Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pathophysiology of HD and the...
Clinical Trials of Lithium and...
Conclusions and Future Directions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(9):1024-1038. doi:10.7150/ijbs.9898 This issue Cite
Review
Preclinical and Clinical Investigations of Mood Stabilizers for Huntington's Disease: What Have We Learned?
Lisa Scheuing, Chi-Tso Chiu, Hsiao-Mei Liao, Gabriel R. Linares, De-Maw Chuang ![]()
Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, 10 Center Drive MSC 1363, Bethesda, MD 20892-1363, USA.
Received 2014-6-13; Accepted 2014-7-8; Published 2014-9-10
Abstract
Huntington's disease (HD) is a lethal, autosomal dominant neurodegenerative disorder caused by CAG repeat expansions at exon 1 of the huntingtin (Htt) gene, which encodes for a mutant huntingtin protein (mHtt). Prominent symptoms of HD include motor dysfunction, characterized by chorea; psychiatric disturbances such as mood and personality changes; and cognitive decline that may lead to dementia. Pathologically multiple complex processes and pathways are involved in the development of HD, including selective loss of neurons in the striatum and cortex, dysregulation of cellular autophagy, mitochondrial dysfunction, decreased neurotrophic and growth factor levels, and aberrant regulation of gene expression and epigenetic patterns. No cure for HD presently exists, nor are there drugs that can halt the progression of this devastating disease. Therefore, the need to discover neuroprotective modalities to combat HD is critical. In basic and preclinical studies using cellular and animal HD models, the mood stabilizers lithium and valproic acid (VPA) have shown multiple beneficial effects, including behavioral and motor improvement, enhanced neuroprotection, and lifespan extension. Recent studies in transgenic HD mice support the notion that combined lithium/VPA treatment is more effective than treatment with either drug alone. In humans, several clinical studies of HD patients found that lithium treatment improved mood, and that VPA treatment both stabilized mood and moderately reduced chorea. In contrast, other studies observed that the hallmark features of HD were unaffected by treatment with either lithium or VPA. The current review discusses preclinical and clinical investigations of the beneficial effects of lithium and VPA on HD pathophysiology.
Keywords: Huntington's disease, Lithium, Glycogen Synthase Kinase-3 Inhibitor, Valproic Acid, Histone Deacetylase Inhibitor, Therapeutic Potential.
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by expanded CAG repeats at exon 1 of the huntingtin (Htt) gene, and results in the aggregate formation of mutant huntingtin protein (mHtt) [1]. HD occurs most often in North America, Europe, and Australia, with a prevalence of roughly six cases per 100,000 [2]. Disease onset typically occurs between ages 30-45, and early age of onset correlates strongly with severity of the polyglutamine expansion [3], where more than 35 repeats is considered disease causing. In contrast, the rate of disease progression is more weakly correlated with CAG repeat number [4]. The major symptoms of HD patients include psychiatric disturbances such as significant changes in personality or mood; cognitive decline, which may gradually become dementia; and motor dysfunction, characterized by uncontrollable chorea movements [5]. Death occurs approximately 15-20 years after initial onset of symptoms. The hallmark pathological feature of HD—identified postmortem—is death/atrophy of medium-sized spiny neurons within the striatum and, to a lesser extent, loss of neurons in the cortex [6].
No cure presently exists for HD. In 2008, the FDA approved the drug tetrabenazine (Xenazine®) [7], a vesicular monoamine transporter inhibitor, to treat the involuntary physical movements observed in HD. While tetrabenazine can reduce chorea [8, 9], it does not halt disease progression, nor does it treat the psychiatric symptoms associated with HD which can be equally debilitating. In addition the Huntington's Disease Group [9], noted that the clinical efficacy of tetrabenazine is approximately 70%, indicating that a subset of HD patients do not respond to the drug. Tetrabenazine can also have serious neurological side effects, including anxiety, neuroleptic malignant syndrome, depression, and suicidal thoughts or suicidal actions [9]. Given that tetrabenazine elevates the risk for depression, it might be beneficial to treat HD patients with mood stabilizers to prevent or reduce this depressive component.
Accumulating evidence indicates that two prominent mood stabilizing drugs—lithium and valproic acid (VPA), both of which have a long history of safe use for the treatment of bipolar disorder—are particularly promising for treating neurological/neurodegenerative diseases via their diverse mechanisms of action [10]. Lithium, a monovalent cation used to treat mania, inhibits glycogen synthase kinase-3 (GSK-3) and inositol monophosphatase (IMPase) [11, 12]. The anticonvulsant VPA is a fatty acid derivative and a class I and class IIa histone deacetylase (HDAC) inhibitor [13, 14], which enables it to regulate the expression of diverse genes. Via these primary targets, lithium and VPA exert neuroprotective effects [10]. In various neurodegenerative paradigms lithium or VPA were shown to upregulate the expression of brain derived neurotrophic factor (BDNF) and pro-survival molecules (such as B-cell lymphoma-2 (Bcl-2), enhance the expression of protein chaperone heat shock protein 70 (HSP-70), modulate inflammatory proteins, and downregulate pro-apoptotic factors (such as Bax and p53). Lithium and VPA treatment has also reduced neuronal death from glutamate excitotoxicity, enhanced clearance of protein aggregate accumulation, normalized transcriptional dysregulation, preserved blood-brain barrier integrity, and improved memory and motor functional outcome in various neurodegenerative diseases. In sum, lithium and VPA exert multiple therapeutic effects across an array of central nervous system disorders, including promising results in preclinical models of HD. This review will explore current knowledge regarding the mechanisms underlying the pathophysiology of HD, and discuss cellular, animal, and clinical studies investigating the neuroprotective effects of lithium and VPA on HD.
Pathophysiology of HD and the Therapeutic Mechanisms of Mood Stabilizers
Although the genetic mutation responsible for HD was discovered in 1993, the exact mechanisms underlying its symptom pathophysiology are not entirely understood due to the complex interactions of the multiple pathways involved. Figure 1 illustrates the major disrupted pathways discussed in this review that contribute to HD pathophysiology. HD animal models are useful for examining disease pathophysiology because they allow investigators to evaluate the efficacy of potential therapeutic agents and study the underlying molecular mechanisms of dysfunctional signaling pathways. This section summarizes the known major pathophysiological mechanisms of HD as well as relevant preclinical studies that have used lithium and VPA to ameliorate HD symptoms.
The major pathophysiological pathways in Huntington's disease (HD). Mutant huntingtin (mHtt) protein disrupts many normal physiological processes and leads to unbalanced homeostasis of apoptotic molecules, deficits in autophagy, axonal transport impairment, transcriptional dysregulation, reduced cellular neurotrophic support, mitochondrial abnormalities, and glutamate excitotoxicity. mHtt disturbs the balance between pro-apoptotic (such as Bax and p53) and cell survival (such as Bcl-2 and Bcl-xl) molecules. Transcriptional regulation is disrupted in HD; as described in the text, mHtt allows REST translocation to the nucleus resulting in repression of genes including BDNF. As a result of decreased BDNF axonal transport and repression of gene transcription by REST, neurotrophic support of cells is diminished in HD. Impaired axonal transport of autophagosomes also increases autophagy deficits observed in HD. Mitochondrial abnormalities in HD include decreased ATP production and PGC-1a expression, as well as increased cytochrome c release which leads to cell apoptosis. Glutamate excitotoxicity, caused by hyperactivation of excitatory amino acid receptors that increase cell ion permeability and lead to intracellular calcium overload and ultimately cell death, is strongly implicated in HD. The pointed arrows indicate that mHtt increases the described physiological pathway; arrows with blocked ends indicate prevention of a physiological event.
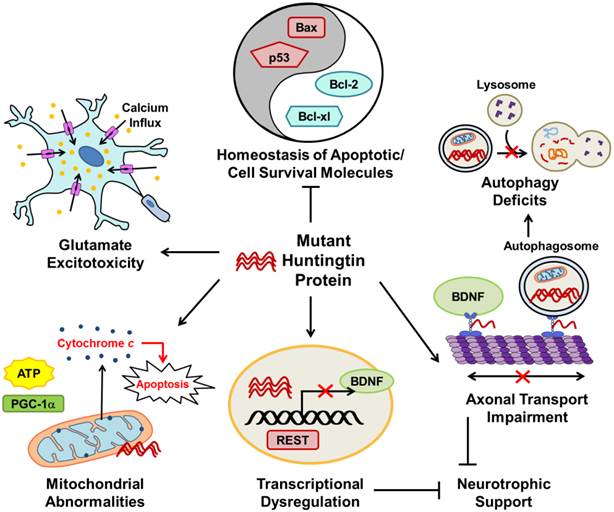
Excitotoxicity: Increased Cell Death in the Striatum and Cortex
One of the first identified pathological features of HD was the progressive neurodegeneration of GABAergic medium-sized spiny neurons in the striatum [6], that project to the substantia nigra and globus pallidus. In HD, striatal neuron death is significantly more extensive than neuronal atrophy within the cerebral cortex, subcortical white matter, certain hypothalamic nuclei, thalamus, and other brain areas [15]. Neurodegeneration of the striatum and cortex coincides with motor dysfunction and psychiatric symptoms, respectively, but the extent to which the symptoms of HD arise from the timing of neuronal death or dysfunction is unknown. Why medium-sized spiny striatal neurons are specifically targeted in HD, even though normal Htt and mHtt proteins are ubiquitously expressed throughout the brain and peripheral tissues in both humans and animals with HD, also remains unknown [16, 17].
