Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(9):1051-1063. doi:10.7150/ijbs.9438 This issue Cite
Research Paper
Transcriptome Characterization Analysis of Bactrocera minax and New Insights into Its Pupal Diapause Development with Gene Expression Analysis
Yongcheng Dong1, Nicolas Desneux2, Chaoliang Lei1 ![]() , Changying Niu1
, Changying Niu1 ![]()
1. Hubei Insect Resources Utilization and Sustainable Pest Management Key Laboratory, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, 430070, China.
2. French National Institute for Agricultural Research (INRA), UMR1355-ISA, 400 Route des Chappes, 06903, Sophia-Antipolis, France.
Received 2014-4-18; Accepted 2014-8-15; Published 2014-9-12
Abstract
Bactrocera minax is a major citrus pest distributed in China, Bhutan and India. The long pupal diapause duration of this fly is a major bottleneck for artificial rearing and underlying mechanisms remain unknown. Genetic information on B. minax transcriptome and gene expression profiles are needed to understand its pupal diapause. High-throughput RNA-seq technology was used to characterize the B. minax transcriptome and to identify differentially expressed genes during pupal diapause development. A total number of 52,519,948 reads were generated and assembled into 47,217 unigenes. 26,843 unigenes matched to proteins in the NCBI database using the BLAST search. Four digital gene expression (DGE) libraries were constructed for pupae at early diapause, late diapause, post-diapause and diapause terminated developmental status. 4,355 unigenes showing the differences expressed across four libraries revealed major shifts in cellular functions of cell proliferation, protein processing and export, metabolism and stress response in pupal diapause. When diapause was terminated by 20-hydroxyecdysone (20E), many genes involved in ribosome and metabolism were differentially expressed which may mediate diapause transition. The gene sets involved in protein and energy metabolisms varied throughout early-, late- and post-diapause. A total of 15 genes were selected to verify the DGE results through quantitative real-time PCR (qRT-PCR); qRT-PCR expression levels strongly correlated with the DGE data. The results provided the extensive sequence resources available for B. minax and increased our knowledge on its pupal diapause development and they shed new light on the possible mechanisms involved in pupal diapause in this species.
Keywords: Bactrocera minax, pupal diapause
Background
Dormancy occurs widely in organisms in order to deal with harsh environments, including flat worms, crustaceans, rotifers, tardigrades, insects, killifish and mammals, which is a status of developmental metabolic depression [1]. It is not a static but dynamic process consisting of several successive phases, including a diapause induction and preparatory period, initiation and maintenance of developmental arrest as well as termination and transition into active development [2]. Regulatory mechanisms responsible for diapause entry and termination are decisive in dodging stressful conditions and seasonal fruiting synchronization [3]. In recent years, interest has been increasingly focusing on comparative studies with regard to global differences in transcript abundance across diapausing-, nondiapausing- and diapause-terminated individuals within facultative [4-7] and obligatory diapause species [8]. Several important components including endocrine regulation, clock mechanism, metabolism depression and stress tolerance were involved in programmed diapause development on insects [8-12]. Studies on molecular regulation events of diapause facilitate better understanding of their physiological responses to physical environmental cues, as well as elusive seasonal adaption.
Winter in temperate zones imposes significant environmental stress on arthropods [13, 14], even challenging Chinese citrus fly Bactrocera minax (Enderlein), a univoltine citrus pest distributed in Asia temperate areas, including China, north-west India and Bhutan [15, 16]. B. minax, like many other species [17-19], mitigates the stresses due to unfavourable seasons and synchronizes with favourable seasons by means of diapause [16, 20, 21]. Within the Tephritidae family, pupal diapause is common in other univoltine insect species in response to harsh environment e.g., Rhagoletis pomonella [22], Rhagoletis cerasi [23], Rhagoletis mendax [24] and Dacus tsuneonis [25]. In most overwintering diapause species, diapause terminates in midwinter rather than in spring [26]. Most diapausing B. minax individuals in the field end their diapause and enter a phase of “quiescence” due to the prolonged winter [20]. The mechanisms governing diapause transitions diverge in univoltine tephritids that are characterized by varying diapause intensity in different ontogenetic trajectories [22, 27, 28]. The diapause developmental transition controlling system manages harnessed nutrient reserves and schedules when diapause will end, as well as complete post-diapause challenges and metamorphosis [29]. However, its underlying molecular mechanism remains poorly understood.
Although B. minax is a biologically interesting and economically important pest, genomic sequence information has not been widely documented. Up to April 18th, 2014, there have been about 20,255 Nucleotide sequences including 186 ESTs deposited in NCBI (http://www.ncbi.nlm.nih.gov/) for Bactrocera genus. The lack of genomic resources still remains a barrier for further study of this fly species. Next-generation DNA sequencing technologies are currently dramatically improving the efficiency of genetic and biological research [30], and will facilitate the study of important biological processes by means of mass expression profile analyses for non-model tephritids that lack genome database [31-33].
In the present study, we have performed de novo transcriptome analysis using short read sequencing technology. A library pool containing all of the developmental stages, i.e., eggs, larvae, pupae and adult flies, was established and sequenced. More than 52 million reads with 4.7 billion nucleotides (nt) were assembled into 47,217 unigenes. Using the transcriptome background, we selected and analysed pupae collected in November (early diapause, ED), January (late diapause, LD), April (post-diapause, PD) and diapause pupae treated with 20-hydroxyecdysone (diapause terminated, DT) for gene expression to evaluate global differences in transcript abundance across different stages. Large amounts of differentially expressed gene sets in the six pairwise comparisons of samples were analysed. Here we have presented comprehensive transcriptome and digital gene expression (DGE) expression profile data that provide sequences of 47,217 unigenes and yield novel insight into the molecular regulation mechanisms on pupal diapause development of an economically important agricultural pest, the Chinese citrus fly.
Methods
Preparation of materials
To obtain an overview of B. minax genetic data, a mixed cDNA sample from different developmental stages was prepared and sequenced using the Illumina sequencing platform, including eggs, larvae, pupae and adults (Additional File 1: Table S1). Samples were field-collected periodically at Sandouping (Latitude: 30.821, Longitude: 111.051) in Yichang, Hubei province, China. Adults were reared on sucrose and brewer's yeast at ~25ºC with a long photoperiod of 14L:10D [34]. As opposed to the facultative diapause species (e.g. S. crassipalpis) [35], pupal diapause in B. minax is obligatory and cannot be terminated if directly exposed to high temperature or if organic chemicals are topically applied [20]. However, diapause individuals can be rescued from developmental arrest with a topical application of 20-hydroxyecdysone, an endocrine trigger known to terminate insect diapause [36], and discriminated from diapause destined individuals by comparing the metabolic rate using a flow-through respirometry system with Expadata data logging software (Sable Systems International Inc. USA) [22]. All the samples were snap frozen in liquid nitrogen and immersed in RNAlater RNA Stabilization Reagent (Qiagen, Hilden) at -80ºC for RNA extraction.
