Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Background
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(9):966-972. doi:10.7150/ijbs.9810 This issue Cite
Research Paper
Metabolic Profiles are Principally Different between Cancers of the Liver, Pancreas and Breast
Anuradha Budhu1 ![]() , Atsushi Terunuma2, Geng Zhang3*, S. Perwez Hussain3, Stefan Ambs2, Xin Wei Wang1
, Atsushi Terunuma2, Geng Zhang3*, S. Perwez Hussain3, Stefan Ambs2, Xin Wei Wang1
1. Liver Carcinogenesis Section,
2. Molecular Epidemiology Section,
3. Pancreatic Cancer Unit, Laboratory of Human Carcinogenesis, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA.
*Current address: Institute of Biophysics, Chinese Academy of Sciences, Beijing, China.
Received 2014-6-4; Accepted 2014-7-14; Published 2014-8-30
Abstract
Molecular profiling of primary tumors may facilitate the classification of patients with cancer into more homogenous biological groups to aid clinical management. Metabolomic profiling has been shown to be a powerful tool in characterizing the biological mechanisms underlying a disease but has not been evaluated for its ability to classify cancers by their tissue of origin. Thus, we assessed metabolomic profiling as a novel tool for multiclass cancer characterization. Global metabolic profiling was employed to identify metabolites in paired tumor and non-tumor liver (n=60), breast (n=130) and pancreatic (n=76) tissue specimens. Unsupervised principal component analysis showed that metabolites are principally unique to each tissue and cancer type. Such a difference can also be observed even among early stage cancers, suggesting a significant and unique alteration of global metabolic pathways associated with each cancer type. Our global high-throughput metabolomic profiling study shows that specific biochemical alterations distinguish liver, pancreatic and breast cancer and could be applied as cancer classification tools to differentiate tumors based on tissue of origin.
Keywords: Breast, Cancer, Liver, Metabolite, Pancreas.
Background
High-throughput genomic studies have been employed to study the alterations that occur in numerous cancer types, including liver, pancreas and breast. These three cancer types combined are estimated to result in approximately 100,000 deaths in the United States in 2013 and are significant contributors to cancer mortality worldwide (SEER: seer.cancer.gov). Globally, breast cancer remains the most frequent cancer among women, while liver and pancreatic cancer both display rising incidence rates and very poor survival (GLOBOCAN, 2008 estimates). A more comprehensive understanding of the initiating and progressive changes that occur in these cancers may allow us to identify better biomarkers and clinical targets to improve outcome.
While patients suffer from primary tumors, a large majority have poor outcome due to metastasis. Among epithelial cancers, 3-5% have a unique phenotype of early and aggressive metastatic dissemination with no identifiable primary tumor. These biologically distinct cases, known as cancer of unknown primary (CUP) are the 7-8th most frequent malignant tumors and a large proportion of such patients have unfavorable outcome with a median survival of 8 months [1]. Diagnosis of CUP patients remains a challenge and the optimal treatment regimens for these individuals is still being explored. Several studies have focused on cytotoxic agents which act broadly, such as gemcitabine, however the survival benefit among CUP patients is modest [2]. In addition, molecular diagnostic tools, mostly based on gene expression analysis, have been used to determine the molecular signatures of CUP in comparison to known alterations in particular types of primary tumors, however they lack precision [3, 4, 5]. Even after a full diagnostic workup, the primary origin of CUP remains unknown in a significant fraction of patients. In some patients where post-mortem examination is performed, primary tumors of the liver, pancreas and breast are among the cancer types most often found [6]. Overall, it remains a clinical challenge to identify which cancer patients are most likely to have a poor outcome under current standard therapy. Therefore, a better understanding of the molecular mechanism of these diseases and discovery of novel therapeutic targets are desperately needed to improve patient outcome.
To more accurately define cancer types, including liver, pancreas and breast, a comprehensive characterization of their genomic status must be made. We and others have identified significantly altered molecular markers, including genes and microRNAs, that are diagnostic and/or prognostic for these cancer types and may serve as candidates for novel therapeutic approaches [7, 8, 9, 10, 11, 12, 13]. Moreover, gene expression and microRNA levels of genomic information have been explored as multiclass molecular cancer classification tools [14, 15]. However, their robust validation and clinical implementation as molecular cancer diagnostic tools to accurately establish tissue of origin remains to be determined.
