Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(10):1116-1127. doi:10.7150/ijbs.10236 This issue Cite
Research Paper
The Steroid Receptor Coactivator-3 Is Required for Developing Neuroendocrine Tumor in the Mouse Prostate
Jean Ching-Yi Tien1, 2, Lan Liao1, Yonghong Liu1, 2, Zhaoliang Liu1, 2, Dong-Kee Lee1, Fen Wang2, Jianming Xu1, 2, 3 ![]()
1. Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas 77030, USA;
2. Institute of Biosciences and Technology, Texas A&M University Health Science Center, Houston, Texas 77030, USA;
3. Insitute for Cancer Medicine, Luzhou Medical College, Luzhou, Sichuan 646000, China.
Received 2014-7-31; Accepted 2014-8-30; Published 2014-10-2
Abstract
Neuroendocrine tumor cells (NETCs) are commonly observed in prostate cancer. Their presence is associated with castration resistance, metastasis and poor prognosis. Cellular and molecular mechanisms for NETC initiation and growth are unknown. TRAMP mice develop heterogeneous adenocarcinomas induced by expression of the SV40-T/t oncogene in prostate epithelial cells. Here, we demonstrate prostate tumors in TRAMP mice with a mixed genetic background are characterized mostly by atypical hyperplasia (AH) containing steroid receptor coactiator-3-positive, androgen receptor-positive and synaptophysin-negative (SRC-3+/AR+/Syp-) cells. Few SRC-3+/AR-/Syp+ NETCs are present in their prostates. We generated TRAMP mice in which SRC-3 was specifically ablated in AR+/Syp- prostatic epithelial cells (termed PE3KOT mice). In these animals, we observed a substantial reduction in SRC-3-/AR+/Syp- AH tumor growth. There was a corresponding increase in SRC-3-/AR+/Syp- phyllodes lesions, suggesting SRC-3 knockout can convert aggressive AH tumors with mostly epithelial tumor cells into less aggressive phyllodes lesions with mostly stromal tissue. Surprisingly, PE3KOT mice developed many more SRC-3+/AR-/Syp+ NETCs versus control TRAMP mice, indicating SRC-3 expression was retained in NETCs. In contrast, TRAMP mice with global SRC-3 knockout did not develop any NETC, indicating SRC-3 is required for developing NETC. Analysis of cell-differentiating markers revealed that these NETCs might not be derived from the mature AR-/Syp+ neuroendocrine cells or the AR+/Syp- luminal epithelial tumor cells. Instead, these NETCs might originate from the SV40-T/t-transformed intermediate/progenitor epithelial cells. In summary, SRC-3 is required for both AR+/Syp- AH tumor growth and AR-/Syp+ NETC development, suggesting SRC-3 is a target for inhibiting aggressive prostate cancer containing NETCs.
Keywords: coactivator, SRC-3/NCOA3, prostate epithelium, carcinogenesis, neuroendocrine cell, transgenic mouse model.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer death in American men [1]. A prostate tumor usually exhibits multifocal lesions with various stages of differentiation patterns, suggesting that this disease is highly heterogeneous and contains multiple neoplastic clones within a single tumor [2]. Prostate adenocarcinoma arises from the prostatic epithelium and is the most common form of PCa [3]. The pathological grading system is established according to the histological differentiation stages of the cancerous epithelial cells. Terms such as well-differentiated (WD), moderately differentiated (MD) and poorly differentiated (PD) adenocarcinomas are used to describe the cancerous stages of prostate adenocarcinomas. Neuroendocrine (NE) PCa is a rare subtype, affecting about 2% of PCa patients. However, this percentage is significantly underestimated according to data from autopsy studies [4]. In fact, focal NE cell differentiation is commonly detected in PCa biopsies, and PCa with NE cells is usually associated with castration resistance, metastasis, resistance to chemotherapy and poor prognosis [5, 6]. The highly malignant small cell lung cancer also consists of NE tumor cells and NE tumor cells are also seen in other types of cancer such as breast cancer. Previous studies have shown androgen depletion transforms prostate adenocarcinoma cells into NE cells in culture and xenograft models, suggesting hormone-targeted therapy may selectively drive the development of more malignant NE PCa [7-9]. However, the origin of neuroendocrine tumor cells in spontaneous PCa and the molecular mechanisms responsible for development of NE tumors are completely unknown.
The transgenic adenocarcinoma of the mouse prostate (TRAMP) is a mouse model in which the rat probasin promoter (PB) drives the expression of the simian vacuolating virus 40 T/t antigens (SV40-T/t) in prostatic epithelial cells for induction of PCa through inhibiting p53, retinoblastoma (RB) and protein phosphatase 2A (PP2A) tumor suppressors [10]. Similar to human PCa, TRAMP mice develop heterogeneous prostate tumors with various differentiation stages, including both types of adenocarcinoma termed atypical hyperplasia (AH) as well as neuroendocrine (NE) tumors [11, 12]. As in men, prostate tumor phenotypes in TRAMP mice may switch from AH to aggressive NE phenotype upon castration [13]. Phyllodes-like lesions (PHY) characterized by staghorn luminal patterns containing hypercellular stroma are also observed in TRAMP mice [14]. The PHY tumor of the prostate is a rare neoplasm with poorly understood pathogenesis [15]. Overall, TRAMP mice offer an advantageous model for studying PCa with different neoplasm types: AH, NE and PHY tumors.
Steroid receptor coactivator-3 (SRC-3, AIB1, ACTR, or NCOA3) is a member of the p160 SRC family. Serving as an adapter to recruit chromatin-remodeling proteins, SRC-3 mediates transcriptional activities of nuclear hormone receptors such as estrogen receptor (ER) and androgen receptor (AR), as well as other transcription factors such as E2F-1, PEA3, AP-1 and NF-kB [16, 17]. Several studies have demonstrated that overexpression of SRC-3 induces breast carcinogenesis, while knockout of SRC-3 inhibits oncogene and chemical carcinogen-induced mammary gland tumorigenesis [18-22]. In human prostate tumors, SRC-3 expression correlates positively with tumor grade and stage [23]. In PCa cell lines, SRC-3 upregulates cell cycle genes, multiple components of the IGF-I/Akt pathway, MMP2 and MMP13 - in all, promoting cell proliferation, migration and invasion [24-26]. These in vitro studies suggest that SRC-3 may play an important role in promoting PCa.