RNA isolation and library preparation for RNA-seq
Each sample was ground in liquid nitrogen and total RNA was isolated using RNeasy RNA Purification Kit (Qiagen, Hilden) according to the manufacturer's protocol. To obtain complete gene expression information, a pooled RNA sample was prepared for transcriptome analysis. cDNA synthesis and libraries construction was performed following Illumina manufacturer's recommendations. In brief, poly (A)+ RNA was purified from 20µg total RNA using oligo (dT) magnetic beads and fragmented in presence of divalent cations under elevated temperature. Those fragmented poly (A)+ RNA were transcribed using random hexamer primers and this was followed by second strand cDNA synthesis using Rnase H and DNA polymerase I. The small fragments were purified using QiaQuick PCR Purification Kit. After that the purified fragments were washed with ethidium bromide buffer for the end-repair, poly (A)+ addition and ligation of sequencing adaptors. Suitable fragments (~200bp) were selected using agarose gel electrophoresis and amplified by PCR to construct a cDNA library.
Illumina sequencing and bioinformatics analysis
The cDNA library was sequenced on Illumina HiSeqTM 2000 using paired-end technology in a single run (2×91bp). The raw reads from the images were generated by Illumina GA Pipeline v 1.6. After filtering raw reads, transcriptome de novo assembly was performed with the transcriptome assembly software Trinity [37]. After assembly, we then aligned those unigenes to database NR, Swiss-Prot, KEGG, and COG using BLASTX (E-value < 0.00001). We performed GO annotation using Blast2GO program, a universal tool for annotation and analysis in functional genomics research [38]. To further study genes' biological functions, KEGG annotation was also conducted [39].
DGE library preparation and sequencing
Total RNA was extracted from four types of pupae including ED, LD, PD and DT using RNeasy RNA Purification Kit (Qiagen, Hilden) following the manufacturer's protocol (Figure 1). DGE libraries were prepared using Illumina Gene Expression Sample Prep Kit. Briefly, poly (A)+ RNA was purified from 6µg total RNA using oligo (dT) magnetic beads. The bound poly (A)+ RNA served as templates and double-stranded cDNA synthesis ensued on the beads. The products were digested with the restriction enzyme NlaIII that cleaves at the CTAG site and the fragments containing 3' ends were deposited by the magnetic beads. The first Illumina adaptor containing the recognition site of MmeI, a type II endonuclease that specially cleaves at 21 bp from CTAG site, was ligated to those cDNA containing 5' ends. As a result, a large number of 21 bp tags were produced after MmeI enzyme digestion and the second Illumina adaptor ligation to the 3' ends. The libraries containing 21 bp tags were linearly amplified in presence of adaptor primers [40] by PCR for 15 cycles. The PCR products were purified by 6% TBE PAGE gel electrophoresis and single-stranded products were added to flowcell for sequencing by synthesis on Illumina Cluster Station and Illumina HiSeqTM 2000.
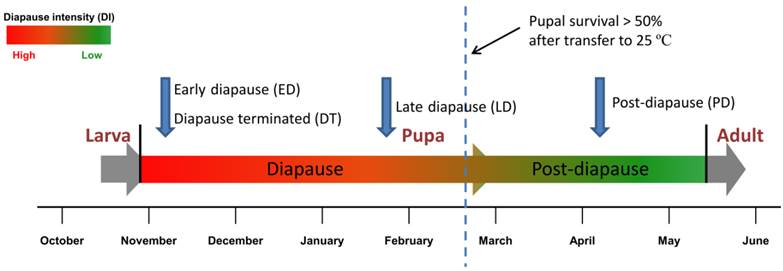
Schema of sampling for digital gene expression analysis. Four DGE libraries, including early diapause pupae (ED), late diapause pupae(LD) and post-diapause pupae (PD) and diapause pupae terminated by 20-Hydroxyecdysone (DT) were selected for digital gene expression analysis during pupal stage. Pupal survival rate under constant 25 ºC temperature was set as an indicator of diapause intensity [20].
Analysis and mapping of DGE tags
The tags were obtained after removing 3' adaptor sequence, empty tags (no tag sequence detected except 3' adaptor), low quality tags (tag sequence containing unknown nucleotide 'N'), and over-long/short tags and tags with only one copy number (perhaps generated by sequencing errors) from the raw data. All tags with CTAG and 17 bp tag sequence were mapped to our transcriptome reference database with no nucleotide mismatch. The unambiguous tags were annotated and calculated for the number of copies which were normalized to TPM (Transcript Per Million tags) following the normalization methods in AC't Hoen P et al. [41].
Evaluation of DGE libraries
The frequency of each DGE library was handled by means of statistics following the rigorous algorithm represented in Audic et al. to identify the differentially expressed genes [42].The false discovery rate (FDR) was used to determine the threshold of P value in multiple tests by manipulating the FDR value. Gene expressions were considered significantly different with a cut-off value FDR < 0.001 and |log2 ratio| > 1. GO and KO enrichment analyses were performed according to a method described by Xue et al [40]. In the GO/KO enrichment analyses, all the P values were rectified by Bonferroni. The corrected P value < 0.05 was set as a threshold to determine significantly enriched GO/KO terms. The cluster analysis was performed using cluster [43] and Java Tree view [44].
Validation of gene expression by qRT-PCR
Total RNA of four types of pupae (ED, LD, PD and DT) were extracted. The concentration of RNA samples were determined using NanoDrop and diluted to 1 µg/µl with Rnase free water. Thereafter, 2µg of total RNA was used for reverse transcription using PrimeScript RT reagent Kit (Takara, Dalian). The specific primers of selected genes were listed in Additional File 2: Table S2. The alpha-tubulin gene was used as an internal gene. qRT-PCR was performed using SYBR Premix EX Tag Kit (Takara, Dalian) following the manufacturer's protocol on a 7300 real-time PCR system (Applied Biosystems, USA). A relative quantitative method (△△Ct) was used to evaluate the relative gene expression data. All qRT-PCR were performed in three biological and three technical replications.
Results
Illumina sequencing and data processing
A total number of 58,334,980 raw reads was generated from the cDNA library (Table 1). The raw trancriptome data were deposited in the GenBank Sequence Read Archive (Accession: SRR1238396). After filtering raw reads, a total of 52,519,948 reads were assembled into 100,498 contigs with an average length of 363 bp. The contigs were then assembled into 47,217 unigenes including 8,981 distinct clusters and 38,236 distinct singletons after paired-end reads and realignment which showed an average length of 815 bp. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GBIT00000000. The version described in this paper is the first version, GBIT01000000. To check the quality of sequencing data, we selected 14 unigenes and amplified their partial sequences by PCR using 14 pairs of primers. The PCR products were consistent between RNA-Seq data and Sanger sequencing (Additional File 3: Table S3).
