Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(10):1138-1149. doi:10.7150/ijbs.10366 This issue Cite
Research Paper
Interaction between HIV-1 Tat and DNA-PKcs modulates HIV transcription and class switch recombination
Shi-Meng Zhang1# ![]() , He Zhang1#, Tian-Yi Yang1, Tian-Yi Ying2, Pei-Xiang Yang3, Xiao-Dan Liu1, Sheng-Jian Tang4, Ping-Kun Zhou1
, He Zhang1#, Tian-Yi Yang1, Tian-Yi Ying2, Pei-Xiang Yang3, Xiao-Dan Liu1, Sheng-Jian Tang4, Ping-Kun Zhou1 ![]()
1. Department of Radiation Toxicology and Oncology; Beijing Institute of Radiation Medicine, 100850, Beijing, China;
2. The State Key Laboratory of NBC Protection for Civilian, 102205, Beijing, China;
3. Beijing Institute of Health Administration and Medical Information, 100850, Beijing, China;
4. Shandong Provincial Key Laboratory of Plastic and Microscopic Repair Technology, Institute of Plastic Surgery, Weifang Medical University, 261053, Weifang, Shandong Province, China.
# These authors contributed equally to this work.
Received 2014-8-19; Accepted 2014-9-17; Published 2014-10-9
Abstract
HIV-1 tat targets a variety of host cell proteins to facilitate viral transcription and disrupts host cellular immunity by inducing lymphocyte apoptosis, but whether it influences humoral immunity remains unclear. Previously, our group demonstrated that tat depresses expression of DNA-PKcs, a critical component of the non-homologous end joining pathway (NHEJ) of DNA double-strand breaks repair, immunoglobulin class switch recombination (CSR) and V(D)J recombination, and sensitizes cells to ionizing radiation. In this study, we demonstrated that HIV-1 Tat down-regulates DNA-PKcs expression by directly binding to the core promoter sequence. In addition, Tat interacts with and activates the kinase activity of DNA-PKcs in a dose-dependent and DNA independent manner. Furthermore, Tat inhibits class switch recombination (CSR) at low concentrations (≤4 µg/ml) and stimulates CSR at high concentrations (≥8 µg/ml). On the other hand, low protein level and high kinase activity of DNA-PKcs promotes HIV-1 transcription, while high protein level and low kinase activity inhibit HIV-1 transcription. Co-immunoprecipitation results revealed that DNA-PKcs forms a large complex comprised of Cyclin T1, CDK9 and Tat via direct interacting with CDK9 and Tat but not Cyclin T1. Taken together, our results provide new clues that Tat regulates host humoral immunity via both transcriptional depression and kinase activation of DNA-PKcs. We also raise the possibility that inhibitors and interventions directed towards DNA-PKcs may inhibit HIV-1 transcription in AIDS patients.
Keywords: HIV-1 tat, DNA-PKcs
Introduction
Tat, an 86- to 101-amino acid regulatory protein encoded by HIV-1, plays a very important role in the efficient transcription and replication of the HIV virus. It binds to the trans-activating region (TAR) and forms a complex with positive transcription elongation factor b (P-TEFb), along with CDK9 and Cyclin T1, to initiate and maintain efficient viral transcription [1-3]. Tat is secreted by infected cells and can enter into bystander cells where it acts as a paracrine molecule. Tat impairs a series of signal pathways by targeting different host-cell proteins. For example, our previous work demonstrated that Tat impairs the cell cycle by targeting Plk1, Cyclin B1 and Tip60 [4].
DNA-dependent protein kinase, a holoenzyme comprised of Ku70, Ku80 and the catalytic subunit DNA-PKcs, is a critical component of the non-homologous end joining pathway (NHEJ) and is responsible for the repair of DNA double-strand breaks [5-7]. When there is a break in the DNA double strand, Ku first binds to the ends of the broken DNA strands. DNA-PKcs is then recruited to the break site in a Ku-dependent manner and is activated via auto-phosphorylation [8, 9]. Following activation, DNA-PKcs phosphorylates a host of downstream targets that function in double-strand break (DSB) repair and DNA damage response (DDR). Moreover, DNA-PKcs also plays an important role in immunoglobulin class switch recombination (CSR) [10-12] and V(D)J recombination, which are a part of the DSB event [13-15]. DNA-PKcs has also been shown to function in regulating mitotic progression and spindle stability in response to DNA damage. Dysfunction of DNA-PKcs could result in mitotic catastrophe of the cell.
