned 52 HCC tissues. Importantly, a significant negative correlation was observed between the expression levels of URHC and ZAK (R2=0.1387, P=0.0066, Fig. 4C). We further assessed the role of URHC in the expression of ZAK. Interestingly, cells transfected with URHC-siRNA increased the ZAK mRNA levels and relative protein expression compared with the controls (Fig. 4D and E). Thus, our findings provided evidence of a negative link between the expression of URHC and ZAK in HCC.
ZAK is a target of URHC, and URHC down-regulates ZAK in SMMC7721 cells. (A) The ZAK gene is located near URHC on chromosome 2. (B) The relative mRNA levels of ZAK in HCC tissues (n=52) and matched normal tissues was determined using qRT-PCR. (C) A negative correlation between the relative mRNA levels of ZAK and URHC was observed in the above 52 HCC tissue samples (R2=0.1387, P=0.0066). (D) SMMC7721 cells were treated with URHC-siRNA, and the mRNA and protein levels of ZAK (E) were examined using qRT-PCR and western blot analyses, respectively. β-actin was used as a reference. The results show the means ± SD from 3 separate experiments. *, P < 0.05.
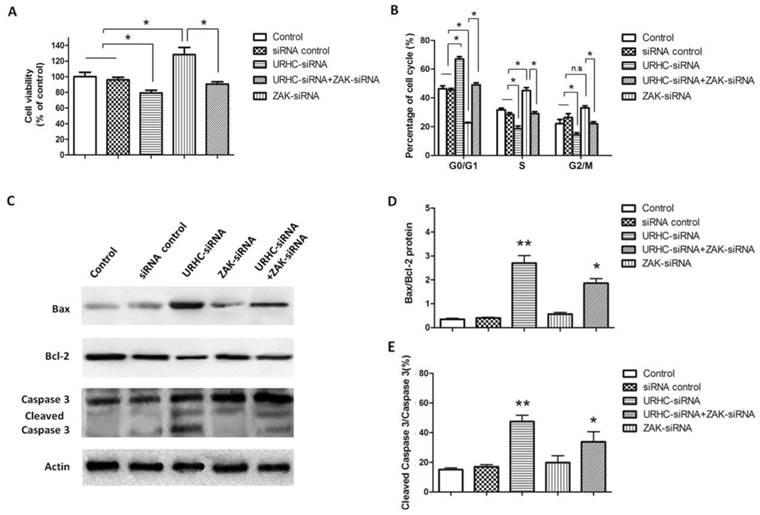
To further assess the contribution of ZAK to the biological effects of URHC, we evaluated the impact of ZAK silencing by RNA interference, and hence ZAK expression, on lncRNA URHC-dependent cell proliferation and apoptosis. In this study, we observed that silencing ZAK increased cell viability in HL-7702 cells by MTT assay and the protein levels of ZAK were confirmed by western blot analysis (Additional file 2: Supplementary Figure S3). Next, we detected whether ZAK was required for URHC-enhanced cell proliferation of SMMC7721 cells. MTT assay showed that SMMC7721 cells transfected with ZAK-siRNA promoted cell growth compared to the cells co-transfected with URHC-siRNA and ZAK-siRNA or the controls, indicating that the inhibitory effects of URHC knockdown on cell proliferation were partially rescued by the presence of siRNA targeting ZAK in SMMC7721 cells (Fig. 5A). In addition, cell cycle results showed that ZAK silencing was able to incompletely abrogate the G0/G1 accumulation induced by URHC knockdown (Fig. 5B). Thus, our data indicated that the effect of URHC on cell proliferation was dependent upon ZAK. We next investigated the ability of ZAK to modulate apoptosis. We observed that ZAK silencing reduced the expression levels of Bax and incompletely attenuated URHC knockdown-mediated apoptosis in SMMC7721 cells (Fig. 5C). In addition, quantitative analyses revealed significant increases in the ratio of Bax/Bcl-2 and cleaved caspase 3/caspase 3 in cells that had been co-transfected with URHC-siRNA and ZAK-siRNA compared with the control or ZAK-siRNA groups (Fig. 5D and E). Based on these findings, we conclude that ZAK mediates the biological effects of URHC on cell proliferation and apoptosis in human HCC.