Excitotoxicity is a mechanism of cell death caused by hyperactivation of excitatory amino acid receptors that increases cell ion permeability and leads to intracellular calcium overload [18]. Excitotoxicity was one of the initial hypotheses proposed to underlie specific neurodegeneration in HD, and was supported by both preclinical [19-21] and clinical [22] evidence. The development of HD excitotoxic animal models was based on the rationale that intrastriatal injection with quinolinic acid (QA), or kainic acid (both are glutamate receptor agonists), or with 3-nitropropionic acid (3-NP, an irreversible succinate dehydrogenase inhibitor) resulted in striatal neurodegeneration patterns similar to those observed in HD patients [23-25]. A more recent study suggested that dysregulation of the striatal GluN3A subunit of the N-methyl-D-aspartate (NMDA) receptors might contribute to synaptic and behavioral impairments in a transgenic mouse model of HD [26]. The excitotoxic features of HD suggest that lithium and VPA—both of which have neuroprotective effects against apoptosis induced by excessive glutamate—are plausible treatments for this disease.
Partly via the inhibition of NMDA receptor-mediated calcium influx, chronic lithium treatment robustly reduced glutamate excitotoxicity in cultured CNS neurons, including rat cerebellar granule cells (CGCs), cerebral cortical, and hippocampal neurons [27]. Later studies found that the ability of lithium to inhibit NMDA receptor-mediated calcium influx resulted from the attenuation of constitutive phosphorylation at Tyr1472 of the NR2B subunit of the NMDA receptor, which is catalyzed by the Src tyrosine kinase Fyn [28, 29]. Lithium's neuroprotective effects against glutamate excitotoxicity also involve the blockade of apoptotic components. In a cellular model of glutamate-induced, NMDA receptor-mediated excitotoxicity, primary CGCs pretreated for seven days with lithium (0.5 mM to 5.0 mM) decreased protein levels of the pro-apoptotic molecules p53 and Bax, inhibited the mitochondrial release of pro-apoptotic protein cytochrome c, and increased protein levels of the pro-survival molecule B-cell lymphoma/leukemia-2 (Bcl-2) compared to non-pretreated controls [30]. Other glutamate-induced apoptotic events were similarly prevented by seven days of treatment with therapeutic concentrations of lithium (0.5-2 mM) in cultured CGCs; these included activation of both c-Jun and N-terminal kinase (JNK) and p38 mitogen-activated protein (MAP) kinase, as well as increased activator protein 1 (AP-1) binding [31].
GSK-3 also plays a role in apoptotic cell death induced by glutamate excitotoxicity [32]. In rat primary cerebral cortical neurons, glutamate-induced excitotoxicity was blocked by RNA interference-induced depletion of GSK-3 isoforms, transfection with isoform-specific dominant-negative mutants of GSK-3, or treatment with other non-selective pharmacological GSK-3 inhibitors [33, 34]. These findings strongly suggest that both isoforms of GSK-3 are initial targets of lithium-induced neuroprotection.
Building on this work, another study found that the in vitro results were supported in a rat QA model of HD. Rats were pretreated for 16 days with lithium at therapeutically relevant levels (1.5 mEq/kg to 3.0 mEq/kg, s.c.). The group pretreated with lithium showed increased striatal cell survival and increased protein levels of Bcl-2, compared to rats treated with only QA [35]. A subsequent study using the same model found that only 24 hours of lithium pretreatment increased striatal Bcl-2 protein levels, decreased lesion size, and increased proliferation of striatal cells near the QA injection site [36]. The authors identified a small population of the proliferating cells as neurons or astrocytes, but were unable to identify the larger proliferating population.
Like QA, 3-NP striatal injection causes striatal lesions and produces excitotoxicity [37], but it also increases oxidative stress because it is an irreversible mitochondrial inhibitor. In a rat 3-NP model, three weeks pretreatment with therapeutic levels of lithium (blood concentration of 0.9 mEq/l) increased lifespan, reduced lesion area, and prevented activation of the calpain/cdk5/p-MEF2 pathway (which is involved in neuronal cell death) compared with untreated 3-NP controls [38]. Taken together, the findings provide compelling evidence that lithium acts as a neuroprotective, anti-apoptotic, and cell-proliferating agent in excitotoxic models of HD.
VPA was also found to be neuroprotective in various excitotoxic cellular and animal models. In rat primary cortical neurons, for instance, pretreatment with VPA significantly increased cell viability and induced the expression of heat shock protein 70 (HSP70), a chaperone protein that helps to regulate protein folding and refolding of partially denatured proteins [39], via HDAC inhibition [40]. Other studies found that HSP70 exerts a wide variety of neuroprotective effects against apoptosis, such as antagonizing apoptosis-inducing factors [41], inhibiting the activation of nuclear factor-kappaB (NF-κB) [42], and preventing mitochondrial cytochrome c release and caspase activation [43]. VPA and four derivatives of the VPA parent compound (with structural modifications) also increased cell survival and increased HSP70 mRNA levels through HDAC inhibition in rat CGCs treated with excessive glutamate [44]. Notably, VPA also robustly induced HSP70 via epigenetic mechanisms, decreased infarction volume, and improved functional recovery in rodents subjected to cerebral ischemia—another in vivo excitotoxic model [45, 46]. It is interesting to note that in a stroke model, the neuroprotective effects of lithium were associated with elevated expression of HSP70 protein in the ischemic brain [47].
Another recent QA model rat study found that three weeks co-treatment with VPA and rosiglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, improved motor dysfunction, increased neuroprotective brain-derived neurotrophic factor (BDNF) in the striatum, and decreased striatal lesion volume, as well as pro-apoptotic tumor necrosis factor-alpha (TNF-α) and caspase-3 levels, compared with control rats [48]. The combined neuroprotective mechanism of rosiglitazone and VPA was thought to be partly due to PPARγ activation and HDAC inhibition pathways, respectively. A different study found that pretreatment with lithium (1.0 mM) combined with VPA (0.5 mM) provided synergistic neuroprotection against glutamate-induced cell death in young rat CGCs [49]. Another study found that potentiation of GSK-3 inhibition and induction of fibroblast growth factor-21 (FGF-21) significantly contributed to the neuroprotective synergy of lithium and VPA combined treatment [50]. A similar study using human neuroblastoma cells (SH-SY5Y) exposed to high glutamate concentrations also found that the same dosage of lithium and VPA pretreatment increased cell viability and increased BDNF protein and mRNA levels [51]. Overall, the different excitotoxic models provide strong evidence that VPA alone and in combination with lithium promotes neuroprotection.
Dysfunction of Autophagy and Protein Quality Regulation
One of the primary neuropathological findings in HD is the formation of protein aggregates (also called inclusion bodies) within brain cells [52], but whether these mHtt aggregates are toxic remains a controversial topic. Some investigators have suggested that cytoplasmic insoluble mHtt aggregates are a beneficial coping response [53], while others have suggested that intracellular aggregates may be directly toxic [54], and thus the function of mHtt aggregates within the cytoplasm is not definitively known. Intranuclear aggregation of soluble mHtt is, in contrast, believed to be toxic [55], most likely due to mHtt's downstream effects on gene expression. Recently mHtt was shown to disrupt the axonal transport of autophagosomes [56], which leads to deficits in autophagy. Several in vivo and in vitro models have demonstrated that lithium and VPA increase the clearance of intracellular protein aggregates related to various neurodegenerative disorders, including HD.
HSPs are a group of molecular chaperones that promote the degradation of abnormally folded proteins through the ubiquitin-proteasome system and autophagy [57-59]; they are also associated with the anti-apoptotic effects, as described above. The expression of HSP70 and its co-chaperone HSP40 were decreased [60, 61] and co-localized with mHtt aggregates [62] in the brains of HD animal models. In contrast, overexpression of HSPs in HD models were found to reduce the formation of mHtt aggregates and suppress the neurodegeneration and toxicity associated with this disease [62-64].