Alterations of metabolites, the biochemical end-products of biological signaling, have also recently been explored as cancer biomarkers. More recent technological advances now allow for high-throughput global analysis of the metabolic changes that occur in cancer [16, 17]. This method has been demonstrated to be highly sensitive to discriminate tumors from non-tumor tissues. Previously, we explored and identified key metabolic alterations that occur in aggressive liver cancer, pancreatic ductal adenocarcinoma and breast cancer [17, 18, 19]. Here, we evaluated whether metabolomic profiling can accurately define cancer types by applying this technique as a multiclass cancer discrimination tool. Using a proof of principle approach, we analyzed global metabolic alterations in tumors and adjacent normal tissues from liver, pancreatic, and breast cancer patients to search for metabolites that are unique to these three diseases. Using this approach, we show that metabolomic profiling is a promising multiclass cancer discrimination tool that can accurately and robustly distinguish cancers by tissue of origin.
Materials and Methods
Study Cohorts and Patient Characteristics
The liver cancer cohort consists of 60 paired fresh-frozen tumor and nontumor hepatic tissues from 30 patients at the Liver Cancer Institute (LCI) and Zhongshan Hospital (Fudan University, Shanghai, China) as previously described [20]. The pancreas cancer cohort consists of 76 paired fresh-frozen primary pancreatic tumor and adjacent nontumor tissues from 38 patients with primary ductal adenocarcinoma at the University of Medicine, Göttingen, Germany, and from the University of Maryland Medical Center at Baltimore, Maryland through the NCI-UMD resource contract as previously described. The breast cancer cohort consists of 130 fresh-frozen paired tumor and adjacent nontumor tissues from 65 patients recruited at hospitals in Baltimore, Maryland, between 1993 and 2003, as previously described [21]. Clinical and pathological information was obtained from medical records and pathology reports and all patients signed a consent form. Tumor histopathology was classified according to the World Health Organization Classification of Tumor system. The studies were approved by the Institutional Review Boards of the participating institutes.
Tissue Samples and Metabolic Profiling
Metabolomic profiling of paired tumor and nontumor tissues was carried out at Metabolon Inc. Liquid chromatography/mass spectrometry in positive and negative modes (LC+/LC-) and gas chromatography/mass spectrometry (GC/MS) were employed. Compound identity was then resolved using Metabolon's reference library to define known or unknown metabolites. A total of 469 metabolites were yielded in the liver specimens, 452 in the pancreatic specimens and 536 in the breast specimens. The total number of metabolites measured includes metabolites with known identities as well as unknown metabolites. To provide the most robust dataset, we applied a seventy-five percent filter to the 469, 452 and 536 metabolites yielded in liver, pancreas and breast cancer, respectively, and only metabolites with known identifiers were used for further analysis. This resulted in 306, 443 and 355 metabolites with abundance values above the detection limit in the liver, pancreas or breast in at least 75% of all of the tissue samples. Among these metabolites, 207 were commonly present among the three cancers.
Statistical Analysis
Analyses of differentially expressed metabolites were performed using BRB-ArrayTools V4.2.1 developed by Dr. Richard Simon and the BRB-ArrayTools Development Team using the Class Comparisons tool applying a paired t-test and comparison of the geometric mean between classes. Principal component analysis (PCA) was performed using Partek V6.5. Hierarchical clustering was performed using Genesis V.1.7.6.
Results
Determining Metabolic Abundance Patterns Among Cancers
The characteristics of the liver, pancreas and breast cancer patients assessed in this study are shown in Additional File 1: Supplemental Table 1. The global metabolic alterations occurring in paired tumor and nontumor tissue specimens of these cancer types were identified using a combination of gas and liquid chromatography and mass spectrophotometry analysis. Of the approximately 3000 metabolites that can be identified with these platforms, 306, 443 and 355 metabolites were at adequate abundance to be detected in the liver, pancreas and breast specimens, respectively. A comparison of these compounds resulted in a composite list of 563 metabolites found among these three cancer types. To assess the overall metabolic patterns among these cancers, we performed unsupervised principal component analysis (PCA) of these 563 metabolites among all of the tumor and nontumor specimens available in this study. In doing so, we found a striking separation by tissue of origin and between cancerous and noncancerous tissue (Figure 1A).
Metabolites patterns can distinguish cancers. (a) A principal component analysis plot is shown comparing the metabolite status of tumor (darker shades) and nontumor (lighter shades) of liver, pancreas and breast tissues among 563 measurable metabolites or (b) 207 commonly yielded metabolites or (c) early stage patients.