Accumulating evidence continues to indicate that AIDS patients with cancer are much more sensitive to radiotherapy than HIV-negative cancer patients. Previously, our group demonstrated that Tat sensitizes cells to radiation by depressing the expression of DNA-PKcs, but the exact mechanism remained elusive [16]. To delineate the mechanism, we conducted a series of in vitro and in vivo assays and demonstrated that Tat depresses DNA-PKcs expression by directly binding to its core promoter region. In addition, Tat also interacts with the FAT domain of DNA-PKcs and activates its kinase activity in a DNA-independent manner. Furthermore, Tat modulates CSR via its dual role in relation to DNA-PKcs. On the converse, DNA-PKcs modulates Tat-mediated HIV transcription by forming a quaternary complex with Tat, Cyclin T1, and CDK9 and also through its kinase activity.
Materials and Methods
Cell lines
The human T-lymphotropic virus type-1-transformed T-cell line MT2 was kindly provided by Dr. Lin Li (Beijing Institute of Microbiology and Epidemiology). CH12F3 cells were kindly provided by Dr. Hua-Dong Pei (Beijing Proteome Research Center). The MT2 and CH12F3 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. HEK293T and Hela cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum.
Plasmids and the recombinant Tat protein
Myc-Tat and flag-Tat were created as described in our previous study [4]. pEYFP-DNA-PKcs was gifted by Dr. David Chen (Southwestern Medical Center). The luciferase reporter plasmid pGL3-DPKPF and its truncation mutants were constructed by inserting the full-length DNA-PKcs promoter or its mutants into the pGL3-basic vector using the Mlu I and Bgl II restriction sites. The full-length DNA promoter sequence was amplified from Hela-cell genomic DNA using the primers DPKPFF (5'CAGACGCGTCGGACGTTCCCGTCGGCTCCG3', Mlu I is underlined) and DPKPFR (5'GGAAGATCTGCCCCTACGCGCGGAGGCGG3' Bgl II is underlined). The recombinant Tat protein was expressed and purified as described in our previous study [17].
In vitro DNA-PKcs kinase assay
The in vitro DNA-PKcs kinase activity was measured using the SignaTECT® DNA-Dependent Protein Kinase Assay System (Promega) according to the manufacturer's protocol. Reactions (25 μl) containing purified MT2-cell nuclear extract, DNA-PK activation buffer, reaction buffer, a DNA-PK biotinylated p53-derived peptide substrate, 0.5 μCi [γ-32P] ATP (10 mCi/ml), double-stranded oligonucleotide (50 bp) and recombinant Tat protein, at the concentrations indicated in the Figure 4, were incubated at 30°C for 5 min. A termination buffer was then added, and 10 μl of each reaction mixture was spotted onto SAM2® capture membranes. After washing with 2 M NaCl, the membranes were dried, and the 32P incorporation was measured using autoradiography.
CSR analysis
The CH12F3 cells were maintained in vitro, and CSR assays were performed as previously described[18]. Briefly, the cells were maintained in RPMI 1640 supplemented with 10% FBS, 10 mM 2-ME, and 5% NCTC-109 (Invitrogen). For the CSR assay, the cells were stimulated with 250 ng/ml of recombinant murine CD40L (PeproTech), 10 ng/ml of recombinant murine IL-4 (R&D Systems), and 1 ng/ml of recombinant human TGF-β (R&D Systems) (CIT) for 72 h and were then analyzed by flow cytometry.