The two main targets of lithium—IMPase and GSK-3β—have opposite effects on autophagy. IMPase inhibition enhances autophagy [65], while GSK-3β inhibition actually downregulates autophagy via the downstream activation of mammalian target of rapamycin (mTOR) [66]. However, lithium inhibits IMPase at lower doses that induce autophagy (Ki ≈ 0.8 mM) [65], but inhibits GSK-3β at higher doses that suppress autophagy (Ki ≈ 2 mM) [67]. Pre- and post-treatment with lithium chloride (2.5-5.0 mM) protected non-neuronal (COS-7 African green monkey kidney) and neuronal (SKNSH human neuroblastoma) cells transfected with mutant Htt exon 1 fragment containing 74 CAG repeats, increased cell survival, and decreased aggregate formation compared with non-treated groups in both cell types [68]. The same group demonstrated that five days treatment with lithium (10 mM) enhanced autophagy in an mTOR-independent manner, by inhibiting IMPase; the subsequent depletion of myo-inositol-1,4,5-trisphosphate (IP3) levels was documented by clearance of mHtt and α-synuclein protein (which forms neuronal protein aggregates in Parkinson's disease) from transfected COS-7 and neuronal SKNSH cells [65]. In an HD Drosophila model, co-treatment with rapamycin and lithium—both of which are mTOR inhibitors—induced autophagy and increased neuroprotection compared to treatment with either agent alone [66]. While these results provide strong evidence for lithium's beneficial effects on autophagy in HD, they need to be replicated at therapeutically relevant doses (in vivo blood serum concentration levels of lithium should be between 0.4-1.0 mM [69]) using primary neuronal cells or, preferably, animal models.
VPA, carbamazepine, and other mood stabilizers can also decrease IP3 levels and induce autophagy [65]. VPA caused autophagy-induced cell death and cell growth inhibition in various human cancer cell lines [70, 71], and increased autophagy in Parkinson's disease animal models [72, 73]. Because so few studies have investigated this issue, it remains unclear whether VPA can induce autophagy of mHtt; further investigation is warranted.
Lithium and VPA have proven efficacy in other non-HD models of neurodegeneration through a variety of routes. Lithium or VPA alone both increased cell viability and enhanced autophagy in a rotenone toxic cellular model of Parkinson's disease [72], as measured by increased lysosome number and microtubule-associated protein 1 light chain 3-II (LC3-II) levels, a marker for autophagy. LC3-II levels correlate with the number of autophagosomes [74], organelles that capture malfunctioning cellular components and transport them to lysosomes for digestion. In a neurotoxic N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease, co-treatment with lithium plus VPA for seven days after MPTP injection improved motor function and enhanced autophagy, as measured by increased LC3-II levels [73]. In an Alzheimer's disease model, a different neurodegenerative disease, treatment with GSK-3β inhibitors VIII and XI (which are more specific than lithium) or transfection with GSK3α/β short interfering RNA (siRNA) in vitro reduced amyloid-beta (Aβ) protein aggregates by increasing degradation of amyloid precursor protein (APP), and reduced its carboxy-terminal fragment by activating the lysosomal/autophagy pathway [75]. In an Alzheimer's disease mouse model, treatment with L803-mts (another GSK-3β inhibitor) reduced Aβ accumulation and improved cognitive function [76]. Lithium alone increased the autophagy of lethal prion protein aggregates in both neuronal (mouse neuroblastoma N2a) and non-neuronal (mouse fibroblast L929) cells, and autophagy was further enhanced by rapamycin co-treatment in an additive fashion [77].
With regard to selective striatal death in HD, a recent, novel hypothesis suggests the involvement of Rhes, a GTP-binding protein that is highly expressed in the striatum [78]. Rhes interacts directly with mHtt to enhance cell toxicity via increased sumoylation, a post-translational protein modification that can alter protein function [79]. However, both Rhes and mHtt are expressed in the striatum years before HD symptoms occur. In the absence of mHtt, Rhes can increase autophagy in an mTOR independent manner, by binding to Beclin-1 and preventing anti-autophagic Bcl-2 from disrupting the Beclin complex [80]; this, in turn, may explain the delay in onset of HD symptoms. Rhes-enhanced autophagy appears to be impaired by mHtt expression, which may sequester and bind to Rhes, thus preventing autophagy. Genetic deletion of Rhes decreased striatal lesion size and improved motor function in a 3-NP model of HD [81], and delayed the onset of HD symptoms in a double transgenic model [82]. In general, Rhes does not harm neurons in the absence of mHtt, but neurotoxic effects have been observed in the presence of mHtt. Interestingly, a knockout mouse model of Rhes resulted in biochemical phenotypes similar to lithium treatment; specifically, Rhes knockout increased striatal phosphorylation of Akt and GSK-3β, thus increasing Akt activity and GSK-3β inhibition [83]. While these results are significant, additional in vivo studies and clinical evidence for Rhes disruption in HD patients are needed to further support the role of Rhes in HD neuropathology.
As the evidence reviewed above underscores, lithium and VPA are excellent tools for studying protein quality control in neurodegenerative diseases such as HD. Further investigation using therapeutic levels of lithium and VPA in in vivo models, may uncover valuable molecular targets for diseases, like HD, characterized by autophagy deficiency.
Abnormal levels of Neurotrophic and Growth Factors
Neurotrophic factors and growth factors are vital for the growth and survival of neurons and glial cells in the developing and mature nervous system. Neurotrophic and growth factors pathologically altered in HD include BDNF, glial cell line-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and transforming growth factor-beta 1 (TGF-β1). Here, we focus on BDNF and TGF-β1expression in HD, because of relevant support from lithium and VPA studies.
BDNF is critical for the growth and survival of maturing neurons, and is implicated in neurogenesis as well as neuronal differentiation and migration. BDNF is highly expressed in the cortex and hippocampus [84], areas of the brain necessary for cognition, emotion, and memory. Striatal BDNF is produced by cortical neurons and supplied by anterograde transport into this brain region [85]. Thus functional BDNF transport is critical for development and support of the striatum. In addition, abnormalities in BDNF expression, signaling, or transport are strongly implicated in many neuropsychiatric disorders, including major depressive disorder, schizophrenia, and bipolar disorder [86]. In vitro evidence suggests that wild-type Htt promotes BDNF vesicular transport, whereas mHtt severely impairs BDNF transport, thus indicating that reduced BDNF transport contributes to the pathophysiology of HD as demonstrated by reduced cellular neurotrophic support and subsequent increases in neurotoxicity [87]. Decreased cortical BDNF mRNA transcript and protein levels were observed in the postmortem brain of HD patients compared with sex- and age-matched controls [88, 89]. One theory suggests that decreased levels of BDNF may make striatal neurons more vulnerable to cell death [90]. In preclinical studies, decreased cortical BDNF exon II, III, and IV mRNA levels were correlated with HD progression in R6/2 transgenic mice [91]; the authors suggested that increased BDNF levels may slow disease development. Relatedly, overexpression of BDNF in the forebrain of R6/1 [92] and YAC128 [93] mouse models of HD improved motor dysfunction and increased brain weight compared to HD mice without BDNF overexpression. These preclinical studies suggest that increased BDNF expression in the brain is neuroprotective against HD, and therefore drugs that increase endogenous levels of BDNF may be necessary to combat HD.
Because BDNF has limited permeability to cross the blood-brain barrier (BBB), pharmacological agents that can easily traverse the BBB and increase endogenous downstream effectors of BDNF are necessary to treat diseases with BDNF deficits, such as HD. Under normal physiological conditions, BDNF binds to post-synaptic tropomyosin-related kinase B (TrkB) receptors and induces signaling cascades such as the phosphatidylinositol 3-kinase (PI3K) pathway, which activates Akt, and/or extracellular signal related kinase (ERK) pathway, thus activating several downstream effectors, such as cAMP response element binding protein (CREB) (for a review of BDNF/TrkB signaling, see [94]). BBB permeable TrkB agonists [95] should increase BDNF downstream effectors within the brain and also provide increased neuroprotection. Treatment of BACHD and R6/2 mouse models of HD, with both intranasal and intraperitoneal administrations of LM22A-4, a small-molecule TrkB agonist, improved motor function in the pole test (but not the rotarod), increased expression of downstream BDNF effectors (Akt, CREB and PLCγ), increased dendritic spine number of medium-sized spiny neurons, and decreased diffuse nuclear mHtt aggregates compared with untreated BACHD or R6/2 mice, respectively [96]. It should be noted, however, that although LM22A-4 successfully reduced neuron and motor impairments, it did not increase the lifespan of R6/2 mice, suggesting that other mechanisms besides TrkB signaling are involved. A preliminary study from our laboratory used similar LM22A-4 administrations in N171-82Q HD mice and found long-term behavioral improvements including normalization of hypolocomotor activity, alleviation of depression-like effects, and amelioration of motor deficits (Chiu et al., Society for Neuroscience abstract #528.04, 2013).