Summary statistics of transcriptome of Bactrocera minax.
| Summary statistics | |
|---|---|
| Total raw reads | 58,334,980 |
| Total reads | 52,519,948 |
| Total nucleotides (nt) | 4,726,795,320 |
| Q20 percentage | 97.43% |
| GC percentage | 43.26% |
| Total number of contigs/ unigenes | 100,498/47,217 |
| Mean length of contigs/ unigenes | 363/815 |
| N50 of contigs/unigenes | 708/1,422 |
| Distinct clusters/ singletons | 8,981/38,236 |
| Sequences with E-value < 10-5 against NR/NT database | 26,843/16,368 |
Function Annotation and classification of assembled transcripts
For the unigene annotation, we aligned the transcript sequences using BLASTX against the non-redundant (nr) NCBI protein database with a cut-off E-value of 10-5. A total of 26,843 (56.85% of all unigenes) sequences provided a BLASTX result (Additional File 4: Table S4). The E-value, similarity and species distribution of the best hits in NR database are shown in Additional File 13: Figure S1. More than 50% mapped sequences have strong homology with E-value less than 1.0×10-50 whereas the remaining homological sequences ranged from 1.0×10-5 to 1.0×10-50 (Figure S1A). The percentage of high similarity sequences i.e., higher than 80% is 18.39% while the other matched sequences (81.61%) showed a similarity ranging from 17% to 80% (Figure S1B). Most matched sequences (60.06%) showed strong homology with Drosophila (Figure S1C).
We performed functional annotations of gene ontology (GO) and of clusters of orthologous groups (COG) to classify the potential functions of the predicted genes. Among 47,217 unigenes, approximately 16.51% and 22.84% of the unigenes were categorized into 50 functional GO groups and 25 COG categories, respectively (Additional File 13: Figure S2). In each of the three main categories of GO classification, i.e., molecular function, biological process and cellular component, the terms “binding and catalytic activity”, “cellular process and metabolic process” and “cell and cell part” were dominant, respectively (Figure S2A, Additional File 5: Table S5). For COG classification, the cluster of 'general function prediction only' category stood for the largest group (3,792, 35.16%) followed by 'Transcription' (1,782, 16.52%), 'Carbohydrate transport and metabolism' (1,774, 16.45%) and 'Translation, ribosomal structure and biogenesis' (1,754, 16.26%) (Figure S2B).
Unigene metabolic pathway analysis
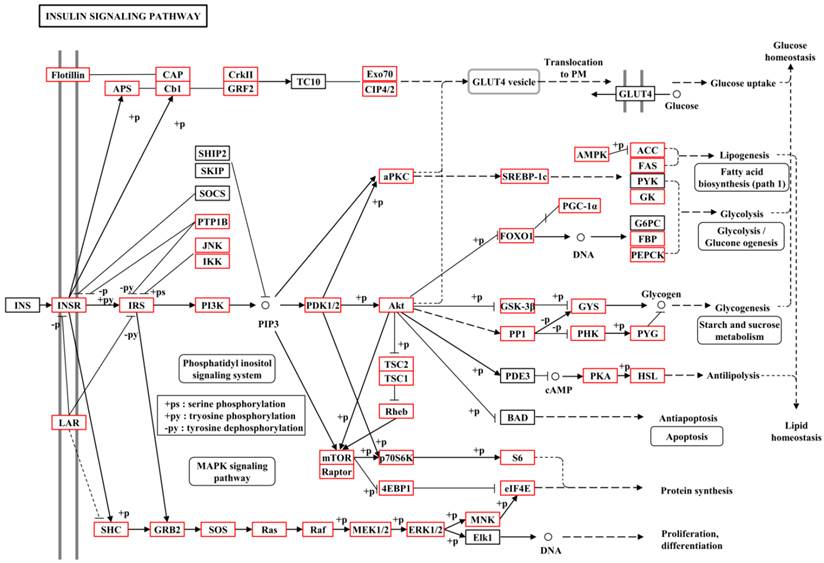
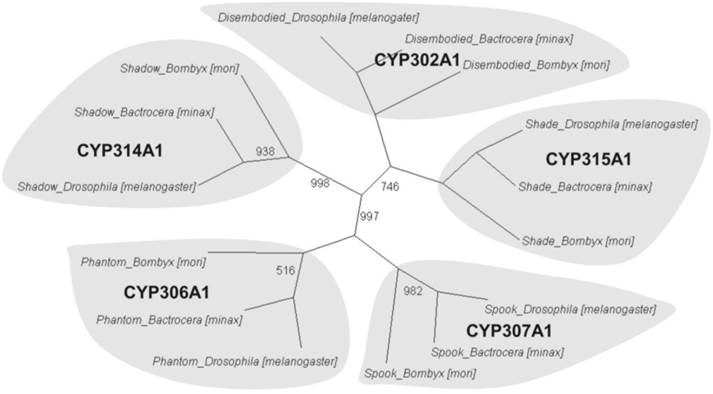
To further understand the gene functions on the biological pathways involved, the assembled unigenes were subjected to the canonical pathways referenced in the Kyoto Encyclopedia of Genes and Genomes (KEGG) for pathway analysis. Based on the comparison with the KEGG database using BLASTX with an E-value cut-off of <10-5, a total of 18,711 unigenes with pathway annotation were assembled into 240 KEGG pathways. The majority of unigenes were clustered in the 'Metabolic pathways' (2,929, 15.65%) followed by 'Pathways in cancer' (746, 3.99%) and 'Regulation of actin cytoskeleton' (666, 3.56%) (Additional File 6: Table S6). Several unigenes identified belonged to prothoracicotropic hormone (PTTH) receptor signalling transduction [45] and steroid hormone biosynthesis pathways (Table 2) [46], as well as circadian rhythm [47]. Some unigenes were identified in the insulin signalling pathway which is a conserved pathway in insect diapause controlling (Figure 2) [48]. The putative identified genes required for ecdysteroidogenesis, i.e., 'Halloween' genes and Neverland, were tabulated in Table 3, which encode the enzymes needed to convert cholesterol to 20E. Halloween P450 genes were reported to be structurally and functionally conserved through insect evolution [49]. These unigenes from B. minax were observed to share descent with Drosophila melanogaster and Bombyx mori by orthologous relationship analysis based on protein sequences alignment (Figure 3). These genes involved in PTTH and steroid hormone biosynthesis provide broad perspectives in the insect diapause [3, 36, 50, 51].
Unigenes annotated to prothoracicotropic hormone (PTTH) receptor signalling transduction and steroid hormone biosynthesis pathways.
| Pathway | All unigenes with pathway annotation | Pathway ID |
|---|---|---|
| MAPK signalling pathway | 403 | Ko04010 |
| Wnt signalling pathway | 382 | Ko04310 |
| MAPK signalling pathway - fly | 120 | Ko04013 |
| mTOR signalling pathway | 155 | Ko04150 |
| Calcium signalling pathway | 334 | Ko04020 |
| Steroid biosynthesis | 47 | Ko00100 |
| Steroid hormone biosynthesis | 70 | Ko00140 |
| Terpenoid backbone biosynthesis | 40 | Ko00900 |
| Insect hormone biosynthesis | 55 | Ko00981 |
| Insulin signalling pathway | 348 | Ko04910 |
Metabolic pathway of insulin signalling pathway for unigenes by KEGG annotation.