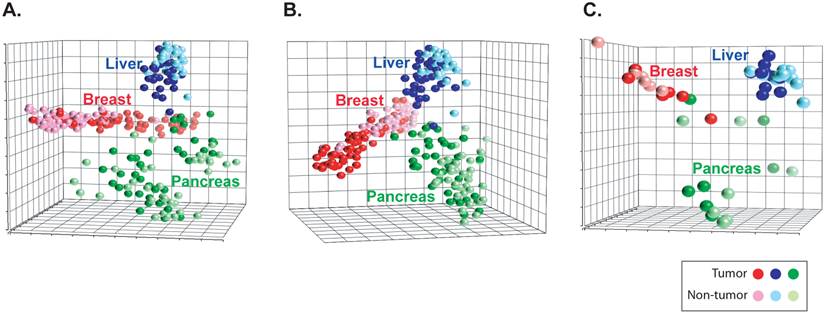
The capacity to distinguish cancer types based on tissue metabolite abundance could be due to the presence of unique metabolite abundance in each cancer type or could be the result of tissue-specific metabolism. Thus, we explored the abundance patterns of the metabolites across the three cancers analyzing cancerous and noncancerous samples. Of the 563 metabolites, 207 were commonly present in the three cancer types and belonged mainly to lipid and amino acid classes. We next determined whether the patterns of abundance among these 207 metabolites were similar or different by tissue type. To do so, we first performed unsupervised PCA among the tumors. The analysis showed that the patterns of abundance for these 207 metabolites largely differed between liver, pancreas and breast tumors (Additional File 1: Supplemental Figure 1A). An analysis of the adjacent nontumor specimens showed a similar separation between cancers (Supplemental Figure 1B). To determine whether these differences were driven by the origin of the tissue (e.g. liver vs breast cancer) or by the specimen type (e.g. tumor versus nontumor), we explored the pattern of these 207 metabolites among the paired tumor and nontumor specimens of all three cancer types using unsupervised PCA. Notably, we found that although metabolite abundance in tumor vs nontumor specimens were evident among liver, pancreas and breast, each cancer type largely segregated from one another (Figure 1B). Thus, although these 207 metabolites are commonly present among each of the three cancers, the patterns of their abundances not only differ between tumor and nontumor tissue, but are significantly different between cancer types.
Since our analysis revealed that certain metabolic alterations were unique to each cancer type, we next determined whether these changes also occur at early stages of cancer development. In this vein, we restricted our analysis to early stage patients diagnosed as TNM stage I or II (single or small tumors with no evidence of metastasis). PCA analysis, restricted to early-stage individuals, showed that the patterns of abundance of the 207 metabolites commonly found in liver, pancreas and breast largely segregated each cancer type and were differentially altered among tumor or nontumor specimens (Figure 1C). Therefore, the abundance patterns of the 207 common metabolites in liver, pancreas and breast largely differ, even at early stages of cancer development.
Identification of Unique Cancer Metabolites
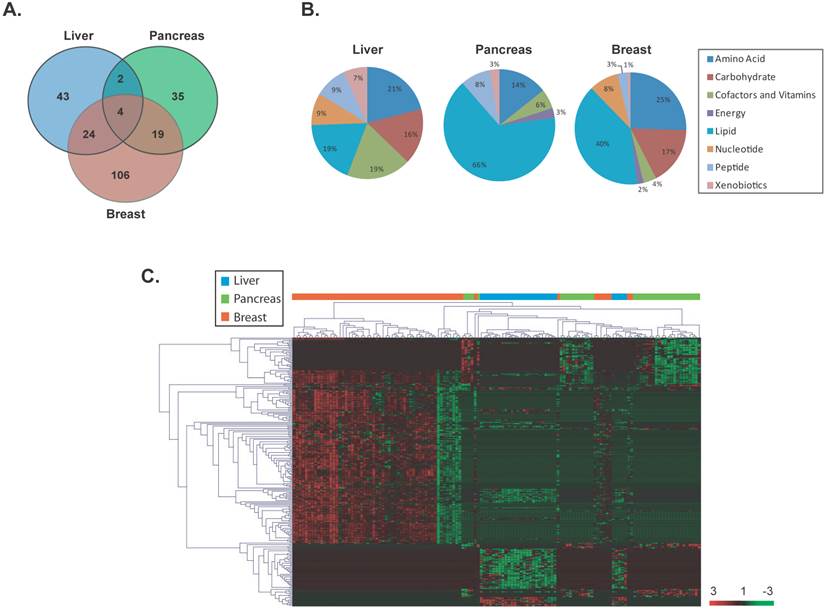
To identify significant differentially altered metabolites that are unique to each cancer type, a supervised class comparison analysis of tumor versus paired nontumor tissue was performed (p<0.01, 10-fold cross-validation. False Discovery Rate (FDR) <0.10) for liver, pancreas or breast specimens (Additional File 1: Supplemental Tables 2, 3 and 4). Among the liver specimens, 43 metabolites were significantly and differentially altered between liver tumors versus paired nontumor liver tissue (Figure 2A). In a similar analysis, 35 metabolites and 106 metabolites were significantly and uniquely altered