We have shown that global knockout of SRC-3 in TRAMP mice significantly inhibits prostate tumor growth and also arrests PCa progression at a well-differentiated stage [27]. Although this study suggests targeting SRC-3 may suppress prostate carcinogenesis, this global SRC-3 knockout model has its pitfalls. First, SRC-3 null mice have a significantly reduced IGF-I level in blood, which may indirectly affect prostate tumorigenesis. Moreover, knockout of SRC-3 in the prostate stromal cells may also indirectly impede prostate epithelial carcinogenesis through paracrine regulatory pathways between epithelium and stroma [28]. In the present study, we developed prostate epithelial cell-specific SRC-3 knockout mice and further illustrated the specific role of SRC-3 in development of different types of prostate adenocarcinoma. We found that specific ablation of SRC-3 mediated by the PB-Cre transgene in the AR-positive (AR+) prostatic epithelial cells, predominantly prostatic luminal epithelial cells (LECs), slows down the growth and progression of AR+ AH tumors in TRAMP mice, but significantly enhances the development of the more aggressive SRC-3-expressing NE tumors and the less aggressive SRC-3-negative PHY tumors. Interestingly, we found that these SRC-3+ NE tumors likely originate from neither the mature NE epithelial cells nor the LEC-like tumor cells, but from the epithelial intermediate/progenitor cells that co-express SRC-3, SV40-T/t antigens, cytokeratin 5 (K5), p63 and Syp. Furthermore, global knockout of SRC-3 in TRAMP mice completely suppresses the development of Syp+ NE tumors, indicating SRC-3 is absolutely required for NE tumor development from these intermediate/progenitor cells.
Materials and Methods
Mice. The floxed SRC-3 mouse line (SRC-3f/f) was generated in our lab as described previously [29] and they are in a mixed 129SvEv/C57BL/6J strain background. The TRAMP (PB-SV40-T/t) transgenic mouse line in C57BL/6J strain background (provided by Dr. Norman Greenberg) and the ARR2PBi-Cre (PB-Cre) transgenic mouse line in FVB strain background were described previously [10, 30]. TRAMP mice were first crossbred with PBi-Cre mice and then, their PB-Cre;PB-SV40-T/t offspring were crossbred with SRC-3f/f mice. The PB-Cre;SRC-3f/+;PB-SV40-T/t offspring were then bred with SRC-3f/f mice again to generate PB-Cre;SRC-3f/f;PB-SV40-T/t testing and SRC-3f/f;PB-SV40-T/t control mice with comparable genetic background for experiments. For convenience, we designated the testing mice as 3PEKOT (SRC-3 prostate epithelial knockout in TRAMP) mice and the control mice as 3FloxT (floxed SRC-3 in TRAMP) mice. For genotype analysis, genomic DNA samples were prepared as DNA templates and PCR reactions were performed using primer pairs specific to PB-Cre or PB-SV40-T/t transgene and to SRC-3 wild type (WT), floxed, or deleted allele as described previously [10, 29, 30]. Animal protocols were approved by the Animal Care and Use Committee of Baylor College of Medicine.
Tissue examination. 18-, 24- and 30-week-old male 3FloxT and 3PEKOT mice were weighed and euthanized. Their lower urogenital tracts were removed en bloc and the weight of urogenital tract was measured. The prostate tissue was then dissected out in a Petri dish containing phosphate-buffered saline (PBS). The prostate tumor tissues were isolated and blotted gently with paper towel to remove excess fluid and PBS. The weight of prostate tumors was then measured, and tumor tissues were then fixed in 4% paraformaldehyde at 4oC overnight for tissue process.
Histological examination and immunohistochemistry (IHC). Fixed prostate tissues and tumors were embedded in paraffin and paraffin sections were processed for H&E staining and immunostaining as described previously [27]. Immunostaining was performed using primary antibodies against SRC-3 (Cell Signaling, #2126), AR (Santa Cruz, sc-816), Syp (Invitrogen, 18-0130), SV40 T antigen (BD Biosciences, 554149), Ki67 (BD Biosceinces, 550609) and K5 (Covance, PRB160P). Secondary antibodies for IHC were biotinylated anti-Rabbit IgG and biotinylated anti-mouse IgG (Vector labs) used at 1:400 dilution. The signal was enhanced using the VECTASTAIN ABC system and visualized with DAB kit. The slides were counterstained with Harris Modified Hematoxylin and mounted with Permount. For double immunohistofluorescence, Tyramide Signal Amplification kit (Life technologies) was used per manufacturer's instructions. For Syp, SV40 T and K5 triple immmunohistofluorescence, slides were incubated with Syp antibody overnight, washed with PBST and then incubated with biotinylated anti-Rabbit monovalent Fab antibodies for 1 hour at room temperature. The slides were further incubated with tertiary streptavidin HRP antibody for 1 hour at room temperature and the green signal was visualized with TSA kit #22 (Life technologies). Prior to incubation with the SV40 T antibody, the slides were blocked with streptavidin/biotin blocking kit (Vector Lab) followed by 3% H2O2 treatment. The slides were then blocked by MOM kit (Vector Lab) for 1 hour at room temperature and incubated with SV40 T antibody overnight at 4oC. The secondary antibody used was biotinylated anti-Mouse IgG followed by the tertiary streptavidin HRP antibody. The red signal was visualized with TSA kit #25 (Life technologies). Finally, the slides were incubated with the streptavidin/biotin blocking reagent, treated with 3% H2O2, incubated with K5 antibody overnight at 4oC followed by incubation with HRP-goat anti-Rabbit IgG for 1 hour at room temperature. The blue signal was then detected with TSA kit #17 (Life technologies).