The genes required for ecdysteroidogenesis identified in insect molting hormone synthesis pathway.
| Gene symbol | Unigene ID | Length (bp) | Accession | Evalue | Species |
|---|---|---|---|---|---|
| Neverland | Unigene21710 | 560 | XP_002070225.1 | 5.00E-46 | Drosophila willistoni |
| Spook | Unigene20374 | 594 | XP_001353744.2 | 7.00E-74 | Drosophila pseudoobscura |
| Phantom | Unigene18345 | 688 | XP_002071819.1 | 2.00E-53 | Drosophila willistoni |
| Disembodied | Unigene7801 | 673 | XP_002047767.1 | 4.00E-61 | Drosophila virilis |
| Shadow | Unigene19056 | 297 | AEO53064.1 | 3.00E-34 | Bactrocera dorsalis |
| Shade | CL2279.Contig1 | 1980 | XP_001956409.1 | 0 | Drosophila ananassae |
Phylogenetic orthologous relationship of putative Halloween genes from Bactrocera minax with Drosophila melanogaster and Bombyx mori. A neighbour-joining tree was constructed based on P450 protein sequences encoded by Halloween genes of B. minax, D. melanogaster, and B. mori. The complete sequences of D. melanogaster and B. mori were provided in KF Rewitz, MB O'Connor and LI Gilbert [57]. Bootstrap values less than 1000 (for 1000 bootstrap trails) were shown.
Digital gene expression (DGE) libraries sequencing
Four DGE libraries were sequenced and generated approximately 6 million raw tags in each library. Four raw DGE data (ED, LD, PD and DT) were deposited in GenBank Sequence Read Archive (Accession: SRR1238397). After removing low quantity tags, the number of tags for each sample ranged from 5.78 to 6.02 million (Additional File 13: Figure S3 and Additional File 7: Table S7). Among those tags, the total number of all tags mapped to genes distributed from 4.87 million to 5.34 million, and the proportion of these tags ranged from 83.27% to 88.60% in libraries. The distribution of total tags and distinct tags with different abundance categories, i.e., different count numbers, were analysed to evaluate DGE data (Additional File 13: Figure S4). The proportion of high-expression tags (with count numbers > 100) exceeded 80% in total tags whereas their percentage in all distinct tags amounted to approximately 6%. By contrast, those low-expression tags (with count numbers < 5) represented a dominant ratio (no less than 53%) in all distinct tags in spite of the fact that they showed a smaller proportion in total tags (less than 3%).
Gene expression variations among different developmental stages
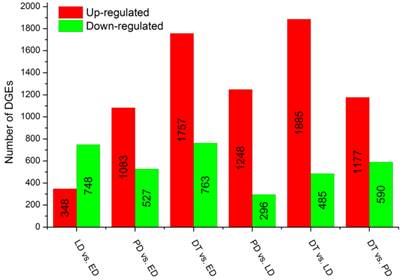
To identify gene expression during pupal diapause developmental stages, the number of tags for each gene was calculated and the differentially expressed genes were identified using an algorithm developed by S Audic and JM Claverie [42]. A total of 15,096 genes were expressed differentially in six pairwise comparisons: DT vs ED, PD vs ED, LD vs ED, DT vs PD, DT vs LT and PD vs LD (Additional File 13: Figure S5). Among these genes, 28.85% (4,355) had significant differences in transcriptional expression level across libraries filtered by the absolute value of log2Ratio ≥ 1 based on the FDR < 0.001 (Figure 4, Additional File 8: Table S8). It was shown that global changes in transcript abundance firstly decreased and then increased with pupal diapause development. However, more differentially expressed transcripts were found in pupae treated by 20-hydroxyecdysone (DT) compared to pupae in diapause status (ED, LD and PD).
Changes in gene expression profile in comparison of different types of pupae. The numbers of upregulated and downregulated genes in different comparisons are summarized. ED: early diapause; LD: late diapause; PD: post-diapause; DT: diapause terminated.
Functional analysis of differentially expressed genes
To fully understand the functions of DGEs involved in pupal diapause development, all the DGEs were mapped to the whole transcriptome background and given GO/KO annotations. GO/KO enrichment analysis results in each comparison were summarized as P value < 0.05 (LD/ED; PD/ED; DT/ED; PD/LD; DT/LD and DT/PD in Additional File 9: Table S9, Additional File 10: Table S10). To simplify the significantly enriched KEGG pathways among comparisons, we concluded KEGG gene sets by limiting corrected P value < 0.01 and removing pathways reported in connection to human diseases (Table 4). GO and KO annotations suggested that transcripts in DT were enriched in stress response, cellular structure, metabolic process and cell proliferation. Metabolic process including protein biosynthesis, endocrine biosynthesis and carbohydrate metabolism was activated for individual development after diapause terminated.
Enrichment of gene set representing KEGG biochemical pathways among different comparisons.
| Comparison | KEGG gene set | Comparison | KEGG gene set |
|---|---|---|---|
| DT vs ED | Ribosome Oxidative phosphorylation Metabolic pathways Proteasome Antigen processing and presentation Protein processing in endoplasmic reticulum Primary bile acid biosynthesis * Steroid biosynthesis * Glyoxylate and dicarboxylate metabolism * Renin-angiotensin system * Phototransduction - fly * Phagosome * Cardiac muscle contraction * Linoleic acid metabolism * Valine, leucine and isoleucine degradation * Cysteine and methionine metabolism * | DT vs LD | Ribosome Metabolic pathways Protein processing in endoplasmic reticulum Phagosome Antigen processing and presentation Proteasome Steroid biosynthesis Lysosome * Collecting duct acid secretion * Oxidative phosphorylation * Butanoate metabolism * Phototransduction - fly * Focal adhesion * Arachidonic acid metabolism * Fructose and mannose metabolism * ECM-receptor interaction * Glyoxylate and dicarboxylate metabolism * |