To analyze the CSR from IgM (IgM+/IgA-) to IgA (IgM-/IgA+), CH12F3 cells were intracellularly stained with PE-conjugated anti-murine IgA clone 1144-2 (12-5994-82; eBioscience) and were assessed for membrane IgM expression using FITC-conjugated anti-murine IgM (11-5890-82; eBioscience). The cells were then analyzed on a Cytomics (Beckman Coulter), and the data were analyzed using CXP software.
Co-immunoprecipitation
For the co-immunoprecipitation, 293T cells were transfected with the plasmids described above. Thirty-six hours after the transfection, the cells were collected and lysed with lysis buffer (50 mM TRIS-HCl; 150 mM NaCl; 1% Nonidet P-40; 0.1% SDS; 1 mM DTT, pH 7.5) containing protease inhibitor cocktail (Roche) at 4°C for 1 h. After centrifugation at 13,000 rpm for 10 min, the supernatant was subjected to immunoprecipitation by incubating with the indicated antibody and was then bound to Protein A/G-agarose beads for 2 h at 4°C. The beads were washed with lysis buffer three times, boiled in Laemmli buffer and analyzed by western blot.
GST pull-down
Tat cDNA was inserted into the pGEX-4T vector and expressed as GST fusion proteins in E. coli Rosetta-gami B(DE3). In preparation, Glutathione (GST)-Sepharose beads were prewashed with TEN100 buffer (20 mM Tris, pH 7.4; 0.1 mM EDTA; and 100 mM NaCl) three times and then equilibrated in the TEN100 buffer. The GST or GST-fusion proteins were then rotated with the GST-Sepharose beads (Pharmacia) at 4°C for 1 h (for immobilization). Next, whole-cell extracts or purified proteins containing the immobilized GST or GST-fusion proteins were rotated at 4°C for 1 h. For the whole-cell extracts, the beads were washed four times with NETN buffer (0.5% Nonidet P-40; 0.1 mM EDTA; 20 mM Tris, pH 7.4; and 300 mM NaCl). The protein-bound beads were boiled in Laemmli sample buffer, and the proteins were verified by western blot.
Luciferase reporter assay
Full-length and different mutants of the DNA-PKcs promoter region were cloned upstream of firefly luciferase in the pGL3-basic plasmid. MT2 cells were then co-transfected with pGL3 and pRL-CMV (Renilla luciferase, internal standard) with or without myc-Tat. The luciferase reporter assay was carried out using the Dual-Luciferase Reporter Assay System (Promega, USA).
Electrophoretic mobility shift assay (EMSA)
The EMSA was performed using the EMSA kit from Beyotime, according to the manufacturer's instructions. The double-stranded synthetic oligonucleotide probes used for the competition EMSAs were prepared by annealing sense and reverse-complement strands at temperatures ranging from 94°C to room temperature. A double-stranded oligonucleotide corresponding to the -1 to -63 region of the DNA-PKcs promoter (DPK63) was labeled by filling-in the sticky ends with T4 Polynucleotide Kinase and [γ-32P]dATP and was used as the DNA probe in the EMSA. In a dose-dependent experiment, 0.5, 1, and 2 µg of recombinant His-tagged Tat protein and the 32P-labeled DPK63 probe were added. In a specificity competition experiment, the 32P-labeled DPK63 probe and a volume equivalent to 100-fold of an unlabeled DPK63 probe were added. In a non-specific competition experiment, the 32P-labeled DPK63 probe and a volume equivalent to 100-fold of an unlabeled scrambled probe were added. In a super-shift assay, the 32P-labeled DPK63 probe, 2 µg of Tat protein and 1 µg of anti-His antibody were added. Following the above reactions, all of the samples were subjected to 4% nondenaturing polyacrylamide gels in 0.5X Tris-borate-EDTA buffer. The gels were then dried and visualized by autoradiography.