URHC regulates cell proliferation and apoptosis by down-regulating ZAK. (A) The proliferation of SMMC7721 cells that had been transfected with URHC-siRNA, ZAK-siRNA, URHC-siRNA plus ZAK-siRNA, or the siRNA control was examined at 48 h using the MTT assay. (B) SMMC7721 cells were transfected as described in (A), and the cell cycle distribution was calculated by flow cytometer after staining with PI. (C) The protein expression levels of Bax, Bcl-2, caspase 3 and cleaved caspase 3 were measured using western blot analyses, and the ratios of Bax/Bcl-2 (D) and cleaved caspase 3/caspase 3 (E) were determined. β-actin was used as a reference. The results show the means ± SD from at least 3 separate experiments. *, P < 0.05; **, P < 0.001; NS, nonsignificant.
URHC promotes HCC growth through ZAK via the ERK/MAPK signaling pathway
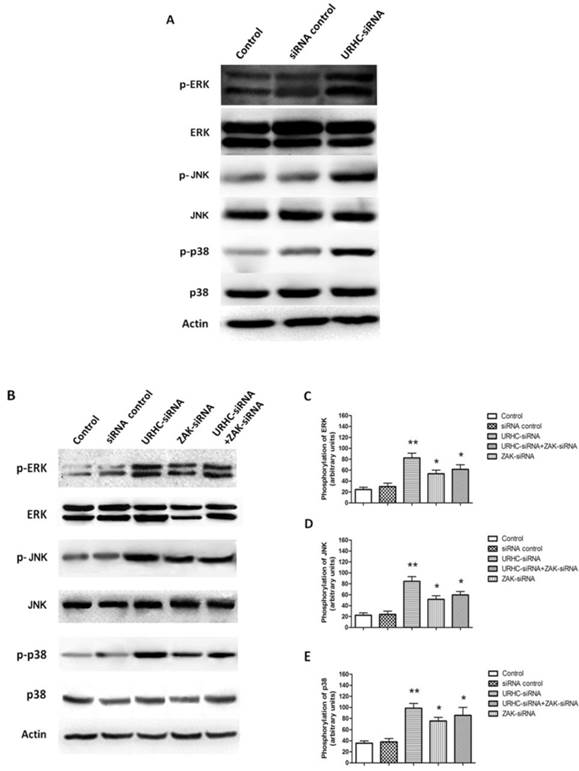
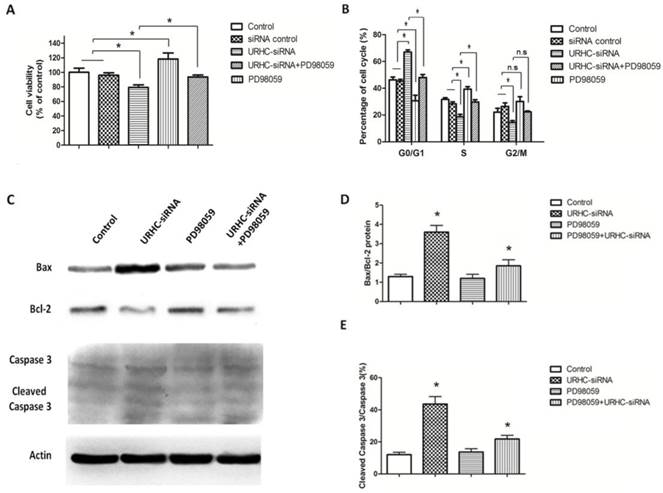
We next attempted to investigate the underlying mechanism by which URHC-ZAK regulated cell growth and apoptosis. Mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38, are mediators of cellular responses to extracellular signals [29]. In the present study, we further assessed whether the expression levels of ERK, JNK, and p38, which are crucial molecules involved in pathways associated with cancer pathogenesis, were altered when URHC was down-regulated. Our results indicated that ERK, JNK and p38 kinase phosphorylation was detected in URHC knockdown cells in contrast to the control cells, indicating that URHC may regulate cell growth and induce apoptosis through the inactivation of the MAPK signaling pathway (Fig. 6A). Next, we examined the levels of ERK, JNK, and p38 in the URHC-silenced plus ZAK-silenced cells. Interestingly, as shown in Fig. 6B, C, D and E, URHC-silencing plus ZAK-silencing attenuated the expression levels of phosphorylated ERK, phosphorylated JNK and p38, which were higher in URHC-silenced cells. Therefore, these data suggest that URHC may inhibit MAPK pathway activation through targeting ZAK. To further demonstrate that cell growth and apoptosis resulted from URHC-ZAK-mediated MAPK induction, we used a specific ERK inhibitor, PD98059 (10 μM), which is an agent that has been widely used for the functional study of this pathway [32, 33]. After treatment of SMMC7721 cells transfected with URHC-siRNA with PD98059, an MTT assay revealed that the inhibition of cell growth induced by URHC down-regulation could be abrogated by PD98059 treatment (Fig. 7A). In addition, we found that the changes of cell cycle distribution, especially S phase, caused by URHC knockdown could be rescued by PD98059 (Fig. 7B). In addition, an apoptosis assay revealed that PD98059 treatment was able to decrease the expression levels of Bax, which were increased in cells that had been transfected with URHC-siRNA (Fig. 7C). As shown in Fig. 7D and E, PD98059 treatment attenuated the increased ratios of Bax/Bcl-2 and cleaved caspase 3/caspase 3 that had been induced by URHC-siRNA. Taken together, our results indicate that URHC regulates cell proliferation and apoptosis by targeting ZAK through the downstream of ERK/MAPK pathway inactivation.