Results
Conditional knockout of SRC-3 in prostatic epithelial cells of TRAMP mice
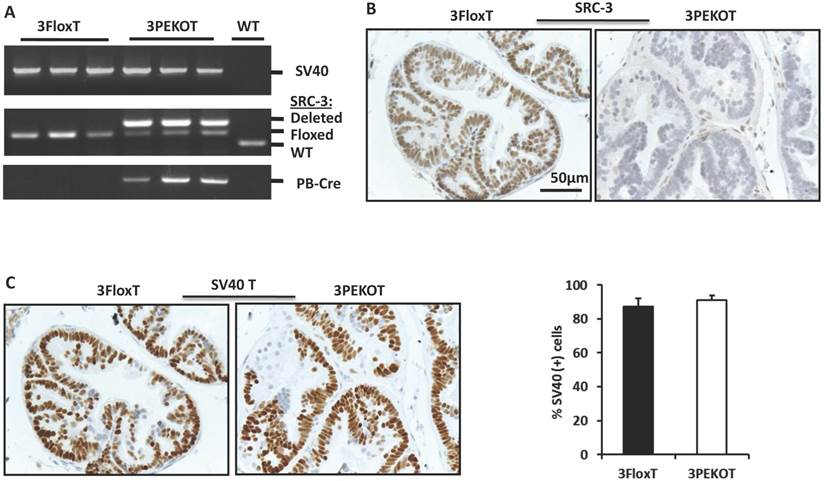
In order to investigate the cell autonomous role of SRC-3 in prostate carcinogenesis, we generated 3PEKOT (PB-Cre;SRC-3f/f;PB-SV40T/t) and 3FloxT (SRC-3f/f;PB-SV40T/t) mice with a comparable mixed strain background by crossbreeding PB-Cre, SRC-3f/f and TRAMP (PB-SV40-T/t) mouse lines [10, 29, 30]. In 3PEKOT male mice, the PB-Cre transgene was only expressed in the prostate epithelial cells [29, 30], and, as such, specified deletion of the floxed SRC-3 alleles only in these cells. Genotype analysis detected a strong band representing the mutant allele with SRC-3 deletion from the prostate DNA samples of 18-week-old 3PEKOT mice, while a much lighter band representing the floxed SRC-3 allele was also detected. This later band indicated the floxed SRC-3 alleles that were not deleted in the prostate stromal cells lacking PB-Cre expression. In 18-week-old 3FloxT control mice harboring the PB-SV40-T/t transgene but not the PB-Cre transgene, only the floxed SRC-3 alleles were detected from the prostate DNA samples (Fig. 1A). SRC-3 protein was detected by IHC in the luminal epithelial cells (LECs) with slightly weaker staining in some basal epithelial cells (BCs) in the prostates of 8-week-old 3FloxT control mice. On the contrary, SRC-3 protein was absent in LECs in the prostates of 3PEKOT mice, although SRC-3 was still present in some BCs (Fig. 1B). In addition, the intensity of SV40-T immunoreactivity and the total number of SV40-T-positive prostatic epithelial cells were comparable between 3FloxT and 3PEKOT mice at 8 weeks of age (Fig. 1C). These results demonstrate that SRC-3 is specifically and efficiently ablated in the prostate LECs of 3PEKOT mice and SRC-3 ablation does not affect the PB-SV40T/t transgene expression during the prostate tumorigenesis induced by SV40-T/t antigens.
Conditional knockout of SRC-3 in prostatic epithelial cells of TRAMP mice. A. Deletion of the floxed SRC-3 alleles in the prostates of 3PEKOT mice. Genomic DNA samples were prepared from prostate tumors of 24-week-old 3FloxT (n=3) and 3PEKOT (n=3) mice or from a WT mouse prostate and used as templates for PCR. DNA fragments representing the PB-SV40-T/t and PB-Cre transgenes and the WT, floxed and deleted SRC-3 alleles were amplified using allele-specific primer pairs. B. IHC for SRC-3 (brown) in the prostate sections prepared from 8-week-old 3FloxT and 3PEKOT mice. C. IHC for SV40 T antigen (brown) in the prostate sections prepared from 8-week-old 3FloxT and 3PEKOT mice. The percentage of SV40 T-positive was calculated by dividing the number of positive epithelial cells by the number of total epithelial cells per imaging field. Three random imaging fields per section and sections from 5 mice were assessed.
Gross weights of prostate tumors in 3PEKOT and 3FloxT mice are similar
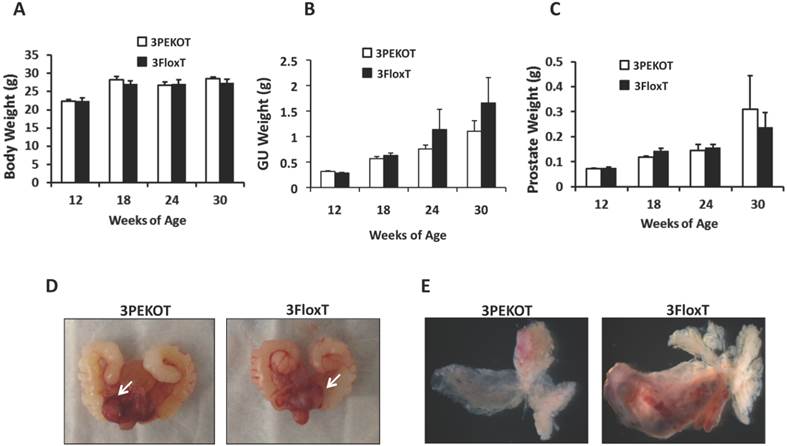
TRAMP mice develop multifocal and aggressive tumors by 24 weeks of age and generally survive up to 30 weeks [27]. Therefore, we evaluated prostate tumor growth in 3PEKOT and 3FloxT mice at 18, 24 and 30 weeks of age. We found that there was no difference in body weight between 3PEKOT and 3FloxT mice at any time point (Fig. 2A). The average genitourinary tract (GU) weight trended lower in 3PEKOT versus 3FloxT mice at 24 and 30 weeks (Fig. 2B). However, these GU weight differences did not reach statistical significance. Indeed, further dissection indicated no weight difference between the prostates of 3PEKOT and 3FloxT mice at 24 and 30 weeks (Fig. 2C). Moreover, morphological examination of GUs and dissected prostates revealed that prostate tumors developed in both 3PEKOT and 3FloxT mice (Fig. 2D and E). Overall, these results demonstrate similar gross tumor weight in both experimental and control groups.
Characterization of prostate tumor growth in 3PEKOT and 3FloxT mice. A. Similar body weights of 3PEKOT and 3FloxT mice (n=10 for each group) at ages indicated. B. The genitourinary tract (GU) weights of 3PEKOT and 3FloxT mice at different ages (n=10 in each group). GU weight trended lower in 3PEKOT versus 3FloxT mice at 24 and 30 weeks, but the difference did not reach statistical significance. C. The average prostate tumor weights of these two groups of mice were also not statistically different. D. The macroscopic gross pictures of the upper genitourinary tracts of 24-week-old 3PEKOT and 3FloxT mice. Arrows indicate prostate tumors. E. The macroscopic gross pictures of dissected prostates with tumors of 24-week-old 3PEKOT and 3FloxT mice. Of note, one of the anterior lobs in this 3FloxT mouse has bigger tumor, which was not consistently observed in all mice at this stage.