Another method to increase endogenous BDNF expression was demonstrated in rat primary cortical neurons primed for two days with therapeutic levels of either lithium (1.0 mM) or VPA (0.5 mM) [97]. Both drug treatments increased BDNF exon IV mRNA transcript levels and BDNF protein levels in vitro compared with untreated control neurons [97]. We extended these findings in vivo using chronic dietary treatment to reach therapeutic levels of lithium (1.0 mM) and VPA (0.5 mM) in N171-82Q mice and YAC128 mice, two transgenic mouse models of HD (see Table 1 for summary of HD mouse models and [98] for review), that differ in their genetic background, behavioral phenotype, symptom progression, and life span [99]. Co-treatment with lithium and VPA more robustly upregulated mRNA and protein levels of BDNF as well as other neuroprotective proteins such as HSP70 in the striatum and cortex than treatment with either mood stabilizer alone. Notably, the mean survival time of N171-82Q mice was prolonged by ten weeks with lithium and VPA co-treatment. Behaviorally, co-treatment with lithium and VPA also more effectively alleviated spontaneous locomotor deficits in the open-field test and depression-like effects in the forced swim and tail suspension tests in both HD mouse models, compared with mono-treatment. In addition, drug co-treatment successfully improved motor skill learning and coordination in the rotarod performance test, and elicited anxiolytic-like effects in YAC128 mice. Interestingly, lithium and VPA's behavioral benefits were associated with GSK-3 and HDAC inhibition, respectively. In a related study, long-term treatment of YAC128 mice with NP03 (a low-dose lithium microemulsion) improved motor function, ameliorated deficits in striatal volume, increased neuronal counts, and enhanced BDNF levels without causing lithium toxicity [100], whereas chronic VPA mono-treatment (100 mg/kg daily) in N171-82Q mice prolonged survival by one month and ameliorated diminished locomotor activity; BDNF levels were not measured [101]. To further understand the synergistic or additive relationship between lithium and VPA, Gupta and colleagues used computer modeling to generate interaction networks based on well-established molecular targets for each drug. They observed enrichment of apoptosis clusters and neurotrophin signaling, and uncovered two key affected molecules that mediate multiple pathways, namely MAPK1/3 and Akt1/2 [102].
Frequently used Huntington's disease transgenic mouse models.
| Mouse Strain | Model Design | CAG Repeat # | Promoter | Appearance of Disease Phenotype |
|---|---|---|---|---|
| R6/1 | Truncated N-terminal fragment models | 116 | Human HTT | 6 weeks |
| R6/2 | 144 | Human HTT | 3 weeks | |
| N171-82Q | 82 | Mouse Prp | After 8 weeks | |
| YAC128 | Full-length HD transgenic models | 128 | Human HTT | 52 weeks |
| BACHD | 97 | Human HTT | 52 weeks |
TGF-β1, a member of the cytokine family, is involved in cell proliferation and differentiation. Although the physiological role of this growth factor in normal behavior and cognition is still largely unknown, recent interest in the possible role of TGF-β1 in HD has grown [103]. TGF-β1 protein levels were decreased in serum samples from asymptomatic HD patients compared to healthy controls and symptomatic patients, a finding confirmed by immunohistochemical analysis of postmortem cortical samples from the three patient groups [104]. The decreased TGF-β1 levels in presymptomatic HD patients led researcher to suggest that serum TGF-β1 levels may serve as a potential HD biomarker during the asymptomatic phase. The same study also found that presymptomatic R6/2 mice exhibited decreased TGF-β1 protein levels in the cortex and had decreased serum levels of TGF-β1 compared with wild-type and symptomatic R6/2 mice. Presymptomatic YAC128 mice also had decreased expression of TGF-β1 only in the cortex, however, serum levels were unchanged compared with wild-type controls. No studies have yet investigated the putative effects of lithium or VPA on TGF-β1 levels and function in HD. However, non-HD studies have shown that TGF-β1 is closely involved in fibrosis (tissue scarring) and lithium exposure was able to reduce harmful scarring by inhibiting TGF-β1 expression in human subconjunctival fibroblasts [105]. Lithium is also known to inhibit the transactivation of Smad3/4, the intracellular transducer of TGF-β1, via protein kinase A and Akt signaling pathways in rat primary cortical neurons [106]. Similarly, VPA treatment in human hepatic stellate cells blocked the TGF-β1autocrine loop [107], indicative of antifibrogenic effects. In both cellular and animal models of HD, lithium alone or in combination with VPA neuroprotectively enhanced BDNF expression and activity, whereas in non-HD models, lithium and VPA have been shown to regulate TGF-β1 expression. Future research should focus on how lithium and VPA regulate the BDNF and TGF-β1 pathways in HD.
Transcriptional Dysregulation
Growing evidence suggests that transcriptional dysregulation is a major pathogenic mechanism in HD [108]. Indeed, transcription factor dysregulation of gene expression is a central facet of the slow, progressive nature of HD pathology. Transcription factors implicated in HD pathophysiology include repressor element-1 silencing transcription factor (REST), CREB, NF-κB, and peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α).
REST, which has a response element within the BDNF promoter II [109], and can regulate BDNF expression is one of the most studied transcription factors in HD. REST co-localizes with wild-type Htt in both neurons and glia from the cortex, striatum, hippocampus, and cerebellum in HD patients and controls [110]. Under normal conditions, wild-type Htt protein associates with REST in the cytoplasm and prevents REST from repressing transcription in the nucleus, thereby allowing BDNF expression [109]. In the pathological state, mHtt dissociates from REST in the cytoplasm, thereby facilitating the translocation of REST to the nucleus where it represses gene transcription, resulting in decreased BDNF expression [109]. Zuccato and colleagues reported that increased REST binding at its DNA binding sites mediated decreased expression of REST target genes, which are involved in neuronal development and differentiation. This phenomenon was observed in both HD patients and in the R6/2 HD mouse model [111], suggesting that targeted downregulation of REST may have therapeutic potential for HD. Non-HD models have provided conflicting results regarding the effects of lithium and VPA on REST function. In a rat fetal alcohol syndrome model, lithium provided neuroprotection by downregulating REST protein levels and decreased its DNA-binding activity [112] compared to only ethanol exposure. Lithium also increased neuronal differentiation and survival in primary rat neural stem cells in the presence of ethanol, compared with untreated controls [112]. In a mouse model of Niemann-Pick Type C disease, a lysosomal storage disease, VPA downregulated REST levels and increased target gene levels such as TrkB and BDNF compared with untreated controls [113]. However in medulloblastoma cells, VPA upregulated REST transcription as demonstrated by increased histone acetylation level in the REST promoter [114].
CREB binds to specific DNA sequences called cAMP response elements (CRE), and either increases or decreases expression of downstream genes. CREB supports cell survival by enhancing expression of cell-protective proteins, such as BDNF and Bcl-2 [115]. Different signaling pathways, including PI3K/Akt and MEK/ERK, can activate CREB through serine phosphorylation [116]. Once activated, CREB recruits its coactivators—CREB-binding protein (CBP) or transducer of regulated CREB activity (TORC)—to directly interact with the transcriptional machinery. CREB dysregulation has been strongly implicated in the pathophysiology of HD. For instance, CREB-regulated genes were downregulated in the brains of HD patients [117], and CREB knockout mice displayed an HD-like phenotype with neurodegeneration in the hippocampus and striatum [118]. In a study focusing on CREB function in HD, YAC128 mice were crossed with dominant-negative CREB mice. The transgenic progeny had accelerated motor impairment compared with YAC128 and wild-type mice [119]. The same study also investigated CREB loss of function in transgenic mice treated with 3-NP, and found that lesion area was greater in the 3-NP group with disruption of CREB relative to wild-type mice. In non-HD models, lithium treatment enhanced CREB activity in hamster insulinoma tumor cells [120] through direct activation of TORC [121]. In CGCs, chronic lithium treatment suppressed the glutamate-induced decrease in phosphorylated CREB and markedly increased CREB-driven gene expression [122].
NF-κB is responsible for the production of cytokines, and is involved in the regulation of immunity, synaptic plasticity, and cell survival [123]. One study found that enhanced NF-κB activation increased astrocyte inflammation and increased HD pathophysiology [124], suggesting that anti-inflammatory treatments may be beneficial for HD. To date, no studies have investigated the effects of lithium or VPA on NF-κB in HD models. In healthy rats, however, chronic VPA administration downregulated NF-κB DNA binding activity [125], suggesting that VPA has anti-inflammatory properties. In human thyroid carcinoma cells, therapeutic levels of VPA decreased the expression of NF-κB regulatory genes and proteins [126]. Another study found that VPA robustly inhibited microglial activation and suppressed NF-κB activation in rats with experimental ischemic cerebral stroke [46, 127]. In general, lithium or GSK-3 inhibitors attenuate NF-κB activation in a variety of cell types. Lithium exposure or GSK-3β siRNA-mediated knockdown inhibited NF-κB activation and induced apoptosis in osteosarcoma cells [128]. Fibroblasts from GSK-3β deficient mouse embryos exhibited decreased NF-κB activation, and lithium treatment of wild-type embryos inhibited transactivation of NF-κB [129]. However, a different study found that knockdown of GSK-3α or β markedly increased NF-κB transactivation in healthy primary cerebral cortical neurons [33]. Also, in human colon cancer cells (HT-29 and Caco-2), lithium enhanced NF-κB activity [130].