URHC regulates MAPK activity through ZAK. (A) The expression and phosphorylation of ERK, JNK, and p38 were examined using western blot analyses in SMMC7721 cells that had been transfected with URHC-siRNA or the siRNA control. (B) The expression and quantitative analyses of phosphorylation of ERK (C), JNK (D), and p38 (E) were examined at 48 h using western blot analyses in SMMC7721 cells that had been transfected with URHC-siRNA, ZAK-siRNA, URHC-siRNA plus ZAK-siRNA, or the siRNA control. β-actin was used as a reference. The results show the means ± SD from 3 separate experiments. *, P < 0.05; **, P < 0.001.
ERK was involved in the URHC-mediated cell proliferation and apoptosis. (A) The growth of SMMC7721 cells that had been treated with URHC-siRNA in the absence or presence of PD98059 (10 μM) or the control was measured using the MTT assay at 48 h. (B) SMMC7721 cells were collected after 48 h, and the cell cycle was analyzed with flow cytometer after staining with PI. (C) SMMC7721 cells were treated as described in (A), western blot analyses were performed to determine the expression levels of Bax, Bcl-2, caspase 3 and cleaved caspase 3, and the ratios of Bax/Bcl-2 (D) and cleaved caspase 3/caspase 3 (E) were determined. β-actin was used as a reference. The results show the means ± SD from at least 3 separate experiments. *, P < 0.05.
Discussion
Recent genome-wide studies have indicated that a greater proportion of the genomic sequence is transcribed into lncRNAs than into protein-coding RNAs. Functional studies have indicated that some lncRNAs, which may act as oncogenes or tumor suppressors, are involved in human cancer pathogenesis, including HCC [11]. Although dysregulation of lncRNAs associated with HCC has been identified, the functions and clinical significance of the majority of dysregulated lncRNAs in the progression and aggressiveness of HCC remain unknown [34-37].
In the current study, we screened for a novel lncRNA, URHC, and found that it was up-regulated in both hepatoma cell lines and clinical HCC tissue samples. Additionally, our data indicated that its high expression was correlated with tumor growth and poor survival in HCC patients, indicating that URHC may display oncogenic activity in HCC. Moreover, we identified the function of URHC in hepatoma cells by applying loss-of-function approaches. Decreased URHC expression inhibited hepatoma cell proliferation and promoted apoptosis in vitro. This is the first report to demonstrate the functional significance of URHC expression in human HCC, and our results indicate that URHC may function as a tumor oncogene.