in pancreatic or breast tumors versus their respective paired nontumor specimen (Figure 2A). These tumor-specific metabolites could effectively discriminate each of the three cancer types (Additional File 1: Supplemental Figure 2). A comparison of the significant and differentially altered metabolites among liver, pancreas and breast tumors versus paired nontumors showed that only 4 metabolites (butyrylcarnitine, hexanocarnitine, glycerol and linolenate) were in common among these cancers and each were involved in lipid metabolism (Figure 2A). Two of these metabolites belonged to the carnitine pathway and were significantly upregulated in tumor versus nontumor specimens in all three cancer types (fold change range: 2.6-13.7). However, the remaining two metabolites were upregulated only in breast tumors. An assessment of the tumor-specific metabolites that were unique to each of the three cancer types revealed that a variety of metabolic pathways are distinctly altered in these diseases with the most evident differences occurring in lipid and amino acid pathways (Figure 2B). Interestingly, metabolites in the carbohydrate category were not significantly altered in pancreatic cancer. Hierarchical clustering of the tissue-of-origin-specific metabolites revealed unique patterns of biochemical abundance in the liver, pancreatic or breast specimens (Figure 2C).
Unique metabolites are differentially altered in liver, pancreas and breast cancers. (a) A VENN diagram is presented for metabolites differentially altered in tumor versus nontumor tissues of the liver, pancreas and breast. (b) Pie charts represent the distribution of cancer-type-specific metabolites according to biochemical sub-pathways. (c) A heat map representing hierarchical clustering of differentially abundant (tumor versus nontumor) metabolites is shown. Metabolites were ordered by euclidian distance and average linkage according to the ratios of abundance in each tumor specimen when compared to its paired nontumor specimen. Each row represents an individual metabolite and each column represents an individual patient. Pseudocolors indicate differential abundance (red, green, black representing metabolite levels above, below or equal to the mean on a log2 scale, respectively).
A further comparison of the patterns of metabolite abundance show that even for metabolites that are commonly measurable in two or more cancer types, the abundance patterns are different between cancer types. For example, certain metabolites present in liver tissue are lowly abundant in tumor specimens whereas their abundance is high in breast tumor specimens (Additional File 1: Supplemental Table 5). To further specify metabolites which could differentiate cancer types, we compared each set of unique tumor-specific metabolites in a given cancer type versus the total metabolic yield for that cancer. This comparison resulted in 9, 16 and 9 metabolites which were uniquely altered and solely present in liver, pancreatic or breast tumors (Additional File 1: Supplemental Figure 3 and Supplemental Table 6). Thus, specific metabolic patterns are evident and can distinguish liver, pancreatic and breast cancer.