In addition to psychiatric, cognitive and motor symptoms, HD patients progressively lose weight despite increased caloric consumption, indicating metabolic dysfunction [131]. Metabolic abnormalities in HD are further suggested by decreased activity of the oxidative phosphorylation pathway in the striatal neurons of late-stage HD patients [132], and reduced ATP production as a function of increased CAG repeats in lymphoblastoid cell lines transfected with mHtt [133]. Interestingly, PGC-1α is a key regulator of mitochondrial biogenesis and oxidative stress [134, 135], and decreased PGC-1α levels have been found in both HD mouse models and HD patients [136, 137]. In the N171-82Q mouse model, overexpression of PGC-1α decreased motor symptoms of HD and significantly decreased mHtt protein aggregation by reducing oxidative stress and by activating transcription factor EB (TFEB), an important regulator of the autophagy-lysosome pathway [138]. In a non-HD model, lithium exposure enhanced PGC-1α expression and increased mitochondrial biogenesis in primary bovine aortic endothelial cells [139].
Aberrant Epigenetic Modifications
Aberrant epigenetic patterns have recently been observed in humans with HD and in transgenic HD mice, though the mechanisms underlying these epigenetic modifications are not well understood, and some of the observed epiphenomena may actually be unrelated to HD. Furthermore, while transcriptional and epigenetic dysregulation are important early events in the pathophysiology of HD, the degree to which altered epigenetics plays a causative role in HD pathology remains unclear [140].
Briefly, the vast amount of DNA is organized by wrapping around histone proteins. Chromatin conformation is further regulated by enzymes known as histone acetyltransferases (HATs), which add charge-neutralizing acetyl groups to the lysine tails of histone proteins, conferring a more transcriptionally active chromatin conformation that generally increases gene expression. In contrast, HDACs are enzymes that remove acetyl groups from histone tails, confer a more transcriptionally inactive form of chromatin, and usually cause decreased gene expression. VPA is a class I and IIa, but not a IIb or class III HDAC inhibitor. It blocks the deacetylation activity of certain HDACs, and overall allows for a more transcriptionally active chromatin shape of target genes [13]. In the study discussed previously, co-treatment with lithium and VPA enhanced histone 3 (H3) acetylation in the striatum and cortex of both YAC128 and N171-82Q transgenic mice, thereby ameliorating the observed hypoacetylation of untreated mice [99]. In preclinical transgenic mouse (R6/2) and mHtt transfected primary neuron models of HD, levels of CBP (an HAT) in the brain are decreased, suggesting that upregulating CBP may ameliorate transcriptional dysregulation [141, 142]. In an amyotrophic lateral sclerosis (ALS) transgenic SOD1 mouse model, VPA treatment exerted neuroprotection by upregulating of CBP but did not improve the overall survival of ALS mice [143].
Wu and colleagues recently reported that lithium treatment in HEK239T, HeLa and SW480 cells downregulated HDAC1 protein, but did not decrease HDAC1 mRNA, suggesting that lithium acts at the translational level to regulate HDAC1 expression [144]. HDAC1 downregulation by lithium was also necessary for the autophagic degradation of long mHtt (Htt590-100Q). Nevertheless this study was associated with several limitations, including inconsistent, high concentrations of lithium (5-40 mM), use of only in vitro approaches, and use of non-neuronal cell types. Future studies should determine if downregulation of HDAC1 by lithium also occurs at therapeutically relevant levels within in vivo models.
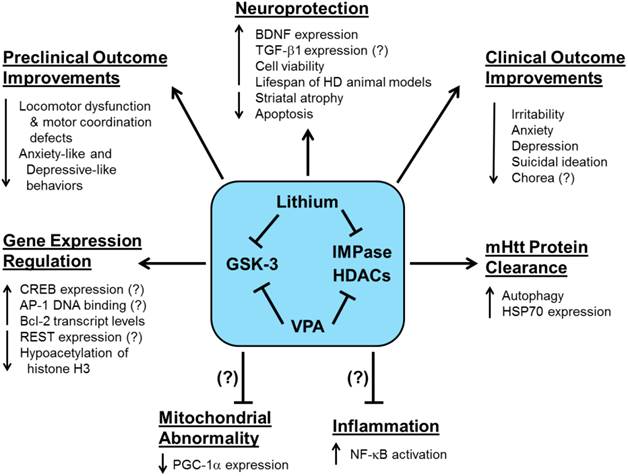
While histone acetylation generally enhances gene expression, DNA methylation, the addition of methyl groups to the cytosine-phosphate-guanine sites, causes a more closed chromatin structure and usually inhibits gene expression [145]. In a unique cell culture study, net DNA demethylation was found in striatal cells expressing 111-CAG repeat mHtt compared with cells expressing normal 7-CAG repeat Htt [146]. The authors also found that increased binding of the AP-1 complex was associated with decreased DNA methylation in striatal cells expressing mHtt, suggesting that decreased AP-1 DNA binding activity may upregulate DNA methylation [147]. In an HD-related model of excessive glutamate excitotoxicity, one week of lithium pretreatment provided neuroprotection by downregulating AP-1 DNA binding activity and enhancing rat CGC viability [31]. This finding contrasts with lithium's upregulation of AP-1 binding activity in healthy rat CGCs [148], as well as in human SH-SY5Y and rat C6 glioma cells [149]. Figure 2 provides a diagram of the preclinical and clinical effects of lithium and VPA treatment in HD.
The preclinical and clinical effects of lithium and valproic acid (VPA) treatment in Huntington's disease (HD). Preclinical and clinical studies have shown that HD pathophysiology involves the dysfunction of multiple complex physiological processes and signaling pathways. More recent preclinical investigations have demonstrated that lithium and VPA exert numerous beneficial effects including enhanced neuroprotection, motor function recovery, mHtt protein clearance, and gene expression regulation. The lithium- or VPA-mediated reduction of mitochondrial abnormality, which is closely related to decreased PGC-1a expression, or inflammation, caused by NF-kB activation, have yet to be determined in HD models. Clinically, HD patients treated with lithium or VPA exhibit improved psychiatric outcomes including reduced irritability, anxiety, depression, and suicidal ideation. It has yet to be validated whether lithium or VPA can reduce chorea in HD patients. Pointed arrows indicate increases or decreases in the respective effects due to lithium- or VPA-induced actions on each physiological process. Arrows with blocked ends indicate lithium- or VPA- mediated prevention of a pathophysiological event. Question marks (?) indicate known effects of lithium or VPA, which have yet to be verified in HD models and patients.
Clinical Trials of Lithium and VPA in HD
Interestingly, both lithium and VPA were separately used in clinical trials of HD years before their neuroprotective characteristics were discovered [150-160]. In two separate non-blind case studies, lithium therapy decreased the intensity of choreiform movements [150], and improved motor function [153] in some of the patients observed. Previous uncontrolled non-blind [150, 151, 153, 154] and controlled blind [155, 157] HD case studies had noted lithium's strong mood stabilizing properties, such as reduced depressive symptoms, irritability, and aggression (see Table 2 for summary). More recent clinical studies also demonstrated lithium's mood stabilizing effects in HD patients of varying ages and duration of disease [161, 162]. One study found that lithium reduced the progression of chorea [161], though another study did not report any changes in chorea [162]. In the latter study, which focused on suicidal HD patients [162], 80% of the patients were also taking tetrabenazine (which increases the risk of depression or suicide [9]). The addition of lithium to the treatment regimen eliminated suicidal symptoms in all patients and improved depressive symptoms, demonstrating that the combination of lithium and tetrabenazine may be beneficial for some HD patients. Taken together, these clinical studies support lithium's potential to improve the psychiatric and possibly motor symptoms of HD. Despite these findings, two placebo-controlled, blind clinical trials reported that lithium was ineffective in the treatment of choreiform movements [156, 158]. However, in both studies, sample size was small, the duration of lithium treatment was relatively short, and standardized measures were not used to report mood changes. It is also possible that an earlier intervention with lithium would be more effective at ameliorating HD symptoms.