Although a variety of lncRNAs have been shown to play important biological roles in many human cancers, the molecular mechanisms by which lncRNAs modulate tumor growth remain largely unknown. The regulatory mechanisms governing lncRNAs are diverse and complicated. LncRNAs may regulate protein-coding gene expression at both the post-transcriptional and transcriptional levels, while transcriptional regulation by lncRNAs may work either in cis or in trans, positively or negatively controlling protein-coding gene expression [38]. Notably, the lncRNA-mediated regulation of the expression of neighboring genes via cis-acting regulation has been widely studied. One example of a well-studied cis-acting lncRNA is HOTTIP, a long intergenic noncoding RNA that is transcribed from the 5' tip of the HOXA locus that coordinates the activation of several 5' HOXA genes. HOTTIP bound the adaptor protein WDR5 directly and targeted WDR5/MLL complexes across HOXA, thereby driving histone H3 lysine 4 trimethylation and gene transcription [39]. Zhang et al. also reported that inactivation of the Malat1 gene resulted in up-regulation of Malat1 neighboring genes, suggesting that Malat1 gene transcription may be important for regulating the expression of nearby genes in cis [40]. Martens et al. found that the expression of the lncRNA SRG1 was negatively correlated with the yeast SER3 gene, indicating that transcriptional interference regulating expression levels in cis may represent a widespread function for lncRNAs [41]. Du et al. reported that HULC, which contributes to HBx-related hepatocarcinogenesis, promoted hepatoma cell proliferation by down-regulating the p18 tumor suppressor gene, which is located near HULC [42]. Thus, we examined whether URHC exerted its effects via the mechanisms outlined above. We were interested in the neighboring gene located near URHC, and URHC was successfully predicted to target ZAK. ZAK, also known as ZAK-α or MLK-like MAP triple kinase-α (MLTK-α), belongs to the mixed lineage kinase family [31]. Yang et al. reported that ZAK may function as a tumor suppressor gene, inhibiting human lung cancer growth by regulating cell proliferation [31]. In addition, ZAK overexpression induced apoptosis in the Hep3B hepatoma cell line, indicating that ZAK expression is cytotoxic [43]. Accordingly, we speculated that ZAK was a potential target of URHC and that URHC regulated cell proliferation and apoptosis via ZAK. Interestingly, our results indicated that ZAK expression was down-regulated in HCC tissues as compared with the surrounding non-tumorous tissues, and URHC was inversely associated with ZAK expression in HCC tissues. Moreover, we observed that URHC down-regulated ZAK at the mRNA and protein levels. The negative correlation between ZAK and URHC expression may be explained by the transcriptional interference regulatory mechanism [38, 41]. The further functional experiments indicated that knockdown of URHC inhibited cell growth and sensitized the cells to apoptosis, which was inversely associated with ZAK-silencing function. In addition, ZAK-silencing partially rescued URHC knockdown-mediated anti-growth effects by reactivating cell proliferation and inhibiting apoptosis, further indicating that URHC is inversely associated with ZAK expression levels. Therefore, we concluded that URHC may promote tumor growth by suppressing the expression of its target gene, ZAK.
MAPK intracellular signaling mediates various biological events in cells, such as gene expression, cell proliferation, differentiation, apoptosis, migration and invasion [44-47]. To date, several reports have demonstrated that deregulated MAPK signaling pathways play critical roles in the pathogenesis of HCC [5, 8]. To elucidate the precise mechanism involved in URHC-induced cell proliferation and apoptosis, the effects of URHC on MAPK activation or inactivation were examined. The ERK pathway is the typical MAPK pathway and its rapid and transient activation is involved in cell proliferation, but some studies have indicated that its growth inhibitory effects are dependent on strong and sustained ERK activation [31]. In addition, many studies support the general view that activation of the ERK pathway delivered a survival signal that counteracted the pro-apoptotic effects associated with JNK and p38 activation [48]. Wang et al. reported that ERK activation played an active role in mediating drug-induced apoptosis and functioned upstream of caspase activation to initiate apoptotic signalling [48]. ERK-dependent Bcl-2 down-regulation and caspase activation was also shown to behave in a similar manner [49]. Therefore, we mainly focused on whether ERK was involved in the cell proliferation and apoptotic processes that were regulated by URHC and ZAK expression. Western blot analyses confirmed that URHC down-regulation significantly increased ERK phosphorylation compared with ZAK-silenced cells and URHC-silenced cells plus ZAK-silenced cells. The opposing regulation of URHC and ZAK suggests that URHC may inactivate the ERK pathway by inhibiting ZAK. To further verify these results, we modulated this signaling pathway using the ERK inhibitor PD98059 in SMMC7721 cells. The results of MTT and cell cycle assay revealed that PD98059 treatment rescued the inhibition of cell proliferation that was induced by URHC down-regulation. Furthermore, western blot analyses revealed that ERK activation was responsible for down-regulating URHC-induced cell apoptosis, the resulting Bax/Bcl-2 modulation and caspase-3 activation, as shown by the reductions in these events when the ERK inhibitor PD98059 was present. In conclusion, our findings suggest that URHC regulates cell proliferation and apoptosis via ERK/MAPK inactivation by targeting ZAK.