Progressive changes of prostate tumor phenotypes from atypical hyperplasia to neuroendocrine and phyllodes-like tumors in 3PEKOT mice
There are three types of tumors that develop in the TRAMP model: atypical hyperplasia (AH), neuroendocrine (NE) tumors, and phyllodes-like (PHY) tumors [12]. We identified each of these tumor types in 3PEKOT and 3FloxT mice. AH regions contained mostly AR+ cells and were devoid of Syp+ cells. NE tumor regions contained exclusively Syp+ tumor cells completely negative for AR. PHY tumor regions were characterized by significant stromal cellularity underlying linearly-distributed epithelial cells. Many AR+ cells were detectable in the stromal compartment, while almost all epithelial cells were AR+ in the PHY tumor regions (Fig. 3A). We detected SRC-3 protein by IHC in the epithelial cells of AH tumor regions and the Syp+ cells of NE tumor regions of 30-week-old 3FloxT mice. We also detected SRC-3 protein in all Syp+ cells of NE tumor regions and the stromal cells of PHY tumor regions, but not in the epithelial tumor cells of AH regions and PHY regions of 30-week-old 3PEKOT mice owning to PB-Cre-mediated SRC-3 knockout in these cell types (Fig. 3A and data not shown).
Increased NE tumor incidence in 3PEKOT mice at 30 weeks of age. A. Three types of tumor lesions developed in TRAMP mice, including atypical hyperplasia (AH), neuroendocrine tumor (NE) and phyllodes (PHY). AH tumor cells express AR and SRC-3 but not Syp. NE tumor cells express Syp and SRC-3 but not AR. PHY tumor cells express AR but not Syp. The images of AH were taken from a tumor section of 3FloxT mouse, while the images of NE and PHY were taken from tumor sections of 3PEKOT mice. B. The frequency of each tumor type in the tumors of 24 and 30-week-old 3FloxT and 3PEKOT mice. See text for the results of statistical analysis.
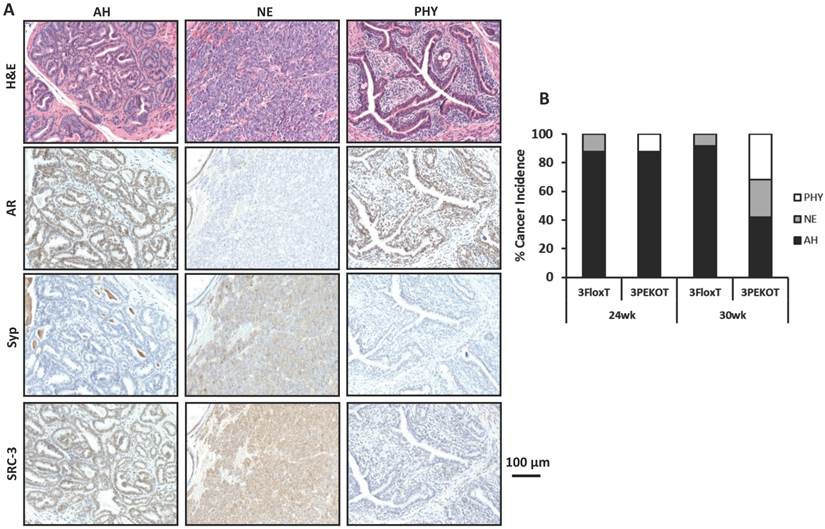
We evaluated the frequency of each tumor type in the prostates of 24 and 30-week-old 3FloxT and 3PEKOT mice. At 24 weeks, AH was the principal prostate cancer type developed in the vast majority of 3FloxT (7/8) and 3PEKOT (7/8) mice. Only one out of eight 3FloxT mice developed NE tumor and one out of eight 3PEKOT mice developed PHY tumor as the dominant histological pattern in the prostate tumor (Fig. 3B). At 30 weeks, we found that 91.7% (11/12) of 3FloxT mice had prostate tumors with a principally AH phenotype, while only a single mouse out of 12 mice (8.3%) had a principally NE tumor phenotype. Moreover, we observed no PHY tumors in 3FloxT mice. On the contrary, only 42% (8/19) 3PEKOT mice had tumors in which the principal histological pattern was AH. Interestingly, 26% (5/19) of these animals developed principally NE tumors, while 32% (6/19) developed principally PHY tumors (Fig. 3B). A Chi-squared test revealed that, while there was no significant difference in cancer type incidence between 3FloxT and 3PEKOT mice at 24 weeks, there was a significant difference between two groups at 30 weeks, χ2 (2, N =31) = 7.967, p=0.019, indicating that 3FloxT mice preferentially develop AH lesions while 3PEKOT mice preferentially develop NE and PHY lesions at 30 weeks. These results demonstrate that 3FloxT control mice initially develop AR+ AH tumors and also maintain the same type of tumors for a long time. However, 3PEKOT mice initially develop AR+ AH tumors in their prostates, but this tumor phenotype in most mice was switched to either AR-/SRC-3+ NE tumors or AR+/SRC-3- PHY tumors. Again, SRC-3 is only expressed in the stromal cells of PHY tumors.
Deletion of SRC-3 slows down prostate AH tumor growth and progression
Because AH tumors have characteristics of LECs that express AR, we decided to first analyze these AH tumors and evaluate the impact of SRC-3 deletion in these AH tumor cells. We compared prostate AH lesions collected from 12-, 18- and 24-week-old 3FloxT and 3PEKOT mice. From histological examination of H&E stained sections, we found that AH tumor progression was slower in 3PEKOT mice (Fig. 4A).
Slower progression of AH tumors in 3PEKOT mice. A. H&E-stained prostate tumor sections prepared from 3PEKOT and 3FloxT mice at different ages. B. IHC for SRC-3 (brown) in AH tumors of 3PEKOT and 3FloxT mice at different ages. C. Representative images of IHC for Ki67 (brown) in 24-week-old 3FloxT and 3PEKOT mice. D. Proliferation indexes of AH tumor cells in 3PEKOT and 3FloxT mice at different ages. Six mice were assayed for each data point. The percentage of Ki67+ cells was calculated by dividing the number of ki67+ tumor cells by total number of tumor cells per imaging field. E. Double immunohistofluorescent staining for SRC-3 (green) and AR (red) in prostate tumors of 24-week-old 3FloxT and 3PEKOT mice. Some SRC-3+ tumor cells are AR-.