Clinical trials with lithium and VPA in Huntington's disease patients.
| Drug Treatment | Duration | Dosage/Interval | Subjects (n) | Major Findings | Treatment Beneficial? | Reference |
|---|---|---|---|---|---|---|
| Lithium + Baclofen | 4 weeks | N.A.*; 15-90 mg Baclofen | 3 | Baclofen, a GABA derivative, worsened motor function, while lithium "further improved" patients | Yes | [150] |
| Lithium | 1.5 weeks + | 24.3 mEq/day | 6 | Lithium reduced hyperkinetic movements, improved voluntary movements, and stabilized irritable mood | Yes | [151] |
| Lithium + Haloperidol | ? | 300 mg lithium; 2 mg haloperidol, both 3x per day | 1 | Patient's motor function improved significantly when lithium was added to haloperidol treatment | Yes | [153] |
| Lithium | 6.5 weeks | N.A.* | 4 | Lithium stabilized mood in 3 out of 4 cases | Yes | [154] |
| Lithium | ? | N.A.* | 2 | Lithium plus unspecified neuroleptics improved mood and hyperkineses, clinicians were blind to treatment | Yes | [155] |
| Lithium | 7 weeks | N.A.* | 9 | In a double-blind trial, lithium did not improve mood or motor function | No | [156] |
| Lithium + Haloperidol | 3 weeks | N.A.* | 6 | In a double-blind trial, lithium plus haloperidol stabilized mood but did not affect motor function | Yes | [157] |
| Lithium | 6 weeks | N.A.* | 6 | In a placebo-controlled trial, lithium did not improve motor function, but authors did not assess mood | No | [158] |
| Lithium | 3-5 years | 150-300 mg/day | 3 | Lithium significantly improved mood and stabilized chorea progression in all patients | Yes | [161] |
| Lithium | 40 weeks - 2 years | 150-300 mg, 2x per day | 5 | Lithium reduced suicidal ideation and improved depression in all patients | Yes | [162] |
| VPA | 4 weeks | 200-600 mg, 3x per day | 3 | VPA did not affect involuntary movements | No | [159] |
| VPA | ? | 600-1200 mg/day | 5 | VPA did not improve choreiform movements; no mention of mood changes or improvement | No | [166] |
| VPA | 13-26 weeks | 500-2400 mg/day | 14 | VPA was ineffective at improving chorea; authors did not discuss mood stabilization | No | [167] |
| VPA | 12 weeks | 600-900 mg, 3x per day | 1 | Development of a state of tolerance for VPA | No | [168] |
| VPA | 9 weeks | 800-2000 mg, 4x per day | 6 | VPA normalized sleep and reduced awakenings, but did not observe change in motor function | Yes | [160] |
| VPA + Olanzapine | 8 weeks | 125-500 mg, 2x per day; 5-10 mg/day | 2 | VPA and olanzapine improved motor function and stabilized mood | Yes | [164] |
| VPA | ? | 900-1200 mg/day | 8 | VPA enhanced mood stabilization and improved basic motor coordination tasks | Yes | [165] |
N.A.* = Lithium within or below therapeutic blood serum concentration; specific dose not available.
VPA has also been suggested as a logical choice for HD treatment [163]. In one case study, VPA reduced the sleep disturbances associated with HD, but had no effect on chorea symptoms [160]. In a more recent study, the lowest effective dose of VPA (125-500 mg) in combination with olanzapine (an atypical antipsychotic; 5-10 mg) improved both psychiatric and motor symptoms in HD patients [164]. Another study of HD patients with severe myoclonic hyperkinesia found increased VPA dosage (from 900 mg to 1200 mg) was correlated with improved motor scores on the Unified Huntington's Disease Rating Scale, as well as mood stabilization [165], for more than half of the patients examined. In contrast, several case studies found that VPA did not improve motor symptoms in HD patients [159, 166, 167]. However, these reports did not measure mood or behavior changes, had small sample sizes, and short treatment durations. A longer VPA treatment period may reveal patient improvement because HDAC inhibition and subsequent transcriptional and translational changes may take more sustained treatment to reach their full effect. Another case study reported that VPA treatment caused a state of tolerance, however this was found in only one late-stage HD patient [168]. While VPA is generally a safe drug [169], it is a known teratogen in both rodents [170] and humans [171], and it should be noted that one case study reported that a single HD patient developed Pisa syndrome, an incredibly rare but dangerous neurological side effect [172], after VPA treatment.
Conclusions and Future Directions
HD disrupts critical cellular mechanisms involved in transcription, translation, epigenetic, cell growth, and metabolic pathways. As highlighted in this review, both lithium and VPA have robust neuroprotective and stabilizing effects that makes them not only useful as therapeutics for treating HD but, perhaps more importantly, makes them efficacious tools for studying disease progression and response, and expanding the translational significance of the results. Furthermore, both agents are FDA-approved medications with a long history of safe use in humans.
As an initial goal, we suggest that clinical studies should explore whether therapeutically relevant levels of lithium or VPA can significantly decrease chorea and ameliorate the psychiatric symptoms of HD. Clinical studies conducted with lithium and VPA in HD patients to date have been plagued by limitations such as small sample size, different stages and burden of disease, absence of a control group, a lack of re-evaluation after drug washout, concurrent drug therapies, non-blind clinician observations, and short treatment duration. In order to determine how effectively lithium and VPA treatment can reduce HD symptoms, large-scale, controlled, double-blind trials are needed in the future. Such studies can also investigate whether lithium and VPA can improve quality of life for HD patients and their caregivers. Given that increasing evidence from HD mouse models supports the notion that combined treatment with lithium and VPA is superior to treatment with either agent alone, we strongly recommend that future clinical trials explore the combined use of these agents.
Abbreviations
3-NP: 3-nitropropionic acid; AP-1: activator protein 1; Bcl-2: B-cell lymphoma/leukemia-2; BDNF: brain-derived neurotrophic factor; CGC: cerebellar granule cell; CREB: cAMP response element binding protein; FGF-21: fibroblast growth factor-21; GSK-3: glycogen synthase kinase 3; HSP70: heat shock protein 70; HDAC: histone deacetylase; HD: Huntington's disease; Htt: huntingtin; mHtt: mutant huntingtin; NF-κB: nuclear factor kappaB; PI3K: phosphatidylinositol 3-kinase; QA: quinolinic acid; REST: repressor element-1 silencing transcription factor; TGF-β1: transforming growth factor-beta 1; TrkB: tropomyosin-related kinase B; VPA: valproic acid.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH). The authors thank Ioline Henter and Peter Leeds of the NIMH, NIH for their excellent editorial assistance.
Competing Interests
The authors have no conflict of interest to disclose, financial or otherwise.
References
1. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971-83
2. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington's disease: a systematic review and meta-analysis. Movement disorders: official journal of the Movement Disorder Society. 2012;27:1083-91
3. Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, International Huntington's Disease Collaborative G. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clinical genetics. 2004;65:267-77
4. Rosenblatt A, Liang KY, Zhou H, Abbott MH, Gourley LM, Margolis RL. et al. The association of CAG repeat length with clinical progression in Huntington disease. Neurology. 2006;66:1016-20
5. Walker FO. Huntington's disease. Lancet. 2007;369:218-28
6. Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E. Regional specificity of brain atrophy in Huntington's disease. Experimental neurology. 1998;154:663-72
7. Yero T, Rey JA. Tetrabenazine (Xenazine), An FDA-Approved Treatment Option For Huntington's Disease-Related Chorea. P & T: a peer-reviewed journal for formulary management. 2008;33:690-4
8. Frank S, Ondo W, Fahn S, Hunter C, Oakes D, Plumb S. et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clinical neuropharmacology. 2008;31:127-33
9. Huntington Study G. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366-72
10. Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacological reviews. 2013;65:105-42
11. Sarkar S, Rubinsztein DC. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy. 2006;2:132-4
12. Chiu CT, Chuang DM. Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacology & therapeutics. 2010;128:281-304
13. Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends in neurosciences. 2009;32:591-601
14. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. The Journal of biological chemistry. 2001;276:36734-41
15. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet neurology. 2011;10:83-98
16. Li SH, Schilling G, Young WS 3rd, Li XJ, Margolis RL, Stine OC. et al. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 1993;11:985-93
17. van der Burg JM, Bjorkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet neurology. 2009;8:765-74
18. Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington's disease. Drug discovery today. 2014 in press
19. Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME. et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. Journal of neuroscience research. 1999;58:515-32
20. Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR. et al. Mutant huntingtin enhances excitotoxic cell death. Molecular and cellular neurosciences. 2001;17:41-53
21. Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P. et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849-60
22. Taylor-Robinson SD, Weeks RA, Bryant DJ, Sargentoni J, Marcus CD, Harding AE. et al. Proton magnetic resonance spectroscopy in Huntington's disease: evidence in favour of the glutamate excitotoxic theory. Movement disorders: official journal of the Movement Disorder Society. 1996;11:167-73
23. Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature. 1976;263:244-6
24. Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM. et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1993;13:4181-92
25. Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168-71
26. Marco S, Giralt A, Petrovic MM, Pouladi MA, Martinez-Turrillas R, Martinez-Hernandez J. et al. Suppressing aberrant GluN3A expression rescues synaptic and behavioral impairments in Huntington's disease models. Nature medicine. 2013;19:1030-8
27. Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2642-7