Although we confirmed that URHC may act as an oncogene to regulate cell proliferation and apoptosis by targeting ZAK, the multiple regulatory mechanisms, including DNA methylation and histone modification [36, 50], that may affect lncRNA expression remain unclear. Consequently, further studies are necessary to elucidate the upstream regulatory mechanisms controlling URHC expression using chromatin immunoprecipitation (ChIP) and/or RNA-binding protein immunoprecipitation (RIP) assays. In addition, construction a gain-of-function URHC model system in more sensitive cells would be optimal to comprehensively assess its influence on HCC cell biology in subsequent studies. Further work is warranted to evaluate the role of URHC and develop therapeutic strategies targeting URHC in vivo.
In summary, we found that URHC was up-regulated in HCC cells and tissues and served as a negative prognostic factor in HCC patients. More importantly, our findings not only indicated that URHC promoted cell proliferation and inhibited apoptosis, but revealed a negative link between URHC and ZAK. Our study further discovered that URHC regulated cell proliferation and apoptosis by down-regulating ZAK expression, and inactivation of the ERK/MAPK pathway was required for increase in HCC growth induced by URHC-ZAK regulation. We primarily confirmed the regulatory mechanism of lncRNA URHC in HCC progression. Therefore, our findings indicate that URHC may act as a tumor oncogene in HCC growth and has the potential to be a therapeutic target for HCC.
Supplementary Materials
Additional File 1Supplementary Tables
Supplementary Figures.
Abbreviations
LncRNA: long non-coding RNA; HCC: hepatocellular carcinoma; URHC: up-regulated in hepatocellular carcinoma; ZAK: sterile alpha motif and leucine zipper containing kinase AZK; KEGG: Kyoto Encyclopedia of Genes and Genomes; qRT-PCR: quantitative real-time polymerase chain reaction; siRNA: small interfering RNA; AJCC: American Joint Committee on Cancer; PVTT: portal vein tumor thrombi; MAPKs: mitogen-activated protein kinases; ERK: extracellular signal-regulated kinase.
Acknowledgements
This work was supported by the Major Program of the National Natural Science Foundation of China (81030010) and Shaanxi Science and Technology Innovation Project (2011KTCL03-15).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Thomas MB, Jaffe D, Choti MM, Belghiti J, Curley S, Fong Y. et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2010;28:3994-4005
3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118-27
4. Yang L, Xu J, Ou D, Wu W, Zeng Z. Hepatectomy for huge hepatocellular carcinoma: single institute's experience. World J Surg. 2013;37:2189-96
5. Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A. et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951-8
6. Greten FR. TAK1: Another mesh in the NF-κB-JNK controlled network causing hepatocellular carcinoma. J Hepatol. 2011;55:721-3
7. Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C. et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972-83
8. Wang SN, Lee KT, Tsai CJ, Chen YJ, Yeh YT. Phosphorylated p38 and JNK MAPK proteins in hepatocellular carcinoma. Eur J Clin Invest. 2012;42:1295-301
9. Wang C, Cigliano A, Delogu S, Armbruster J, Dombrowski F, Evert M. et al. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis: implications for the treatment of human liver cancer. Cell Cycle. 2013;12:1999-2010
10. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629-41
11. Mitra SA, Mitra AP, Triche TJ. A central role for long non-coding RNA in cancer. Front Genet. 2012;3:17
12. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354-61
13. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298-307
14. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155-9
15. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494-504
16. Tano K, Akimitsu N. Long non-coding RNAs in cancer progression. Front Genet. 2012;3:219
17. Li X, Wu Z, Fu X, Han W. Long noncoding RNAs: insights from biological features and functions to diseases. Med Res Rev. 2013;33:517-53
18. Mourtada-Maarabouni M, Hedge VL, Kirkham L, Farzaneh F, Williams GT. Growth arrest in human T-cells is controlled by the non-coding RNA growth-arrest-specific transcript 5 (GAS5). J Cell Sci. 2008;121:939-46
19. Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28:195-208
20. Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295-300
21. Xu D, Yang F, Yuan JH, Zhang L, Bi HS, Zhou CC. et al. Long noncoding RNAs associated with liver regeneration 1 accelerates hepatocyte proliferation during liver regeneration by activating Wnt/β-Catenin signaling. Hepatology. 2013;58:739-51