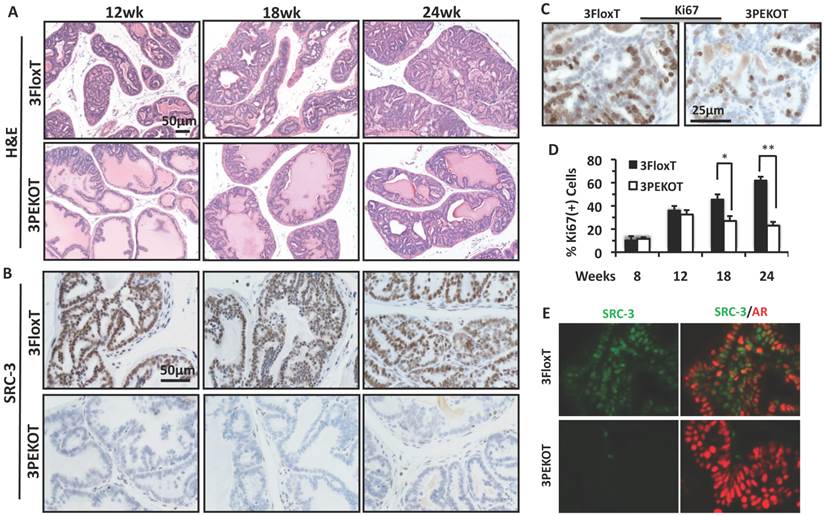
At 12 weeks, 3FloxT mice developed PIN lesions, while 3PEKOT mice contained both hyperplasia and prostate intraepithelial neoplasia (PIN) lesions. At 18 weeks, 3FloxT mice developed well-differentiated adenocarcinoma (WDA), while tumors in 3PEKOT mice were still at PIN stage. At 24 weeks, high-grade moderately differentiated adenocarcinoma (MDA) was observed in 3FloxT mice, while tumors in 3PEKOT were arrested at WDA stage. SRC-3 protein was detected in all LEC-like AH tumor cells in 3FloxT mice throughout stages from 12 to 24 weeks (Fig. 4B). However, in the prostates of 3PEKOT mice SRC-3 protein was ablated in most AH tumor regions. In the regions where SRC-3 was efficiently ablated in LECs, the tumor progression was halted at WDA stage (Fig. 4B). To determine if cell proliferation was reduced in the AH regions of 3PEKOT mice compared to 3FloxT mice, we performed IHC for Ki67. We found that the percentage of Ki67+ cells in the AH tumor regions in 3PEKOT mice was not significantly changed at 8 and 12 weeks of age, but did significantly decreased at 18 and 24 weeks of age when compared with that of 3FloxT mice (Fig. 4C and D). Interestingly, in the AH tumors of 3FloxT mice, the proliferation index increased incrementally as age increased. On the contrary, in the AH tumors of 3PEKOT mice, the proliferation index decreased as age increased from 12 weeks (Fig. 4D). Furthermore, double immunohistofluorescent staining of AR and SRC-3 revealed that SRC-3 was expressed in all AR+ AH tumor cells in 3FloxT mice. Knockout of SRC-3 in these cells caused higher AR immunoreactivity, suggesting that the AH tumors in 3PEKOT mice might be more differentiated (Fig. 4E). Moreover, in some AH tumors of 3FloxT mice, a small number of SRC-3+/AR- cells can be identified, supporting the notion that tumors with SRC-3 expression have a less differentiated phenotype. Taken together, these results demonstrate that ablation of SRC-3 in AH tumor cells decreased cell proliferation and prevented the progression of these tumors.
SRC-3 is required for the development of prostate neuroendocrine tumor
In agreement with the results shown in Fig. 3B, IHC for AR and Syp identified AR+/Syp- AH tumors in all of the 3FloxT mice at 30 weeks of age, but frequent AR-/SyP+ NE tumors in 3PEKOT mice at 30 weeks of age (Fig. 5A). To define the general contribution of SRC-3 in development of NE tumors, we analyzed AR and Syp expression in prostate tumors isolated from 24- and 30-week-old TRAMP;SRC-3-/- mice with global knockout of SRC-3 (6 mice at each age group). Importantly, we only found AR+/Syp- AH tumors in these mice (Fig. 5A and data not shown), indicating that SRC-3 is required for the development of NE tumor in TRAMP mice.
SRC-3+ NE tumors are commonly observed in 30-week-old 3PEKOT mice. A. IHC for AR and Syp (brown) in tumor sections of 30 week-old 3FloxT, 3PEKOT and SRC-3-/-;TRAMP mice. AH, atypical hyperplasia; NE, neuroendocrine tumor. B. IHC for SV40 T antigen, AR, SRC-3 and Syp (brown) in adjacent sections of a NE tumor from a 30-week-old 3PEKOT mouse.
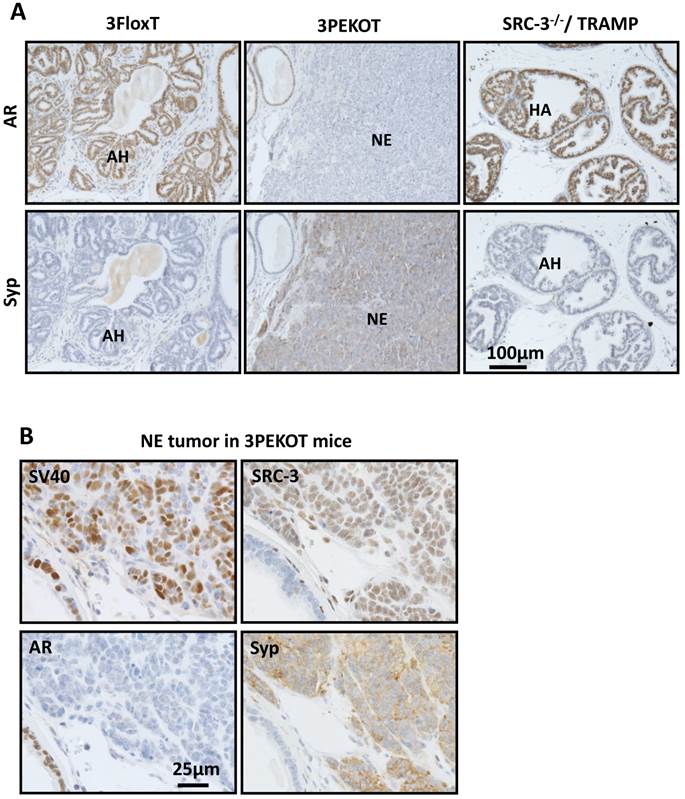
Since Cre is not expressed in NE cells in the prostates of PB-Cre transgenic mice [30], we hypothesized that the floxed SRC-3 alleles in the NE tumors developed in 3PEKOT mice at 30 weeks were not deleted. Indeed, IHC revealed that almost all of these AR-/Syp+ NE tumor cells still expressed SRC-3 protein, as well as the SV40-T antigen (Fig. 5B). These results clearly indicate that SRC-3 is essential for the development of NE tumors induced by inhibition of p53, Rb and PP2A by SV40-T/t antigens in the mouse prostate and this essential role of SRC-3 in the development of NE tumor is independent of AR.