| DT vs PD | Ribosome Protein processing in endoplasmic reticulum Metabolic pathways Antigen processing and presentation Complement and coagulation cascades Cysteine and methionine metabolism * Glycolysis/Gluconeogenesis * Proteasome * Phagosome * Steroid biosynthesis * Oxidative phosphorylation * Butanoate metabolism * Primary bile acid biosynthesis * Collecting duct acid secretion * Glyoxylate and dicarboxylate metabolism * Protein digestion and absorption * Pentose phosphate pathway * Linoleic acid metabolism * Spliceosome * Valine, leucine and isoleucine degradation * Phototransduction - fly * | PD vs ED | Renin-angiotensin system Protein processing in endoplasmic reticulum * ECM-receptor interaction * Protein export * Complement and coagulation cascades * Tyrosine metabolism * |
| PD vs LD | ECM-receptor interaction Phagosome Protein export Complement and coagulation cascades Riboflavin metabolism * Protein processing in endoplasmic reticulum * | ||
| LD vs ED | Proteasome * ECM-receptor interaction * Oxidative phosphorylation * Galactose metabolism * |
All P-values<0.001 except for those with an asterisk (*P<0.01)
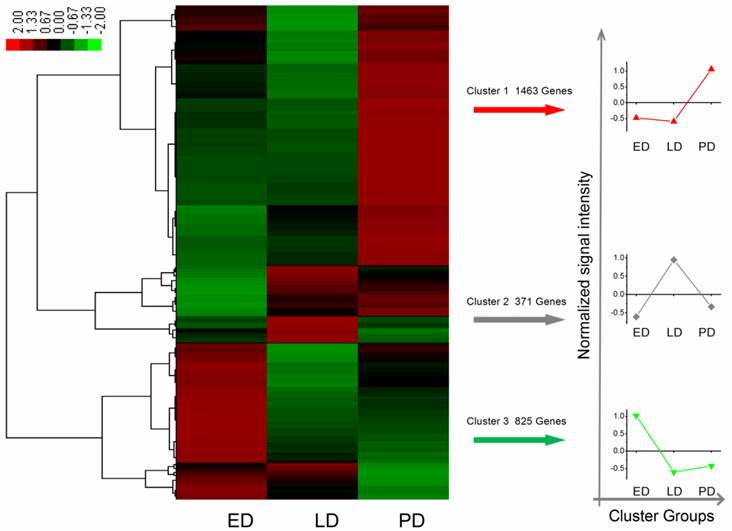
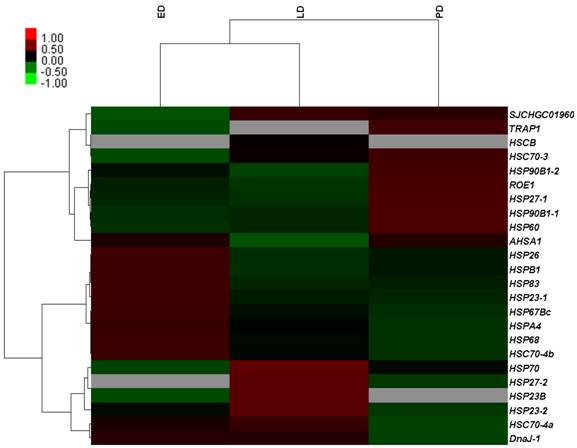
A total of 12,713 genes were detected during B. minax pupal diapause development, i.e., ED, LD and PD. Among these, 2,659 differentially expressed genes were filtered according to the absolute value of log2Ratio ≥ 1 based on the FDR < 0.001 (Additional File 11: Table S11). These genes were divided into three groups using Genesis based on K-means method (Figure 5) [52]. Apart from few stress-associated transcripts, e.g., binding, structure molecular activity, molecular transducer activity and enzyme regulator activity, over-expressed in late diapause clustered together, clusters 1 and 3 represented two reverse expression patterns for catalytic, transporter, transcription regulator, translation regulator and antioxidant activities, which related to diapause entry and preparation to resume development. Moreover, 24 unigenes classified as heat shock proteins (HSPs, annotated in Additional File 12: Table S12) contributed to cold hardiness and dehydration tolerance which showed diverse responsive expression patterns to diapause (Figure 6).
Differential expression genes were filtered with the absolute value of log2Ratio ≥ 1 based on the FDR < 0.001 among ED, LD and PD. Each column stands for an experimental sample i.e., ED, LD, and PD for early, late, and post-diapause respectively, and each row represents a gene. Red stands for high expression while green stands for low expression.
Differential expression genes of identified heat shock proteins related to diapause development in Bactrocera minax early, late, and post-diapause stage. Each column stands for an experimental sample i.e., ED, LD, and PD for early, late, and post-diapause respectively, and each row represents a gene. Red stands for high expression while green stands for low expression (Grey denotes undetected expression).
Validation of gene expression profile
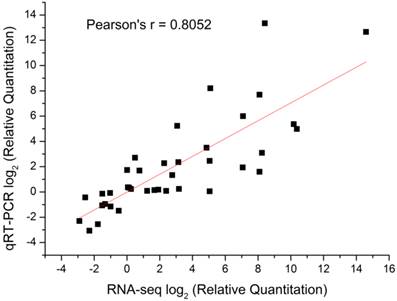
To validate the gene expression profiles, 15 genes were examined using qRT-PCR. The results of qRT-PCR expression strongly correlated with the DGE data (Pearson's r correlation coefficient > 0.80, Figure 7). Therefore, DGE profiling can be used as a tool to investigate specific diapause associated genes which show comparative expression levels among the different stages of pupae.
Correlation analysis of qRT-PCR results and digital gene expression data for selected genes among different developmental stages
Discussion
Transcriptome data analysis and genes identified in diapause related pathway
From Illumina sequencing, we obtained 58,334,980 reads and assembled into 47,217 unigenes in B. minax. Among them, 56.85% (26,843) unigenes were found homologous to functional genes encoding specific proteins in GenBank. As a member of Diptera, B. minax showed a close relationship with Drosophila, a model Diptera species whose genomic data is well documented [53]. A similar distribution was found in another Tephritidae fruit fly B. dorsalis [31]. From our research results, only 11 unigene sequences were annotated with recorded sequences of B. minax in GenBank, which suggested that the transcriptome sequences provided a meaningful contribution to genetic data on B. minax.
Many genes linked to insect diapause regulation were identified in reported pathways. Crosstalk between insulin signalling and FOXO i.e., forkhead transcription factor, mediated the diapause response in Diptera and Lepidoptera [7, 48, 54]. Wnt and TOR (target of rapamycin) signalling were considered as candidates for early regulation of diapause termination in the apple maggot [8]. It is well known that induction and termination of diapause are controlled by endocrine hormones secreted by endocrine glands and mediated by environment stimuli [36]. Endocrine signalling and hormone biosynthesis cascades, including PTTH receptor signalling transduction [6, 50, 51, 55] and ecdysone biosynthesis [49, 56], are closely linked to insect diapause termination. Halloween gene family, encoding cytochrome P450 enzymes that mediated the biosynthesis of 20E from cholesterol, have been structurally and functionally conserved for 400 million years throughout insect evolution [49]. Spook (CYP307A1), a rate-limiting enzyme for 20E biosynthesis [45], is required for 20E biosynthesis in Drosophila melanogaster [57], which is involved in step catalysing 7-dehydrocholesterol to diketol and was named the “Black Box”. Moreover, these sequences classified in ERK/MAPK pathway cascades also provided an insightful perspective on signal transduction for further gene mining [58-60]. Therefore, de novo transcriptome sequencing provided comprehensive genetic data that substantially improved the genomic background for this Tephritid fly species.