Results
Identification of the core sequence of the DNA-PKcs promoter
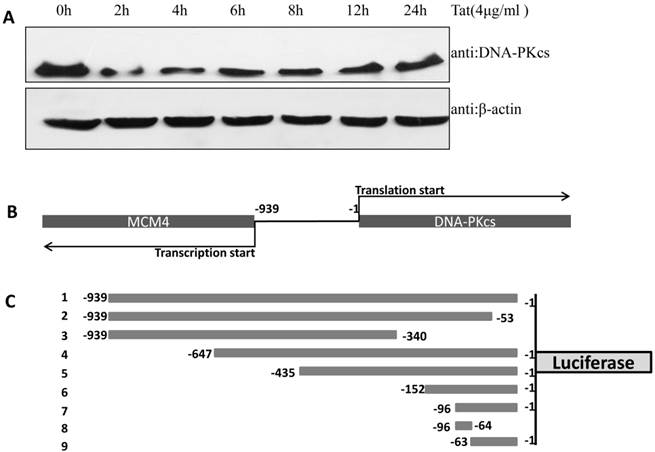
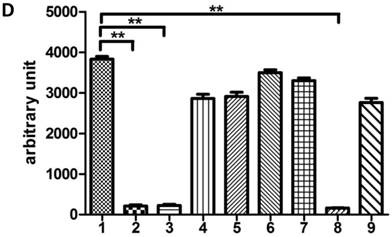
In our previous study, we demonstrated that DNA-PKcs was depressed in HIV-1 Tat-expressing cells. This down-regulation of PRKDC (encoding DNA-PKcs) was also confirmed by microarray [16]. However, the detailed mechanism of DNA-PKcs down-regulation remained, for the most part, unclear. To confirm that DNA-PKcs is down-regulated directly by Tat, we treated MT2 cells with recombinant Tat protein (4 µg/ml), and the cells were harvested at different time points. As shown in Fig. 1A, Tat drastically down-regulates DNA-PKcs. It is well documented that PRKDC localizes head to head with MCM4, though both share a common promoter sequence of less than 1 kb, as shown in Fig. 1B. The common promoter region lacks a TATA box, and PRKDC has no CAAT box, which is characteristic of house-keeping genes [19, 20]. To further characterize the core region of PRKDC, we cloned the sequence between the PRKDC translation start site and the MCM4 transcription start site (939 bp) and its mutants, as shown in Fig. 1C, into the luciferase reporter plasmid pGL3-basic. A dual luciferase assay demonstrated that the sequences containing the region between -63 and -1 have strong promoter activity, while sequences lacking this region have 100-fold lower activity (Fig. 1D). Our results definitively showed that the -63 to -1 region is the core sequence of the PRKDC promoter.
Identification of a key region in the DNA-PKcs promoter. A, E. coli HIV-1 Tat protein was added into the culture medium of MT2 cells, and DNA-PKcs was detected at different time points. B, A schematic representation of the divergent MCM4 and DNA-PKcs transcripts in chromosome 8. C, A schematic representation of the promoter sequence we cloned and its mutants. The longest sequence represents the sequence between the MCM4 transcription start site and the DNA-PKcs translation start site. All of the sequences were cloned into the firefly luciferase reporter plasmid pGL3-basic using the Mlu I and Bgl II restriction sites. D, All of the pGL3 luciferase-reporter constructs were transfected into HEK293 cells to determine their promoter activity. The numbers represent the average and SD from at least 3 replicates of each constructs. **P ≤0.01.