Neuroendocrine tumor cells may be derived from the progenitor cell subpopulation
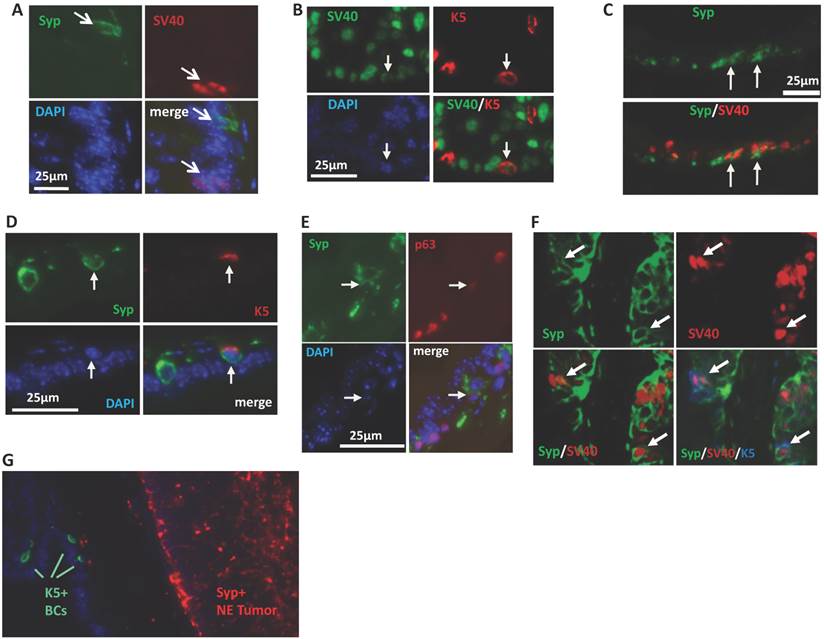
To explore the cellular origin of the SRC-3-dependent NE tumors, we performed double immunofluorescent staining for SV40-T antigen that induces tumorigenesis and the Syp that marks the neuroendocrine cell linage to identify the cell type that NE tumors may come from. At the early stage (8 weeks of age) of prostate tumorigenesis in TRAMP mice when the prostate still had normal morphology, we only detected Syp+ NE cells in three out of seven 3PEKOT mice, indicating these NE cells are extremely rare in the prostate epithelium at this stage. Importantly, these well-scattered NE cells did not express SV40-T, indicating these individual mature NE cells are normal NE cells and not the originating cells of NE tumors in 3PEKOT mice. At this stage, SV40-T was only detected in some of the Syp- epithelial cells (Fig. 6A). These observations raised the possibility that NE tumor cells might be derived and differentiated from progenitor-like epithelial cells that express SV40-T, since there are stem cells residing in the p63+ BC population in the prostate of adult mouse and these stem cells are capable of generating basal, luminal and NE epithelial cells [31-34]. To investigate this possibility, we immunostained Syp, SV40-T, p63 and K5 in prostate sections prepared from 8-12-week-old 3PEKOT and 3FloxT mice. p63 and K5 are prostate BC markers that are also expressed in epithelial progenitor and intermediate progenitor cells [31-34]. We found SV40-T expression in a small subpopulation of K5+ BCs in the prostate epithelium of both 3PEKOT and 3FloxT mice (n=6 for each group), suggesting that K5+ basal/progenitor cells may be transformed by the SV40-T/t oncogenic proteins (Fig. 6B). We detected small clusters of Syp+ cells, and some of these cells co-expressed SV40-T or K5 with Syp in all examined 3PEKOT mice (Fig. 6C and D). However, Syp and K5 double positive cells were not observed in the prostates of 3FloxT mice at the same stage (data not shown). Moreover, we also identified cells with weaker Syp and p63 immunostaining signals, another feature of intermediate cells, in the prostates of 3PEKOT mice (Fig. 6E). More importantly, by performing triple immunofluorescent staining we confirmed a small subpopulation of cells that indeed co-express Syp, K5 and SV40-T in the prostates of 3PEKOT mice (Fig. 6F). Taken together, these results indicate that Syp and SV40-T begin to express in some of the epithelial intermediate progenitor cells derived from p63+/K5+ progenitor cells. These SV40-T/t-transformed intermediate progenitor cells but not the mature NE cells may be the originating cells of NE tumors observed in TRAMP mice. As expected, the Syp+ cells in the large NE tumor regions no longer express K5 and p63 (Fig. 6G and data not shown), supporting the notion that these tumor cells are differentiated from SV40-T, p63 and K5 triple positive tumor progenitor cells.
NE tumors may be derived from intermediate progenitor cells expressing SV40-T antigen. A. Double immunostaining for Syp (green) and SV40-T antigen (red) in the prostate tissue section of an 8-week-old 3PEKOT mouse. Arrows indicate immunostaining signals for Syp or SV40-T. B. Double immunostaining for SV40-T (green) and K5 (red) in a prostate tissue section of a 3PEKOT mouse. Arrows indicate the same cell positive for SV40-T, K5 and DAPI staining. C. Double immunostaining for Syp (green) and SV40-T (red) in a prostate tissue section of a 3PEKOT mouse. Arrows indicate double positive cells in the cluster of NE cells. D. Double immunostaining for Syp (green) and K5 (red) in the prostate section of a 3PEKOT mouse. Arrows indicate the double positive cells. E. Double immunostaining for Syp (green) and p63 (red). Arrows indicate the Syp+ cell with weak p63 immunoreactivity. F. Triple immunostaining for Syp (green), SV40-T (red) and K5 (blue) in a prostate section of 12-week-old 3PEKOT mouse. Arrows indicate the triple positive cells. G. Double immunostaining for K5+ BCs and Syp+ NE tumor cells in a prostate of 30-week-old 3PEKOT mouse with a large NE tumor.
Discussion
In this study, we generated 3FloxT and 3PEKOT mice and demonstrated that specific ablation of SRC-3 in the AR+ prostate epithelial cells does not affect SV40-T/t expression driven by the androgen-responsive rat probasin promoter, which is consistent with our previous study showing SRC-3 is not essential for androgen-regulated normal prostate regeneration and gene expression in mice [27]. We found that deletion of SRC-3 significantly reduced the proliferation and growth of the AR+ AH tumor cells in 3PEKOT mice versus 3FloxT mice. This result is consistent with our previous study showing global knockout of SRC-3 in TRAMP mice did not affect AH tumor initiation but restricted its growth and progression [27]. Because TRAMP;SRC-3-/- mice used in the previous study had multiple other phenotypes such as significantly decreased body weight and circulating levels of IGF-I and IGFBP-3 [27, 35, 36], we were unable to distinguish the intrinsic role of SRC-3 in the prostate tumor cells from the systemic or local stromal influence. In the current study, inhibition of AH tumor growth by specific knockout of SRC-3 in the AR+ prostate epithelial cells in 3PEKOT mice clearly indicates the cell-autonomous role of SRC-3 in promotion of AR+ tumor cell growth. Since androgen signaling is normal in the prostate of SRC-3-/- mice [27], the inhibited AH tumor growth in 3PEKOT mice is unlikely attributed to a changed AR function. Instead, previous in vitro studies have shown that SRC-3 promotes prostate cancer cell proliferation through enhancing the activity of PI3K-Akt signaling pathway and upregulating cell cycle genes [24, 25].