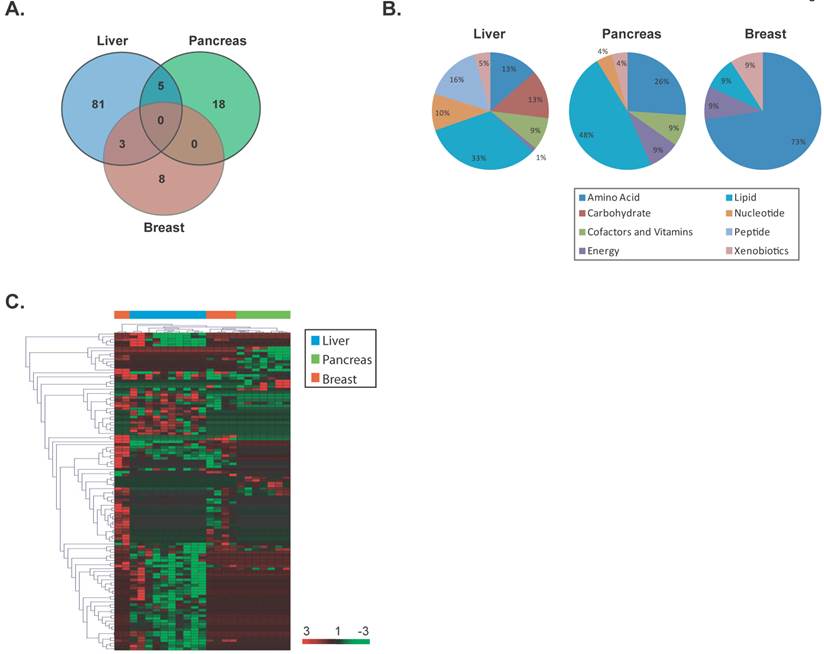
To determine whether specific metabolite changes also occur at early stages of cancer development, we restricted our analysis to early stage individuals as defined above and identified significant differentially altered metabolites in each cancer type using a supervised class comparison analysis of tumor versus paired nontumor specimens (p<0.05, 10-fold cross-validation, FDR <0.20) (Additional File 1: Supplemental Tables 7, 8 and 9). Among the liver specimens, 81 metabolites were significantly and uniquely altered between liver tumors versus paired nontumors (Figure 3A). In a similar analysis, 18 and 8 metabolites were significantly and uniquely altered in pancreatic and breast tumors, respectively. A comparison of the significant and differentially altered metabolites among early stage liver, pancreas and breast tumors versus paired nontumor specimens showed that no metabolites were in common among these cancer types. While several metabolic pathways were found to be altered in these diseases, larger alterations in lipid pathways occur in early stage liver and pancreatic cancers, while amino acid pathways are mainly perturbed in early stage breast cancer (Figure 3B). Hierarchical clustering of the tissue-of-origin-specific early stage metabolites demonstrated that unique patterns of biochemicals are found in either liver, pancreatic or breast specimens at early disease stage (Figure 3C). In a similar fashion to the analysis performed in all cancers, when a further comparison of the patterns of metabolite abundance in early stage cancers was performed, for the majority of metabolites commonly measurable in two or more cancer types, the abundance patterns are different between cancer types (Additional File 1: Supplemental Table 10). Thus, palpable tissue specific metabolic changes are present at early stages of cancer that can distinguish tissue of origin.
Unique metabolites are differentially altered in early stage liver, pancreas and breast cancers. (a) A VENN diagram is presented for metabolites differentially altered in tumor versus nontumor tissues of early stage liver, pancreas and breast. (b) Pie charts represent the distribution of cancer-type-specific metabolites according to biochemical sub-pathways. (c) A heat map representing hierarchical clustering of differentially abundant (tumor versus nontumor) metabolites among early stage patients is shown. Metabolites were ordered by euclidian distance and average linkage according to the ratios of abundance in each tumor specimen when compared to its paired nontumor specimen. Each row represents an individual metabolite and each column represents an individual patient. Pseudocolors indicate differential abundance (red, green, black representing metabolite levels above, below or equal to the mean on a log2 scale, respectively).
Discussion
In recent years, a large scientific effort, with the goal of improving patient outcomes through precision cancer medicine, has been placed on identifying molecular markers to aid in better diagnosis, prognosis and treatment prediction of cancer patients. Several levels of genomic information have been explored for this purpose, including gene expression, microRNA expression and epigenetic alterations. We and others have recently begun to use information gathered at the biochemical level of signaling by assessing the global metabolic alterations that occur in cancer. This has allowed us to discern the end result of composite complex genomic changes that regulate cancer initiation and outcome which more accurately reflects disease biology than a gene expression signature. In this study, we have used a global approach to assess the significant alterations of metabolites across multiple cancer types. By comparing and contrasting the global metabolic profiles of paired tumor and nontumor specimens, we have identified both common and unique biochemical biomarkers of liver, pancreas and breast cancer.
Metabolites which are the end products of transcriptome pathways could be more relevant markers of biological phenotypes than gene expression and could be superior in identifying the origin of a tissue. The findings from our current study indicate that certain metabolites or metabolic pathways are distinct in particular cancer types and may be useful to differentiate primary tumors. It would be of interest in the future to compare these results with metabolic profiles of other cancers to determine their similarities or differences. In this regard, specific metabolites might prove useful as diagnostic biomarkers or as targets for therapeutic intervention. In line with other genomics-based studies, these results could be expanded in the future to explore the utility of metabolites as cancer-specific prognostic biomarkers or integrated with other levels of genomic information to more specifically define the molecular pathways specific to each cancer. Thus, there seems to be great potential of metabolic profiling, on its own or perhaps in combination with genomic and pathological information, to aid in the classification of tumor classes and clinical management.