Different genes expressed in DT compared to ED, LD and PD
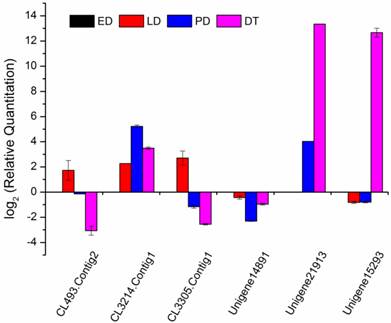
Compared to the different diapause status (ED, LD and PD), diapause terminated (DT) individuals shifted largely to aerobic metabolism which mainly focused on protein processing, carbohydrate and lipid metabolism favouring the developmental resumption. Numerous genes were selectively upregulated and downregulated in global transcript expression level (Figure 4). The GO/KO enrichment analysis revealed that large-scale gene sets were enhanced in metabolism and oxidative phosphorylation pathways in transcript abundance of DT compared to the other landmarks, i.e., ED, LD and PD (Table S9, Table S10). The KO enrichment in DT vs ED indicated that pupae treated with 20-hydroxyecdysone significantly clustered into amino acid and carbohydrate metabolism, as well as lipid metabolism (Table 4, Table S10). This points to large scale protein remodelling and energy preparation for the resumption of development releasing from metabolism depression. Linoleic acid, which is necessary for emergence and normal formation of the scales on the wings [61], was upregulated in its biosynthesis pathway in DT vs ED/LD (Table 4, Table S10). Four top pathways were identical when comparing DT to ED/LD/PD with P value < 0.001, “Ribosome”, “Protein processing in endoplasmic reticulum”, “Metabolic pathways”, “Antigen processing and presentation”. Ribosome which served as the primary site of biological synthesis with regard to protein rearrangement and enzyme biosynthesis was involved in the top enriched pathway when diapause terminated. The sequences highly enriched in ribosome showed its significant role in diapause transition when diapause terminated, despite the fact it appeared to slightly lag after upstream regulatory genes expression. These genes especially expressed in DT were presumably associated with diapause switching mechanism and not solely for metabolic machinery. The phosphorylation of the ribosomal protein S6 can result in increased ecdysone production [46]. RpS3a (Unigene15293) coding ribosomal protein S3a was found diapause-specifically expressed only in DT and verified by qRT-PCR (Figure 8), which showed a consistent expression pattern in C. pipiens diapause individuals [62]. Phenylalanine and tyrosine, the crucial aromatic amino acids used to produce the new adult cuticle that will form at the onset of adult morphogenesis [8], were significantly upregulated in biosynthesis in DT vs ED/LD. Serine/threonine-specific protein kinase (Akt, Unigene21913), which is one of the major upstream genes in insulin signalling pathway associated with multiple cellular processes such as cell growth [8], was specifically upregulated in the high expression level in DT involved in controlling cell proliferation (Figure 8). It was surprising that the 20-hydroxyecdysone application positively stimulated hormone precursor biosynthesis such as “steroid biosynthesis” and “primary bile acid biosynthesis”. Further testing is required to determine whether exogenous 20-hydroxyecdysone application acts on the brain in the similar way tricarboxylic acid (TCA) intermediates mediated hormonal control [50] or just replenishes the deficiency of ecdysteroids [63] to interrupt diapause.
qRT-PCR analysis for selected genes among different developmental stages. Gene relative expression levels are calculated by real-time PCR using alpha-tubulin as the standard and are shown on ordinate axis. Early diapause, ED; late diapause, LD; post-diapause, PD; diapause terminated, DT.
Gene expression pattern in ED-LD-PD
Various stress inducible sequences, coding heat shock protein chaperones, were detected during diapause development. Through diapause development and termination in the natural environment, B. minax was strongly influenced by low temperature [20]. HSPs are a group of highly conserved molecules that play vital roles in insect diapause subjected to cold stress [10, 64]. In Rhagoletis pomonella, HSPs specifically expressed during pupal diapause acted a protectant against low temperature injury [65]. In our study, several putative identified HSPs exhibited a diverse expression pattern through the ED-LD-PD developmental axis (Figure 6), which was closely linked to concurrent typical physiological characteristics such as elevated stress tolerance and suppressed metabolism. Two HSPs Unigene14891 and CL493.Contig2 expressions were verified by qRT-PCR (Figure 8). A similar finding reported that the hardiness of the onion maggot Delia antiqua to cold was enhanced by HSP60 [66]. Suppressing the expression of HSP23 and HSP70 did not change the diapause transition or duration; it reduced overwintering cold tolerance of S. crassipalpis [10]. Apart from the function of freeze-resistance, HSPs also played an important role in diapause development. HSP26 prevented diapause termination and protected encysting Artemia embryos from stress [67] and HSP23 was considered a normal component of the diapause syndrome and played an essential role during overwintering development arrest [68].
Metabolisms, controlling protective molecules conversion and substantial preparation for metamorphosis, responded and adapted to environmental changes. Glycerol and sorbitol, as the most common cryoprotectant molecules, lower the supercooling points and protect against protein denaturation [69]. Glycerol kinase (CL3214.Contig1) is a rate-limiting enzyme in glycerol utilization [70], increasingly expressed during diapause to promote glycerol biosynthesis which was stress-induced and related to high cold resistance (Figure 8) [71]. In a specific way, sorbitol dehydrogenase (CL3305.Contig1), related to diapause termination in eggs of B. mori [72], was upregulated in LD (Figure 8). This gene was presumably involved in catalyzing biological carbohydrate conversion from sorbitol which inhibited the development of the diapausing egg of B. mori [73]. Compared to ED, genes involved in metabolism and hormone biosynthesis in LD were largely suppressed except for the enhanced ribosome pathway (Table S10) when exposed to an intensive cold pressure. During the post-diapause development, amino acid metabolism and insect hormone biosynthesis were positively enhanced, in a similar way to that reported in the apple maggot fly R. pomonella [8]. Tyrosine metabolism increasingly expressed in PD suggested it had already been prepared for new adult morphogenesis despite the natural harsh environment imposed (Table 4, Table S10).
Conclusions
In the present study, a transcriptome database has been produced on a large scale for B. minax using Illumina sequencing. Through our results, we have gained new insights into the genomics of this univoltine fruit fly and they can be used as a reference to provide comparative studies within the tephritids. Based on the transcriptome background, DGE analysis has contributed significantly to speedy disclosure diapause-specific candidate genes/pathways for this organism without complete genome sequences or other genetic tools/resources. It provides valuable data with regard to further investigation on molecular mechanism underlying the pupal diapause termination in B. minax. In addition, our data provides a substantial contribution to many other meaningful biological processes in molecular research on B. minax and other related pests.
Supplementary Material
Additional File 1Table S1.
Table S2.
Table S3.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Table S10.
Table S11.
Table S12.
Figures S1-S5.
Acknowledgements
We are grateful to Professor Xiaoguang Ma (Institute of Health Quarantine, Chinese Academy of Inspection and Quarantine) for his assistance in measuring pupal respiration rate. We also thank the technical support for Illumina sequencing and initial data from Beijing Genome Institute (BGI) at Shenzhen, China. This study was funded by the National Natural Science Foundation of China (31371945 and 31071690), Crop disease and insect pest monitoring and control program (2014), Hubei Research and Development Funds (2012IHA01302), and International Atomic Energy Agency (via Research Contract No. 17153 and CRP 18269).
Authors' contributions
YD and CN conceived and designed the experiments. YD performed the experiments. YD and ND analysed the data. YD, CN and CL contributed reagents/materials/analysis tools. YD, ND and CN shared in the scoping and writing responsibilities.
Competing Interests
The authors have declared that no competing interest exists.