PHY tumors are fibroepithelial tumors composed of an epithelial and stromal component. Approximately 70% of PHY tumors are benign [37]. We did not find PHY tumors in 3FloxT mice, but observed PHY tumors in 32% of 3PEKOT mice. In these PHY tumors, SRC-3 was present in the stromal cells but absent in the SRC-3 knockout epithelial tumor cells. It is currently unclear whether SRC-3 expressed in these stromal cells plays a role in PCa development. Furthermore, the increased incidence of SRC-3-/AR+ epithelial tumor cells in PHY lesions also correlated with decreased incidence of SRC-3-/AR+ AH tumors in 3PEKOT mice. These observations suggest knockout of SRC-3 may help to convert the more malignant AH tumors into the less malignant PHY tumors, or just give them more space to grow.
The most interesting finding of this study is the extensive development of neuroendocrine tumors in 3PEKOT mice and the fact that these tumor cells still express SRC-3 owning to their escape from PB-Cre-mediated deletion of the floxed SRC-3 alleles in this cell lineage. Together with our results showing TRAMP;SRC-3-/- mice do not develop neuroendocrine tumor, we conclude that SRC-3 plays a permissive role in neuroendocrine tumor development in 3PEKOT mice. Since the evolution to a neuroendocrine phenotype in human PCa is associated with particularly aggressive castration-resistant prostate cancer (CRPC) [38], our findings that SRC-3 promotes neuroendocrine tumor development in TRAMP mice should aid in understanding mechanisms that promote human PCa progression into aggressive CRPC in the setting of androgen ablation therapy.
Because NE cells are observed in more than 30% of human primary PCa and the number of NE cells positively correlates with Gleason Score and CRPC cases, it is important to find the origin of these cancer-associated NE cells. In general, NE cells are thought to originate from neural crest-derived progenitors [39]. However, clinical analyses of the benign prostate implicate local epithelial stem cells as NE precursors [40, 41]. Moreover, in vitro studies have suggested NE tumor cells in PCa arise from trans-differentiation of epithelial adenocarcinoma with a luminal phenotype [42, 43]. In TRAMP mice, it has been shown that the BrdU-retaining cells after androgen deprivation express SV40-T antigen and Syp, suggesting NE tumor cells arise independent of androgen-deprivation [44]. In TRAMP mice with a FVB strain background, NE precursor lesions can be detected at 4 weeks of age [12]. These studies suggest that NE tumor cells can be derived either from AR+/Syp- adenocarcinomas through trans-differentiation or from unknown progenitor cells.
In this study, we showed that differentiated individual Syp+ NE cells do not express SV40-T antigen and thus, these NE cells are not the parent cells of the Syp+/SV40-T+ NE tumor cells in TRAMP mice. We also clearly showed that SRC-3 is efficiently knocked out in AR+/Syp- AH and PHY tumor cells in 3PEKOT mice, while the Syp+/AR- NE tumor cells maintain SRC-3 expression. These findings prove that the SRC-3+ NE tumor cells cannot be trans-differentiated from the SRC-3 knockout AR+/Syp- tumor cells. Instead, our data indicate that the AR-/Syp+/SRC-3+ NE tumor cells originate from SV40-T/t-transformed progenitor cells. First, at early stages of PCa development in adult 3PEKOT mice, we found that Syp+/SV40-T+ NE cells always form clusters containing several such cells, but singly scattered normal NE cells never express SV40-T antigen, suggesting these clustered NE cells might be the transformed pioneers of NE tumors. Second, we found certain cells in the Syp+/SV40-T+ clusters co-express p63, a marker of BCs that contain prostate epithelial progenitors in adult mice. A previous study also identified SV40-T expression in a subset of p63+ BCs in TRAMP mice [45]. However, p63 is not expressed in bigger Syp+ NE lesions seen at later stages in tumor development. These results suggest the p63+/Syp+/SV40-T+ cells are progenitors derived from the p63+ stem-like cells in the BC layer. Finally, the progenitor feature of these p63+/Syp+/SV40-T+ cells is further supported by their relatively weak expression of K5, which is a marker for basal and intermediate epithelial cells and cancer stem cells in the prostate [46]. The aforementioned analysis was made possible by co-expression of Cre and SV40-T/t in a subpopulation of basal/intermediate cells. It is noteworthy that overlap between expression of these transgenes was not 100% given sequence difference in the upstream androgen response elements and the stochastic nature of promoter activation in different transgenic mouse lines.
In summary, our results suggest that SV40-T/t antigens start to express in a subset of p63+/K5+ intermediate cells derived from adult prostate epithelial stem-like cells in TRAMP mice. The inhibition of p53, RB and PP2A tumor suppressors by SV40-T/t antigens in these intermediate cells causes uncontrolled expansion of these cancer stem-like cells. In the presence of SRC-3, the majority of these cells undergo partial differentiation into AR+/Syp-/SRC-3+ luminal-like AH tumor cells, while a small population of these cells undergo partial differentiation into AR-/Syp+/SRC-3+ NE tumor cells. When SRC-3 is conditionally knocked out by the AR-dependent expression of the PB-Cre transgene in the AR+/Syp- AH tumor cell lineage of 3PEKOT mice, the proliferation of these tumor cells is inhibited, leading to a switch to PHY tumors with dominant stromal accumulation. However, SRC-3 cannot be knocked out in AR-/Syp+ NE tumor cell lineage in 3PEKOT mice, allowing NE tumors rapidly develop and progress into large malignant NE tumors. Because SRC-3 is required for AH tumor growth and NE tumor development, global knockout of SRC-3 in TRAMP mice can effectively inhibit the growth and progression of AH tumors [27] and the development of NE tumors (data in this study). These findings have important basic and translational scientific implications. More specifically, our model suggests that the highly aggressive PCa with NE tumor cells may be originated from undifferentiated or poorly differentiated intermediate progenitor cells that can generate both AH and NE tumor cells. Thus, antiandrogen therapy may only inhibit the growth of partially differentiated AR+ AH tumor cells, but not the AR- progenitors and the AR-/Syp+ NE tumor cells, allowing these cells to progress into metastatic CRPC. Furthermore, our findings also suggest that SRC-3 may serve as a molecular target to control both AH and NE tumor growth and progression.