HIV-1 Tat down-regulates DNA-PKcs expression by directly binding to the core promoter sequence
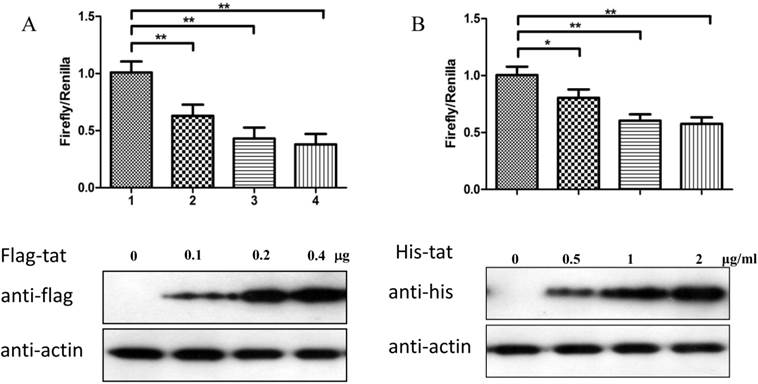
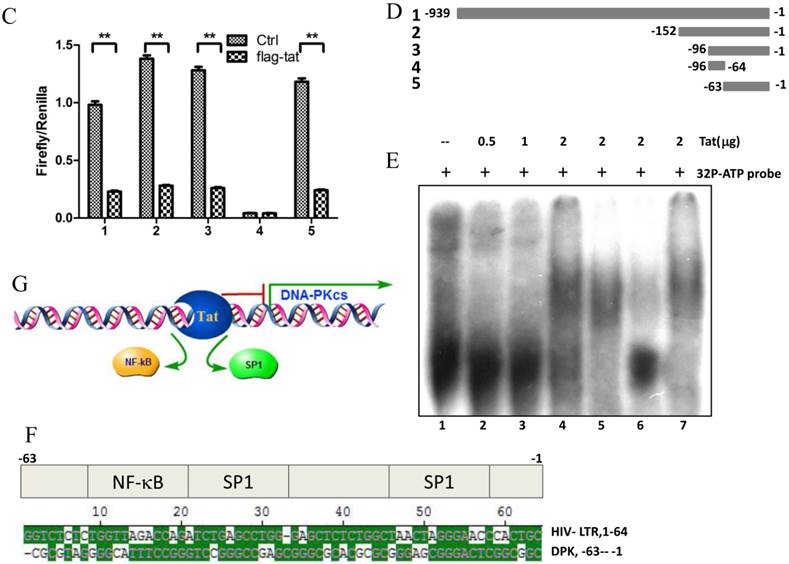
To explore whether Tat regulates PRKDC at the transcriptional level, HEK293 cells transfected with pGL3-DPKPF (containing the full-length PRKDC promoter) and RL-CMV were co-transfected with a flag-Tat plasmid or treated with recombinant Tat protein. As shown in Fig. 2A and B, both gene transfection and administration of the Tat protein were capable of suppressing the PRKDC promoter activity in a dosage dependent manner. To further investigate the Tat-targeted region, we conducted a luciferase assay using pGL3-DPKPF and its truncation mutants (Fig. 2D). The results showed that Tat targets the core promoter region to suppress the PRKDC promoter (Fig. 2C). Bioinformatics methods predicted that there are two SP1 binding sites and one NF-κB binding site in this core region (Fig. 2F), and there is high homology between the core region and the HIV 5'-LTR sequence (>45%). It is well known that Tat activates HIV transcription by targeting SP1. However, the actual location of physical interaction between Tat and SP1 remains controversial [21, 22]. In addition, Tat binds to the TAR (Trans-Activating Response) element of its own RNA, which is encoded by LTRs. Based on the LTR similarities, we speculated that the core promoter region could be bound by the Tat protein. Not surprisingly, in vitro EMSA results showed that Tat binds to the core promoter region of PRKDC (Fig. 2E). Therefore, Tat depresses PRKDC expression by directly binding to the core promoter region.
HIV-1 Tat down-regulates DNA-PKcs at the transcriptional level. A, The firefly luciferase reporter plasmid pGL3-basic containing the full-length DNA-PKcs promoter insertion (pGL3-DPKPF, Figure 1C, construct number 1) was transfected together with or without the renilla luciferase plasmid RL-CMV (internal standard) and flag-Tat into HEK293 cells. The dual luciferase activity and expression of Tat were assayed 48 h- post-transfection. B, pGL3-DPKPF and RL-CMV were transfected into HEK293 cells and recombinant Tat protein was added 24 h after the transfection. The dual luciferase activity and intracellular levels of Tat were measured 48 h- post-transfection. C,D, pGL3-DPKPF and its mutants, as indicated, were transfected with or without RL-CMV and flag-tat into HEK293 cells. The dual luciferase activity was assayed 48 h after transfection. E, A gel shift assay was performed to detect the direct interaction between Tat and the -63 to -1 region of the DNA-PKcs promoter. 1, Negative control, 32P-labeled oligo; 2, 32P-labeled oligo+0.5 µg of Tat protein; 3, 32P-labeled oligo+1 µg of Tat protein; 4, 32P-labeled oligo+2 µg of Tat (His-tagged) protein; 5, 32P-labeled oligo+2 µg of Tat protein+100-fold of unlabeled scrambled oligo (nonspecific competitor); 6, 32P-labeled oligo+2 µg of Tat protein+100-fold of unlabeled oligo (specific competitor); 7, 32P-labeled oligo+2 µg of Tat protein+anti-His antibody. F, Alignment of the HIV-1 5' LTR 1-63 region and the DNA-PKcs promoter -63 to -1 region. Upper panel: a bioinformatic prediction of transcription factor binding for the DNA-PKcs promoter -63 to -1 region. G, A model describing Tat protein binding to the core promoter region of DNA-PKcs. Please see the text for details. In A, B and C, three independent transfection were performed and values were shown with average and SD. *P ≤0.05, **P ≤0.01.