22. Xu ZY, Yu QM, Du YA, Yang LT, Dong RZ, Huang L. et al. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int J Biol Sci. 2013;9:587-97
23. Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38
24. Maruyama R, Suzuki H. Long noncoding RNA involvement in cancer. BMB Rep. 2012;45:604-11
25. Petrovics G, Zhang W, Makarem M, Street JP, Connelly R, Sun L. et al. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004;23:605-11
26. Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D. et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011;6:1984-92
27. Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y. et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366-83
28. Chehrehasa F, Meedeniya AC, Dwyer P, Abrahamsen G, Mackay-Sim A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J Neurosci Methods. 2009;177:122-30
29. Li S, Dong P, Wang J, Zhang J, Gu J, Wu X. et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via a ROS/JNK-dependent mitochondrial pathway. Cancer Lett. 2010;298:222-30
30. Tsang WP, Wong TW, Cheung AH, Co CN, Kwok TT. Induction of drug resistance and transformation in human cancer cells by the noncoding RNA CUDR. RNA. 2007;13:890-8
31. Yang JJ, Lee YJ, Hung HH, Tseng WP, Tu CC, Lee H. et al. ZAK inhibits human lung cancer cell growth via ERK and JNK activation in an AP-1-dependent manner. Cancer Sci. 2010;101:1374-81
32. Wu R, Duan L, Ye L, Wang H, Yang X, Zhang Y. et al. S100A9 promotes the proliferation and invasion of HepG2 hepatocellular carcinoma cells via the activation of the MAPK signaling pathway. Int J Oncol. 2013;42:1001-10
33. Li T, Lu YY, Zhao XD, Guo HQ, Liu CH, Li H. et al. MicroRNA-296-5p increases proliferation in gastric cancer through repression of Caudal-related homeobox 1. Oncogene. 2014;33:783-93
34. Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM. et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330-42
35. Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY, Zhang F. et al. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol. 2012;29:1810-6
36. Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S. et al. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750-6
37. Yang Z, Zhou L, Wu LM, Lai MC, Xie HY, Zhang F. et al. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol. 2011;18:1243-50
38. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59
39. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y. et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120-4
40. Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G. et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111-23
41. Martens JA, Laprade L, Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature. 2004;429:571-4
42. Du Y, Kong G, You X, Zhang S, Zhang T, Gao Y. et al. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J Biol Chem. 2012;287:26302-11
43. Liu TC, Huang CJ, Chu YC, Wei CC, Chou CC, Chou MY. et al. Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem Biophys Res Commun. 2000;274:811-6
44. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37-40
45. Wang X, Lu N, Niu B, Chen X, Xie J, Cheng N. Overexpression of Aurora-A enhances invasion and matrix metalloproteinase-2 expression in esophageal squamous cell carcinoma cells. Mol Cancer Res. 2012;10:588-96
46. Dong Y, Han Q, Zou Y, Deng Z, Lu X, Wang X. et al. Long-term exposure to imatinib reduced cancer stem cell ability through induction of cell differentiation via activation of MAPK signaling in glioblastoma cells. Mol Cell Biochem. 2012;370:89-102
47. Wynne S, Djakiew D. NSAID inhibition of prostate cancer cell migration is mediated by Nag-1 Induction via the p38 MAPK-p75(NTR) pathway. Mol Cancer Res. 2010;8:1656-64
48. Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435-43
49. Chang GC, Hsu SL, Tsai JR, Wu WJ, Chen CY, Sheu GT. Extracellular signal-regulated kinase activation and Bcl-2 downregulation mediate apoptosis after gemcitabine treatment partly via a p53-independent pathway. Eur J Pharmacol. 2004;502:169-83
50. Yang F, Huo XS, Yuan SX, Zhang L, Zhou WP, Wang F. et al. Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol Cell. 2013;49:1083-96
Author contact
![]() Corresponding authors: Ke-Feng Dou and Wen-Jie Song, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, No. 127 West Changle Road, Xi'an, 710032, Shaanxi, People's Republic of China. Phone: 86-29-84775255; Fax: 86-29-84775255; E-mail: gdwkgwxedu.cn (Ke-Feng Dou); wjsongedu.cn (Wen-Jie Song).
Corresponding authors: Ke-Feng Dou and Wen-Jie Song, Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, No. 127 West Changle Road, Xi'an, 710032, Shaanxi, People's Republic of China. Phone: 86-29-84775255; Fax: 86-29-84775255; E-mail: gdwkgwxedu.cn (Ke-Feng Dou); wjsongedu.cn (Wen-Jie Song).