Acknowledgements
We thank Ms. Suoling Zhou for experimental assistance. This work is supported by NIH grants R01DK058242 and R01CA112403 to J.X., US Department of Defense Pre-doctoral CaP Research Scholarship W81XWH11-1-0194 to J.C.Y.T., NIH grants CA96824 and CA140388 and CPRIT grant RP110555 to F.W., and NIH grant P30CA125123 to Dan L. Duncan Cancer Center Genetically Engineered Mouse Core.
Competing Interests
All authors have no conflict of interests to disclose.
References
1. Jemal A, Siegel R, Xu J. et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300
2. Ruijter ET, van de Kaa CA, Schalken JA. et al. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol. 1996;180:295-9
3. Dickinson SI. Premalignant and malignant prostate lesions: pathologic review. Cancer Control. 2010;17:214-22
4. Brawn PN, Speights VO. The dedifferentiation of metastatic prostate carcinoma. Br J Cancer. 1989;59:85-8
5. Komiya A, Suzuki H, Imamoto T. et al. Neuroendocrine differentiation in the progression of prostate cancer. Int J Urol. 2009;16:37-44
6. Beltran H, Tagawa ST, Park K. et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386-9
7. Ismail AH, Landry F, Aprikian AG. et al. Androgen ablation promotes neuroendocrine cell differentiation in dog and human prostate. Prostate. 2002;51:117-25
8. Yuan TC, Veeramani S, Lin FF. et al. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13:151-67
9. Wright ME, Tsai MJ, Aebersold R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol Endocrinol. 2003;17:1726-37
10. Greenberg NM, DeMayo F, Finegold MJ. et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439-43
11. Hurwitz AA, Foster BA, Allison JP. et al. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol. 2001 Chapter 20: Unit 20 5
12. Chiaverotti T, Couto SS, Donjacour A. et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. Am J Pathol. 2008;172:236-46
13. Tang Y, Wang L, Goloubeva O. et al. The relationship of neuroendocrine carcinomas to anti-tumor therapies in TRAMP mice. Prostate. 2009;69:1763-73
14. Kaplan-Lefko PJ, Chen TM, Ittmann MM. et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55:219-37
15. McCarthy RP, Zhang S, Bostwick DG. et al. Molecular genetic evidence for different clonal origins of epithelial and stromal components of phyllodes tumor of the prostate. Am J Pathol. 2004;165:1395-400
16. Xu J, Wu RC, O'Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615-30
17. Walsh CA, Qin L, Tien JC. et al. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int J Biol Sci. 2012;8:470-85
18. Anzick SL, Kononen J, Walker RL. et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965-8
19. Torres-Arzayus MI, Font de Mora J, Yuan J. et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263-74
20. Kuang SQ, Liao L, Wang S. et al. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005;65:7993-8002
21. Kuang SQ, Liao L, Zhang H. et al. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875-85
22. Fereshteh MP, Tilli MT, Kim SE. et al. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res. 2008;68:3697-706
23. Gnanapragasam VJ, Leung HY, Pulimood AS. et al. Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer. 2001;85:1928-36
24. Zhou HJ, Yan J, Luo W. et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976-83
25. Zou JX, Zhong Z, Shi XB. et al. ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate. 2006;66:1474-86
26. Yan J, Erdem H, Li R. et al. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460-8
27. Chung AC, Zhou S, Liao L. et al. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Res. 2007;67:5965-75
28. Niu YN, Xia SJ. Stroma-epithelium crosstalk in prostate cancer. Asian J Androl. 2009;11:28-35
29. Liu Z, Liao L, Zhou S. et al. Generation and validation of a mouse line with a floxed SRC-3/AIB1 allele for conditional knockout. Int J Biol Sci. 2008;4:202-7
30. Jin C, McKeehan K, Wang F. Transgenic mouse with high Cre recombinase activity in all prostate lobes, seminal vesicle, and ductus deferens. Prostate. 2003;57:160-4
31. Kurita T, Medina RT, Mills AA. et al. Role of p63 and basal cells in the prostate. Development. 2004;131:4955-64
32. Lawson DA, Xin L, Lukacs RU. et al. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci U S A. 2007;104:181-6
33. Ousset M, Van Keymeulen A, Bouvencourt G. et al. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat Cell Biol. 2012;14:1131-8
34. Lee D-K, Liu Y, Liao L. et al. The Prostate Basal Cell (BC) Heterogeneity and the p63-Positive BC Differentiation Spectrum in Mice. Int J Biol Sci. 2014;10(9):1007-1017 doi:10.7150/ijbs.9997
35. Xu J, Liao L, Ning G. et al. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379-84
36. Liao L, Chen X, Wang S. et al. Steroid receptor coactivator 3 maintains circulating insulin-like growth factor I (IGF-I) by controlling IGF-binding protein 3 expression. Mol Cell Biol. 2008;28:2460-9
37. Guillot E, Couturaud B, Reyal F. et al. Management of phyllodes breast tumors. Breast J. 2011;17:129-37
38. Mosca A, Berruti A, Russo L. et al. The neuroendocrine phenotype in prostate cancer: basic and clinical aspects. J Endocrinol Invest. 2005;28:141-5
39. Nelson EC, Cambio AJ, Yang JC. et al. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:6-14
40. Bonkhoff H. Neuroendocrine differentiation in human prostate cancer. Morphogenesis, proliferation and androgen receptor status. Ann Oncol. 2001;12(Suppl 2):S141-4
41. Rumpold H, Heinrich E, Untergasser G. et al. Neuroendocrine differentiation of human prostatic primary epithelial cells in vitro. Prostate. 2002;53:101-8
42. Burchardt T, Burchardt M, Chen MW. et al. Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J Urol. 1999;162:1800-5
43. Bang YJ, Pirnia F, Fang WG. et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc Natl Acad Sci U S A. 1994;91:5330-4
44. Huss WJ, Gray DR, Tavakoli K. et al. Origin of androgen-insensitive poorly differentiated tumors in the transgenic adenocarcinoma of mouse prostate model. Neoplasia. 2007;9:938-50
45. Reiner T, de Las Pozas A, Parrondo R. et al. Progression of prostate cancer from a subset of p63-positive basal epithelial cells in FG/Tag transgenic mice. Mol Cancer Res. 2007;5:1171-9
46. Wang S, Garcia AJ, Wu M. et al. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103:1480-5
Author contact
![]() Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine. E-mail: jxuedu.
Corresponding author: Jianming Xu, PhD, Department of Molecular and Cellular Biology, Baylor College of Medicine. E-mail: jxuedu.