28. Hashimoto R, Fujimaki K, Jeong MR, Christ L, Chuang DM. Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-D-aspartate receptor-mediated excitotoxicity. FEBS letters. 2003;538:145-8
29. Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. Journal of neurochemistry. 2002;80:589-97
30. Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. The Journal of biological chemistry. 1999;274:6039-42
31. Chen RW, Qin ZH, Ren M, Kanai H, Chalecka-Franaszek E, Leeds P. et al. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. Journal of neurochemistry. 2003;84:566-75
32. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Progress in neurobiology. 2001;65:391-426
33. Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. The Journal of biological chemistry. 2006;281:30479-84
34. Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. The Journal of biological chemistry. 2007;282:3904-17
35. Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y. et al. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington's disease. Neuroscience. 2001;106:603-12
36. Senatorov VV, Ren M, Kanai H, Wei H, Chuang DM. Short-term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, an excitotoxic model of Huntington's disease. Molecular psychiatry. 2004;9:371-85
37. Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT. et al. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000;20:119-29
38. Crespo-Biel N, Camins A, Pallas M, Canudas AM. Evidence of calpain/cdk5 pathway inhibition by lithium in 3-nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology. 2009;56:422-8
39. Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences: CMLS. 2005;62:670-84
40. Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S. et al. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. Journal of neurochemistry. 2009;111:976-87
41. Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N. et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nature cell biology. 2001;3:839-43
42. Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:53-63
43. Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T. et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature cell biology. 2000;2:469-75
44. Leng Y, Marinova Z, Reis-Fernandes MA, Nau H, Chuang DM. Potent neuroprotective effects of novel structural derivatives of valproic acid: potential roles of HDAC inhibition and HSP70 induction. Neuroscience letters. 2010;476:127-32
45. Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. Journal of neurochemistry. 2004;89:1358-67
46. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. The Journal of pharmacology and experimental therapeutics. 2007;321:892-901
47. Ren M, Senatorov VV, Chen RW, Chuang DM. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6210-5
48. Mishra J, Chaudhary T, Kumar A. Rosiglitazone Synergizes the Neuroprotective Effects of Valproic Acid Against Quinolinic Acid-Induced Neurotoxicity in Rats: Targeting PPARgamma and HDAC Pathways. Neurotoxicity research. 2014 [Epub ahead of print]
49. Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:2576-88
50. Leng Y, Wang Z, Tsai LK, Leeds P, Fessler EB, Wang J. et al. FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is markedly elevated in neurons by mood stabilizers. Molecular psychiatry. 2014 [Epub ahead of print]
51. Croce N, Mathe AA, Gelfo F, Caltagirone C, Bernardini S, Angelucci F. Effects of lithium and valproic acid on BDNF protein and gene expression in an in vitro human neuron-like model of degeneration. Journal of psychopharmacology. 2014 [Epub ahead of print]
52. Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R. et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549-58
53. Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805-10
54. Perutz MF, Windle AH. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature. 2001;412:143-4
55. Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Human molecular genetics. 2008;17:345-56
56. Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:1293-305
57. Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852-8
58. Ma YJ, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827-30
59. Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nature reviews Molecular cell biology. 2005;6:891-8
60. Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R. et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Human molecular genetics. 2004;13:1389-405
61. Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H. et al. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. The EMBO journal. 2008;27:827-39
62. Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Human molecular genetics. 2000;9:2009-18
63. Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Human molecular genetics. 2000;9:2811-20
64. Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S. et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. The Journal of biological chemistry. 2005;280:34908-16
65. Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M. et al. Lithium induces autophagy by inhibiting inositol monophosphatase. The Journal of cell biology. 2005;170:1101-11
66. Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Human molecular genetics. 2008;17:170-8
67. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Current biology: CB. 1996;6:1664-8
68. Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. The Journal of biological chemistry. 2002;277:33791-8
69. Malhi GS, Tanious M, Gershon S. The lithiumeter: a measured approach. Bipolar disorders. 2011;13:219-26
70. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro-oncology. 2010;12:328-40
71. Dong LH, Cheng S, Zheng Z, Wang L, Shen Y, Shen ZX. et al. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. Journal of hematology & oncology. 2013;6:53
72. Xiong N, Jia M, Chen C, Xiong J, Zhang Z, Huang J. et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience. 2011;199:292-302
73. Li XZ, Chen XP, Zhao K, Bai LM, Zhang H, Zhou XP. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: possible mediation through enhanced autophagy. The International journal of neuroscience. 2013;123:73-9
74. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542-5
75. Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-beta precursor protein. Molecular and cellular biology. 2012;32:4410-8
76. Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. The Journal of biological chemistry. 2013;288:1295-306
77. Heiseke A, Aguib Y, Riemer C, Baier M, Schatzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. Journal of neurochemistry. 2009;109:25-34
78. Spano D, Branchi I, Rosica A, Pirro MT, Riccio A, Mithbaokar P. et al. Rhes is involved in striatal function. Molecular and cellular biology. 2004;24:5788-96
79. Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327-30
80. Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a Striatal-selective Protein Implicated in Huntington Disease, Binds Beclin-1 and Activates Autophagy. The Journal of biological chemistry. 2014;289:3547-54
81. Mealer RG, Subramaniam S, Snyder SH. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:4206-10
82. Baiamonte BA, Lee FA, Brewer ST, Spano D, LaHoste GJ. Attenuation of Rhes activity significantly delays the appearance of behavioral symptoms in a mouse model of Huntington's disease. PloS one. 2013;8:e53606
83. Harrison LM, Muller SH, Spano D. Effects of the Ras homolog Rhes on Akt/protein kinase B and glycogen synthase kinase 3 phosphorylation in striatum. Neuroscience. 2013;236:21-30
84. Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511-26
85. Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL. et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856-60
86. Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacological reviews. 2012;64:238-58
87. Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP. et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127-38
88. Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008;18:225-38
89. Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain research. 2000;866:257-61
90. Zuccato C, Cattaneo E. Huntington's Disease. Handbook of experimental pharmacology. 2014;220:357-409
91. Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M. et al. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacological research: the official journal of the Italian Pharmacological Society. 2005;52:133-9
92. Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington's disease phenotypes in mice. Journal of neurochemistry. 2008;105:369-79
93. Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:14708-18
94. Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Developmental neurobiology. 2010;70:304-22
95. Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN. et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. The Journal of clinical investigation. 2010;120:1774-85
96. Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M. et al. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:18712-27
97. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Molecular psychiatry. 2009;14:51-9
98. Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nature reviews Neuroscience. 2013;14:708-21
99. Chiu CT, Liu G, Leeds P, Chuang DM. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington's disease. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011;36:2406-21
100. Pouladi MA, Brillaud E, Xie Y, Conforti P, Graham RK, Ehrnhoefer DE. et al. NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiology of disease. 2012;48:282-9
101. Zadori D, Geisz A, Vamos E, Vecsei L, Klivenyi P. Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington's disease. Pharmacology, biochemistry, and behavior. 2009;94:148-53
102. Gupta A, Schulze TG, Nagarajan V, Akula N, Corona W, Jiang XY. et al. Interaction networks of lithium and valproate molecular targets reveal a striking enrichment of apoptosis functional clusters and neurotrophin signaling. The pharmacogenomics journal. 2012;12:328-41
103. Martinez-Canabal A. Potential Neuroprotective Role of Transforming Growth Factor beta1 (TGFbeta1) in the Brain. The International journal of neuroscience. 2014 [Epub ahead of print]
104. Battaglia G, Cannella M, Riozzi B, Orobello S, Maat-Schieman ML, Aronica E. et al. Early defect of transforming growth factor beta1 formation in Huntington's disease. Journal of cellular and molecular medicine. 2011;15:555-71
105. Chung EJ, Sohn YH, Kwon SH, Jung SA, Lee JH. Lithium chloride inhibits TGF-beta1-induced myofibroblast transdifferentiation via PI3K/Akt pathway in cultured fibroblasts from Tenon's capsule of the human eye. Biotechnology letters. 2014;36:1217-24