References
1. MacRae TH. Gene expression, metabolic regulation and stress tolerance during diapause. Cellular and Molecular Life Sciences. 2010;67(14):2405-2424
2. Kostál V. Eco-physiological phases of insect diapause. Journal of Insect Physiology. 2006;52(2):113-127
3. Denlinger DL. Regulation of diapause. Annual Review of Entomology. 2002;47(1):93-122
4. Ragland GJ, Denlinger DL, Hahn DA. Mechanisms of suspended animation are revealed by transcript profiling of diapause in the flesh fly. Proceedings of the National Academy of Sciences. 2010;107(33):14909-14914
5. Rinehart JP, Robich RM, Denlinger DL. Isolation of diapause-regulated genes from the flesh fly, Sarcophaga crassipalpis by suppressive subtractive hybridization. Journal of Insect Physiology. 2010;56(6):603-609
6. Zhang QR, Denlinger DL. Dynamics of diapause hormone and prothoracicotropic hormone transcript expression at diapause termination in pupae of the corn earworm, Helicoverpa zea. Peptides. 2012;34(1):120-126
7. Poelchau MF, Reynolds JA, Elsik CG, Denlinger DL, Armbruster PA. Deep sequencing reveals complex mechanisms of diapause preparation in the invasive mosquito, Aedes albopictus. Proceedings of the Royal Society B: Biological Sciences. 2013;280(1759):20130143
8. Ragland GJ, Egan SP, Feder JL, Berlocher SH, Hahn DA. Developmental trajectories of gene expression reveal candidates for diapause termination: a key life-history transition in the apple maggot fly Rhagoletis pomonella. Journal of Experimental Biology. 2011;214(23):3948-3960
9. Reynolds JA, Poelchau MF, Rahman Z, Armbruster PA, Denlinger DL. Transcript profiling reveals mechanisms for lipid conservation during diapause in the mosquito, Aedes albopictus. Journal of Insect Physiology. 2012;58(7):966-973
10. Rinehart JP, Li A, Yocum GD, Robich RM, Hayward SA, Denlinger DL. Up-regulation of heat shock proteins is essential for cold survival during insect diapause. Proceedings of the National Academy of Sciences. 2007;104(27):11130-11137
11. Teets NM, Kawarasaki Y, Lee RE Jr, Denlinger DL. Expression of genes involved in energy mobilization and osmoprotectant synthesis during thermal and dehydration stress in the Antarctic midge, Belgica antarctica. Journal of Comparative Physiology B. 2013;183(2):189-201
12. Meuti ME, Denlinger DL. Evolutionary links between circadian clocks and photoperiodic diapause in insects. Integrative and Comparative Biology. 2013;53(1):131-143
13. Denlinger DL. Dormancy in tropical insects. Annual Review of Entomology. 1986;31(1):239-264
14. Kang L, Chen B, Wei J-N, Liu T-X. Roles of thermal adaptation and chemical ecology in Liriomyza distribution and control. Annual Review of Entomology. 2009;54:127-145
15. Wang X, Luo L. Research progress in the Chinese citrus fruit fly. Entomological Knowledge. 1995;32:310-315
16. Dorji C, Clarke AR, Drew RAI, Fletcher BS, Loday P, Mahat K, Raghu S, Romig MC. Seasonal phenology of Bactrocera minax (Diptera: Tephritidae) in western Bhutan. Bulletin of Entomological Research. 2006;96(5):531
17. Ragland GJ, Sim SB, Goudarzi S, Feder JL, Hahn DA. Environmental interactions during host race formation: host fruit environment moderates a seasonal shift in phenology in host races of Rhagoletis pomonella. Functional Ecology. 2012;26(4):921-931
18. Robbins HM, Van Stappen G, Sorgeloos P, Sung YY, MacRae TH, Bossier P. Diapause termination and development of encysted Artemia embryos: roles for nitric oxide and hydrogen peroxide. Journal of Experimental Biology. 2010;213(9):1464-1470
19. Benoit JB, Denlinger DL. Meeting the challenges of on-host and off-host water balance in blood-feeding arthropods. Journal of Insect Physiology. 2010;56(10):1366-1376
20. Dong YC, Wang ZJ, Clarke AR, Pereira R, Desneux N, Niu CY. Pupal diapause development and termination is driven by low temperature chilling in Bactrocera minax. Journal of Pest Science. 2013;86(3):429-436
21. Zhou XW, Niu CY, Han P, Desneux N. Field evaluation of attractive lures for the fruit fly Bactrocera minax (Diptera: Tephritidae) and their potential use in spot sprays in Hubei Province (China). Journal of Economic Entomology. 2012;105(4):1277-1284
22. Ragland GJ, Fuller J, Feder JL, Hahn DA. Biphasic metabolic rate trajectory of pupal diapause termination and post-diapause development in a tephritid fly. Journal of Insect Physiology. 2009;55(4):344-350
23. Papanastasiou SA, Nestel D, Diamantidis AD, Nakas CT, Papadopoulos NT. Physiological and biological patterns of a highland and a coastal population of the European cherry fruit fly during diapause. Journal of Insect Physiology. 2011;57(1):83-93
24. Teixeira LAF, Polavarapu S. Phenological differences between populations of Rhagoletis mendax (Diptera: Tephritidae). Environmental Entomology. 2002;31(6):1103-1109
25. Yasuda T, Narahara M, Tanaka S, Wakamura S. Thermal responses in the citrus fruit fly, Dacus tsuneonis: evidence for a pupal diapause. Entomologia Experimentalis et Applicata. 1994;71(3):257-261
26. Tauber MJ, Tauber CA. Insect seasonality: diapause maintenance, termination, and postdiapause development. Annual Review of Entomology. 1976;21(1):81-107
27. Teixeira LAF, Polavarapu S. Diapause development in the blueberry maggot Rhagoletis mendax (Diptera: Tephritidae). Environmental Entomology. 2005;34(1):47-53
28. Feder JL, Powell THQ, Filchak K, Leung B. The diapause response of Rhagoletis pomonella to varying environmental conditions and its significance for geographic and host plant-related adaptation. Entomologia Experimentalis et Applicata. 2010;136(1):31-44
29. Hahn DA, Denlinger DL. Energetics of insect diapause. Annual Review of Entomology. 2011;56:103-121
30. Mardis ER. Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics. 2008;9:387-402
31. Shen GM, Dou W, Niu JZ, Jiang HB, Yang WJ, Jia FX, Hu F, Cong L, Wang JJ. Transcriptome Analysis of the Oriental Fruit Fly (Bactrocera dorsalis). PloS One. 2011;6(12):e29127
32. Gomulski LM, Dimopoulos G, Xi Z, Soares MB, Bonaldo MF, Malacrida AR, Gasperi G. Gene discovery in an invasive tephritid model pest species, the Mediterranean fruit fly, Ceratitis capitata. BMC Genomics. 2008;9:243
33. Schwarz D, Robertson HM, Feder JL, Varala K, Hudson ME, Ragland GJ, Hahn DA, Berlocher SH. Sympatric ecological speciation meets pyrosequencing: sampling the transcriptome of the apple maggot Rhagoletis pomonella. BMC Genomics. 2009;10:633
34. Köppler K, Kaffer T, Vogt H. Substantial progress made in the rearing of the European cherry fruit fly, Rhagoletis cerasi. Entomologia Experimentalis et Applicata. 2009;132(3):283-288
35. Fujiwara Y, Denlinger DL. High temperature and hexane break pupal diapause in the flesh fly, Sarcophaga crassipalpis, by activating ERK/MAPK. Journal of Insect Physiology. 2007;53(12):1276-1282
36. Denlinger D, Yocum G, Rinehart J. Hormonal control of diapause. Comprehensive Molecular Insect Science. 2005;3:615-650
37. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma FD, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology. 2011;29:644-652
38. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674-3676
39. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T. KEGG for linking genomes to life and the environment. Nucleic Acids Research. 2008;36(Database issue):D480-4
40. Xue J, Bao YY, Li BL, Cheng YB, Peng ZY, Liu H, Xu HJ, Zhu ZR, Lou YG, Cheng JA, Zhang CX. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PloS One. 2010;5(12):e14233
41. AC't Hoen P, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, de Menezes RX, Boer JM, van Ommen GJB, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Research. 2008;36(21):e141-e141
42. Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Research. 1997;7(10):986-995
43. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences. 1998;95(25):14863-14868
44. Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246-3248
45. Rewitz KF, Larsen MR, Lobner-Olesen A, Rybczynski R, O'Connor MB, Gilbert LI. A phosphoproteomics approach to elucidate neuropeptide signal transduction controlling insect metamorphosis. Insect Biochemistry and Molecular Biology. 2009;39(7):475-483
46. Huang X, Warren JT, Gilbert LI. New players in the regulation of ecdysone biosynthesis. Journal of Genetics and Genomics. 2008;35(1):1-10
47. Emerson KJ, Bradshaw WE, Holzapfel CM. Complications of complexity: integrating environmental, genetic and hormonal control of insect diapause. Trends in Genetics. 2009;25(5):217-225
48. Sim C, Denlinger DL. Insulin signaling and FOXO regulate the overwintering diapause of the mosquito Culex pipiens. Proceedings of the National Academy of Sciences. 2008;105(18):6777-6781
49. Rewitz K, Rybczynski R, Warren JT, Gilbert LI. The Halloween genes code for cytochrome P450 enzymes mediating synthesis of the insect moulting hormone. Biochemical Society Transactions. 2006;34:1256-1260
50. Xu WH, Lu YX, Denlinger DL. Cross-talk between the fat body and brain regulates insect developmental arrest. Proceedings of the National Academy of Sciences. 2012;109(36):14687-14692
51. Mizoguchi A, Ohsumi S, Kobayashi K, Okamoto N, Yamada N, Tateishi K, Fujimoto Y, Kataoka H. Prothoracicotropic hormone acts as a neuroendocrine switch between pupal diapause and adult development. PloS One. 2013;8(4):e60824
52. Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18(1):207-208
53. Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, Ruby JG, Brennecke J; Harvard FlyBase curators; Berkeley Drosophila Genome Project, Hodges E, Hinrichs AS, Caspi A, Paten B, Park SW, Han MV, Maeder ML, Polansky BJ, Robson BE, Aerts S, van Helden J, Hassan B, Gilbert DG, Eastman DA, Rice M, Weir M, Hahn MW, Park Y, Dewey CN, Pachter L, Kent WJ, Haussler D, Lai EC, Bartel DP, Hannon GJ, Kaufman TC, Eisen MB, Clark AG, Smith D, Celniker SE, Gelbart WM, Kellis M. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450(7167):219-232
54. Bao B, Hong B, Feng QL, Xu WH. Transcription factor fork head regulates the promoter of diapause hormone gene in the cotton bollworm, Helicoverpa armigera, and the modification of SUMOylation. Insect Biochemistry and Molecular Biology. 2011;41(9):670-679
55. Young SC, Yeh WL, Gu SH. Transcriptional regulation of the PTTH receptor in prothoracic glands of the silkworm, Bombyx mori. Journal of Insect Physiology. 2012;58(1):102-109
56. Kidokoro K, Wata K, Fujiwara Y, Takeda M. Effects of juvenile hormone analogs and 20-hydroxyecdysone on diapause termination in eggs of Locusta migratoria and Oxya yezoensis. Journal of Insect Physiology. 2006;52(5):473-479
57. Rewitz KF, O'Connor MB, Gilbert LI. Molecular evolution of the insect Halloween family of cytochrome P450s: phylogeny, gene organization and functional conservation. Insect Biochemistry and Molecular Biology. 2007;37(8):741-753
58. Gu SH, Young SC, Tsai WH, Lin JL, Lin PL. Involvement of 4E-BP phosphorylation in embryonic development of the silkworm, Bombyx mori. Journal of Insect Physiology. 2011;57(7):978-985
59. McKay M, Morrison D. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26(22):3113-3121
60. Fujiwara Y, Denlinger DL. p38 MAPK is a likely component of the signal transduction pathway triggering rapid cold hardening in the flesh fly Sarcophaga crassipalpis. Journal of Experimental Biology. 2007;210(Pt 18):3295-3300
61. Fraenkel G, Blewett M. Linoleic acid and arachidonic acid in the metabolism of two insects, Ephestia kuehniella (Lep.) and Tenebrio molitor (Col.). Biochemical Journal. 1947;41(3):475
62. Kim M, Sim C, Denlinger DL. RNA interference directed against ribosomal protein S3a suggests a link between this gene and arrested ovarian development during adult diapause in Culex pipiens. Insect Molecular Biology. 2010;19(1):27-33
63. Bodnaryk RP. Ecdysteroid levels during post-diapause development and 20-hydroxyecdysone-induced development in male pupae of Mamestra configurata Wlk. Journal of Insect Physiology. 1985;31(1):53-58
64. Wang X, Kang L. Molecular mechanisms of phase change in locusts. Annual Review of Entomology. 2014;59:225-244
65. Lopez-Martinez G, Denlinger DL. Regulation of heat shock proteins in the apple maggot Rhagoletis pomonella during hot summer days and overwintering diapause. Physiological Entomology. 2008;33(4):346-352
66. Kayukawa T, Ishikawa Y. Chaperonin contributes to cold hardiness of the onion maggot Delia antiqua through repression of depolymerization of actin at low temperatures. PloS One. 2009;4(12):e8277
67. King AM, MacRae TH. The small heat shock protein p26 aids development of encysting Artemia embryos, prevents spontaneous diapause termination and protects against stress. PloS One. 2012;7(8):e43723
68. Yocum G, Joplin K, Denlinger D. Upregulation of a 23kDa small heat shock protein transcript during pupal diapause in the flesh fly, Sarcophaga crassipalpis. Insect Biochemistry and Molecular Biology. 1998;28(9):677-682
69. Yamashita O, Yaginuma T. Silkworm eggs at low temperatures: implications for sericulture. Insects at low temperature. Springer. 1991:424-445
70. Kihara F, Itoh K, Iwasaka M, Niimi T, Yamashita O, Yaginuma T. Glycerol kinase activity and glycerol kinase-3 gene are up-regulated by acclimation to 5 degrees C in diapause eggs of the silkworm, Bombyx mori. Insect Biochemistry and Molecular Biology. 2009;39(11):763-769
71. Yoder JA, Benoit JB, Denlinger DL, Rivers DB. Stress-induced accumulation of glycerol in the flesh fly, Sarcophaga bullata: evidence indicating anti-desiccant and cryoprotectant functions of this polyol and a role for the brain in coordinating the response. Journal of Insect Physiology. 2006;52(2):202-214
72. Fujiwara Y, Tanaka Y, Iwata KI, Rubio RO, Yaginuma T, Yamashita O, Shiomi K. ERK/MAPK regulates ecdysteroid and sorbitol metabolism for embryonic diapause termination in the silkworm, Bombyx mori. Journal of Insect Physiology. 2006;52(6):569-575
73. Horie Y, Kanda T, Mochida Y. Sorbitol as an arrester of embryonic development in diapausing eggs of the silkworm, Bombyx mori. Journal of Insect Physiology. 2000;46(6):1009-1016
Author contact
![]() Corresponding author: ioirhzau.edu.cn (CL); niuchangying88com (CN).
Corresponding author: ioirhzau.edu.cn (CL); niuchangying88com (CN).