Direct interactions between Tat and DNA-PKcs
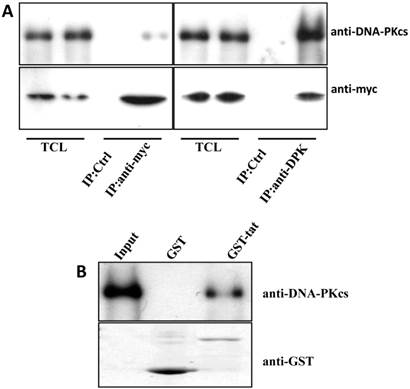
The Tat protein localizes predominantly to the cell nucleus and interacts with a number of nuclear proteins, including Cyclin T1, CDK9, Cyclin B1, etc. [1-3]. DNA-PKcs also resides primarily in the nucleus to maintain genomic integrity, especially under irradiating condition [9]. Based on this evidence, we presumed that there is a direct interaction between Tat and DNA-PKcs. To prove this, we performed a reciprocal co-immunoprecipitation (Co-IP) experiment using MT2 cell lysates containing ectopically expressed myc-tagged Tat, followed by a subsequent GST pull-down assay. The results demonstrated that Tat and DNA-PKcs strongly interact with each other (Fig. 3A, B).
Direct interaction between Tat and DNA-PKcs. A, MT2 cells were transfected with a myc-Tat plasmid. The total cell lysates were immunoprecipitated with normal IgG, anti-myc or anti-DNA-PKcs antibody, and a western blot was performed using the indicated antibodies. B, GST and GST-Tat proteins were expressed in E. coli Rosetta-strain cells, conjugated to glutathione beads and incubated with MT2 cell lysates. C, A schematic representation of the major domains of DNA-PKcs. D, The different DNA-PKcs truncation mutant plasmids with myc-tags were co-transfected with flag-Tat into HEK293 cells. The immunoprecipitation was performed with an anti-myc antibody and the WB was analyzed with the indicated antibodies.
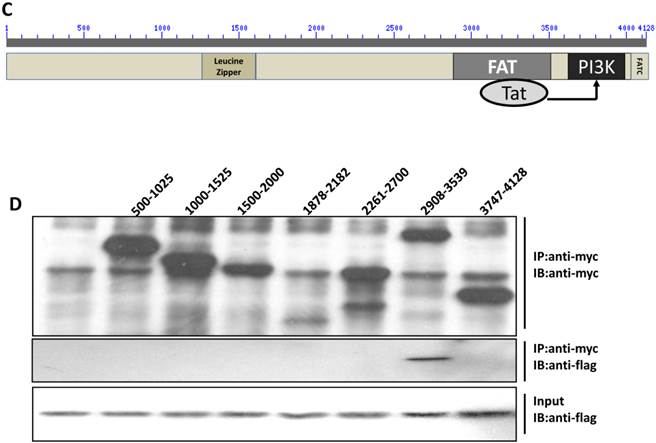
To further delineate the interaction domain of DNA-PKcs, we constructed a series of truncation-mutant expression vectors of DNA-PKcs. We co-transfected the myc-tagged truncation mutants along with flag-tagged Tat into 293T cells to determine the interacting domains. As shown in Fig. 3D, Tat strongly binds to the FAT domain of DNA-PKcs, just upstream of the PI3K domain.