106. Liang MH, Wendland JR, Chuang DM. Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Molecular and cellular neurosciences. 2008;37:440-53
107. Watanabe T, Tajima H, Hironori H, Nakagawara H, Ohnishi I, Takamura H. et al. Sodium valproate blocks the transforming growth factor (TGF)-beta1 autocrine loop and attenuates the TGF-beta1-induced collagen synthesis in a human hepatic stellate cell line. International journal of molecular medicine. 2011;28:919-25
108. Riley BE, Orr HT. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes & development. 2006;20:2183-92
109. Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L. et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nature genetics. 2003;35:76-83
110. Schiffer D, Caldera V, Mellai M, Conforti P, Cattaneo E, Zuccato C. REST is Present in Human Control and Huntington's Disease Neurons. Neuropathology and applied neurobiology. 2014 [Epub ahead of print]
111. Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E. et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:6972-83
112. Ishii T, Hashimoto E, Ukai W, Tateno M, Yoshinaga T, Saito S. et al. Lithium-induced suppression of transcription repressor NRSF/REST: effects on the dysfunction of neuronal differentiation by ethanol. European journal of pharmacology. 2008;593:36-43
113. Kim SJ, Lee BH, Lee YS, Kang KS. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochemical and biophysical research communications. 2007;360:593-9
114. Taylor P, Fangusaro J, Rajaram V, Goldman S, Helenowski IB, MacDonald T. et al. REST is a novel prognostic factor and therapeutic target for medulloblastoma. Molecular cancer therapeutics. 2012;11:1713-23
115. Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11-4
116. Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annual review of biochemistry. 1999;68:821-61
117. Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience. 2000;97:505-19
118. Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A. et al. Disruption of CREB function in brain leads to neurodegeneration. Nature genetics. 2002;31:47-54
119. Choi YS, Lee B, Cho HY, Reyes IB, Pu XA, Saido TC. et al. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's disease. Neurobiology of disease. 2009;36:259-68
120. Boer U, Eglins J, Krause D, Schnell S, Schofl C, Knepel W. Enhancement by lithium of cAMP-induced CRE/CREB-directed gene transcription conferred by TORC on the CREB basic leucine zipper domain. The Biochemical journal. 2007;408:69-77
121. Heinrich A, Boer U, Tzvetkov M, Oetjen E, Knepel W. Stimulation by lithium of the interaction between the transcription factor CREB and its co-activator TORC. Bioscience reports. 2009;29:77-87
122. Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116:425-35
123. Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harbor perspectives in biology. 2009;1:a001271
124. Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington's disease. Human molecular genetics. 2013;22:1826-42
125. Rao JS, Bazinet RP, Rapoport SI, Lee HJ. Chronic treatment of rats with sodium valproate downregulates frontal cortex NF-kappaB DNA binding activity and COX-2 mRNA. Bipolar disorders. 2007;9:513-20
126. Schwertheim S, Worm K, Schmid KW, Sheu-Grabellus SY. Valproic acid downregulates NF-kappaB p50 activity and IRAK-1 in a progressive thyroid carcinoma cell line. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2014;46:181-6
127. Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:52-7
128. Tang QL, Xie XB, Wang J, Chen Q, Han AJ, Zou CY. et al. Glycogen synthase kinase-3beta, NF-kappaB signaling, and tumorigenesis of human osteosarcoma. Journal of the National Cancer Institute. 2012;104:749-63
129. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86-90
130. Nemeth ZH, Deitch EA, Szabo C, Fekete Z, Hauser CJ, Hasko G. Lithium induces NF-kappa B activation and interleukin-8 production in human intestinal epithelial cells. The Journal of biological chemistry. 2002;277:7713-9
131. Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RA. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506-13
132. Browne SE, Beal MF. The energetics of Huntington's disease. Neurochemical research. 2004;29:531-46
133. Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M. et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Human molecular genetics. 2005;14:2871-80
134. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V. et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115-24
135. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S. et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397-408
136. Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59-69
137. Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L. et al. Impaired PGC-1alpha function in muscle in Huntington's disease. Human molecular genetics. 2009;18:3048-65
138. Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA. et al. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Science translational medicine. 2012;4:142ra97
139. Struewing IT, Barnett CD, Tang T, Mao CD. Lithium increases PGC-1alpha expression and mitochondrial biogenesis in primary bovine aortic endothelial cells. The FEBS journal. 2007;274:2749-65
140. Valor LM, Guiretti D. What's wrong with epigenetics in Huntington's disease? Neuropharmacology. 2013
141. Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H. et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6763-8
142. Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW. et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiology of disease. 2006;23:543-51
143. Rouaux C, Panteleeva I, Rene F, de Aguilar JLG, Echaniz-Laguna A, Dupuis L. et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. Journal of Neuroscience. 2007;27:5535-45
144. Wu S, Zheng SD, Huang HL, Yan LC, Yin XF, Xu HN. et al. Lithium down-regulates histone deacetylase 1 (HDAC1) and induces degradation of mutant huntingtin. The Journal of biological chemistry. 2013;288:35500-10
145. Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6-21
146. Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ. et al. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2354-9
147. Lauterbach EC. Neuroprotective effects of psychotropic drugs in Huntington's disease. International journal of molecular sciences. 2013;14:22558-603
148. Ozaki N, Chuang DM. Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. Journal of neurochemistry. 1997;69:2336-44
149. Yuan PX, Chen G, Huang LD, Manji HK. Lithium stimulates gene expression through the AP-1 transcription factor pathway. Brain research Molecular brain research. 1998;58:225-30
150. Anden NE, Dalen P, Johansson B. Baclofen and lithium in Huntington's chorea. Lancet. 1973;2:93
151. Dalen P. Lithium therapy in Huntington's chorea and tardive dyskinesia. Lancet. 1973;1:107-8
152. Manyam NV. Serotonin, lithium, and Huntington's chorea. Lancet. 1973;2:331
153. Manyam NV, Bravo-Fernandez E. Lithium carbonate in Huntington's chorea. Lancet. 1973;1:1010
154. Mattsson B. Huntington's chorea and lithium therapy. Lancet. 1973;1:718-9
155. Mattsson B, Persson SA. Huntington's chorea, lithium, and G.A.B.A. Lancet. 1973;2:684
156. Aminoff MJ, Marshall J. Treatment of Huntington's chorea with lithium carbonate. A double-blind trial. Lancet. 1974;1:107-9
157. Leonard DP, Kidson MA, Brown JG, Shannon PJ, Taryan S. A double blind trial of lithium carbonate and haloperidol in Huntington's chorea. The Australian and New Zealand journal of psychiatry. 1975;9:115-8
158. Vestergaard P, Baastrup PC, Petersson H. Lithium treatment of Huntington's chorea. A placebo-controlled clinical trial. Acta psychiatrica Scandinavica. 1977;56:183-8
159. Symington GR, Leonard DP, Shannon PJ, Vajda FJ. Sodium valproate in Huntington's disease. The American journal of psychiatry. 1978;135:352-4
160. Puca FM, Genco S, Minervini MG, Specchio LM, Napoletano V, Brancasi B. et al. [Sleep in chronic chorea patients after therapy with sodium valproate]. Bollettino della Societa italiana di biologia sperimentale. 1984;60:989-92
161. Danivas V, Moily NS, Thimmaiah R, Muralidharan K, Purushotham M, Muthane U. et al. Off label use of lithium in the treatment of Huntington's disease: A case series. Indian journal of psychiatry. 2013;55:81-3
162. Raja M, Soleti F, Bentivoglio AR. Lithium treatment in patients with huntington disease and suicidal behavior. Journal of clinical psychopharmacology. 2013;33:819-21
163. Tremolizzo L, Rodriguez-Menendez V, DiFrancesco JC, Sala G, Galbussera A, Appollonio I. et al. Huntington's disease and HDACi: would sulpiride and valproate be of therapeutic value? Medical hypotheses. 2007;69:964-5
164. Grove VE Jr, Quintanilla J, DeVaney GT. Improvement of Huntington's disease with olanzapine and valproate. The New England journal of medicine. 2000;343:973-4
165. Saft C, Lauter T, Kraus PH, Przuntek H, Andrich JE. Dose-dependent improvement of myoclonic hyperkinesia due to Valproic acid in eight Huntington's Disease patients: a case series. BMC neurology. 2006;6:11
166. Lenman JA, Ferguson IT, Fleming AM, Herzberg M, Robb JE, Turnbull MJ. Sodium valproate in chorea. British medical journal. 1976;2:1107-8
167. Pearce I, Heathfield KW, Pearce MJ. Valproate sodium in Huntington chorea. Archives of neurology. 1977;34:308-9
168. Tan BK, Leijnse-Ybema HJ, Zee MF. Sodium valproate in Huntington's chorea. Clinical neurology and neurosurgery. 1976;79:62-5
169. DeVane CL. Pharmacokinetics, drug interactions, and tolerability of valproate. Psychopharmacology bulletin. 2003;37(Suppl 2):25-42
170. Vorhees CV. Teratogenicity and developmental toxicity of valproic acid in rats. Teratology. 1987;35:195-202
171. Information for Healthcare Professionals: Risk of Neural Tube Birth Defects following prenatal exposure to Valproate. US Food and Drug Administration. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm192649.htm
172. Salazar Z, Tschopp L, Calandra C, Micheli F. Pisa syndrome and parkinsonism secondary to valproic acid in Huntington's disease. Movement disorders: official journal of the Movement Disorder Society. 2008;23:2430-1
Author contact
![]() Corresponding author: De-Maw Chuang, Ph.D. Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, Building 10, Room 3D38, 10 Center Drive, MSC 1363, Bethesda, MD 20892-1363, USA. Tel: 301-496-4915 Fax: 301-480-9290 E-mail: chuangnih.gov.
Corresponding author: De-Maw Chuang, Ph.D. Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, Building 10, Room 3D38, 10 Center Drive, MSC 1363, Bethesda, MD 20892-1363, USA. Tel: 301-496-4915 Fax: 301-480-9290 E-mail: chuangnih.gov.