Our understanding of the specific aberrant metabolic patterns that occur in cancers may allow us to more accurately define the molecular portrait of each cancer type. In fact, tissue-specific gene expression-based profiles have been developed and applied to CUP specimens in order to aid in the identification of tumor origin for which other robust classification methods do not currently exist. However, non-specificity and inaccuracy have made it difficult to identify CUP subsets based on their gene expression patterns with a molecular profile that is consistent with a certain primary tumor. Future studies should include CUP patients for whom current diagnosis and prognosis remains a significant clinical challenge. Future studies may also explore the metabolic alterations that occur in blood products from patients with known primary cancers with that of CUP patients in order to achieve less invasive means of cancer classification. However, such investigations will require extensive validation and prospective studies.
In conclusion, we have identified subsets of metabolites that show distinct alterations in liver, pancreas and breast cancer. These findings provide a conceptual framework to validate cancer-type specific metabolite levels in future prospective studies. Thus, specific metabolite abundance patterns may allow for the classification of tissue of origin for specimens such as CUP. Such distinction of particular patient groups with common signaling events may allow for patient stratification and the application of specific treatment regimens.
Supplementary Material
Additional File 1Supplementary Figures and Tables.
Abbreviations
CUP: cancer of unknown primary
PCA: principal component analysis
Acknowledgements
We thank Karen Yarrick for bibliographic assistance. This work was supported by the Intramural Research Program of the Center for Cancer Research, the US National Cancer Institute and NCI Director's Innovation Awards to Stefan Ambs and Anuradha Budhu.
Conflict of Interests
The authors declare no financial or non-financial competing interests.
References
1. Levi F, Te VC, Erler G. et al. Epidemiology of unknown primary tumours. Eur J Cancer. 2002;38:1810-1812
2. Amela EY, Lauridant-Philippin G, Cousin S. et al. Management of "unfavourable" carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84:213-223
3. Varadhachary GR, Talantov D, Raber MN. et al. Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol. 2008;26:4442-4448
4. Tothill RW, Li J, Mileshkin L. et al. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol. 2013.
5. Horlings HM, van Laar RK, Kerst JM. et al. Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol. 2008;26:4435-4441
6. Mayordomo JI, Guerra JM, Guijarro C. et al. Neoplasms of unknown primary site: a clinicopathological study of autopsied patients. Tumori. 1993;79:321-324
7. Budhu A, Jia HL, Forgues M. et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897-907
8. Budhu A, Forgues M, Ye QH. et al. Prediction of venous metastases, recurrence and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99-111
9. Zhang G, Schetter A, He P. et al. DPEP1 Inhibits Tumor Cell Invasiveness, Enhances Chemosensitivity and Predicts Clinical Outcome in Pancreatic Ductal Adenocarcinoma. PLoS One. 2012;7:e31507
10. Lee EJ, Gusev Y, Jiang J. et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046-1054
11. Van de Vijver MJ, He YD, Van't Veer LJ. et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999-2009
12. Boersma BJ, Reimers M, Yi M. et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer. 2008;122:1324-1332
13. Hudson RS, Yi M, Esposito D. et al. MicroRNA-106b-25 cluster expression is associated with early disease recurrence and targets caspase-7 and focal adhesion in human prostate cancer. Oncogene. 2012.
14. Ramaswamy S, Tamayo P, Rifkin R. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A. 2001;98:15149-15154
15. Volinia S, Calin GA, Liu CG. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257-2261
16. Sreekumar A, Poisson LM, Rajendiran TM. et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910-914
17. Budhu A, Roessler S, Zhao X. et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066-1075
18. Zhang G, He P, Tan H. et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983-4993
19. Terunuma A, Putluri N, Mishra P. et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398-412
20. Roessler S, Jia HL, Budhu A. et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Research. 2010;70:10202-10212
21. Boersma BJ, Howe TM, Goodman JE. et al. Association of breast cancer outcome with status of p53 and MDM2 SNP309. J Natl Cancer Inst. 2006;98:911-919
Author contact
![]() Corresponding author: A. Budhu, Laboratory of Human Carcinogenesis, National Cancer Institute, NIH, 37 Convent Drive, Building 37, Room 3050A, Bethesda, MD 20892, USA; Email: budhuanih.gov.
Corresponding author: A. Budhu, Laboratory of Human Carcinogenesis, National Cancer Institute, NIH, 37 Convent Drive, Building 37, Room 3050A, Bethesda, MD 20892, USA; Email: budhuanih.gov.