HIV-1 Tat activates DNA-PKcs in a DNA-independent manner
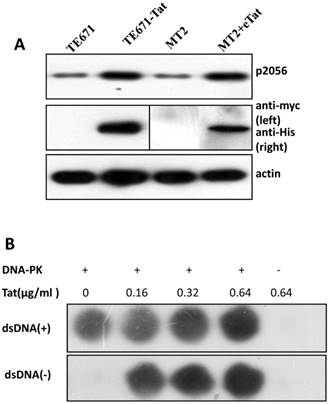
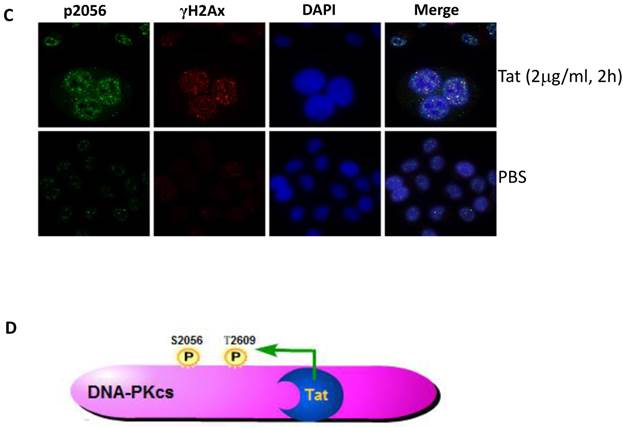
It is well established that DNA-PKcs plays a major role in non-homologous end joining, the primary pathway in mammals for repairing double-strand breaks. Following irradiation, DNA-PKcs is activated and auto-phosphorylates its Ser2056 [9, 23] near the ends of the double-stranded DNA breaks. However, the mechanism of DNA-activated auto-phosphorylation remains largely unknown. Because Tat interacts with DNA-PKcs, we questioned if it also modulates the kinase activity of DNA-PKcs. We detected the phosphorylation of Ser2056 in cells stably expressing Tat (TE671-Tat) and in recombinant Tat protein treated MT2 cells. As shown in Fig. 4A, both endogenous and exogenous Tat strongly stimulated DNA-PKcs auto-phosphorylation at Ser2056. To verify that Tat activates DNA-PKcs by direct interaction rather that via other signaling pathways, we conducted an in vitro DNA-PKcs kinase assay. The results showed that in the presence of DNA, Tat activates DNA-PKcs in a dose-dependent manner. Interestingly, Tat was capable of activating DNA-PKcs even in the absence of DNA. Immunofluorescence staining also confirmed that exogenous Tat activates DNA-PKcs to phosphorylate H2Ax (Fig. 4C). Overall, our results demonstrated that Tat activates DNA-PKcs in a DNA-independent and dosage-dependent manner.
HIV-1 Tat activates DNA-PKcs in a DNA-independent manner. A, DNA-PKcs S2056 phosphorylation in TE671, TE671-Tat (stably expressing Tat), MT2 and recombinant-Tat-treated MT2 cells was assayed with anti-S2056-p WB analysis. B, The in vitro kinase activity of DNA-PKcs was conducted with a DNA-Dependent Protein Kinase Assay Kit (Promega). Upper panel: DNA-PKcs activity in the presence of double-stranded DNA; Lower panel: DNA-PKcs activity in the absence of double-stranded DNA. Different concentrations of recombinant Tat were added as indicated. C, Tat stimulates the phosphorylation of H2Ax. Hela cells were seeded into six-well plates with coverslips and treated with recombinant Tat protein for 2 h and were then fixed and stained as described in the Materials and Methods. Anti-γH2Ax (red), anti-S2056-p (green) and nuclear staining by DAPI were performed. D, A model describing how Tat binds to DNA-PKcs and activates it in a DNA-independent manner.
HIV-1 Tat regulates class switch recombination
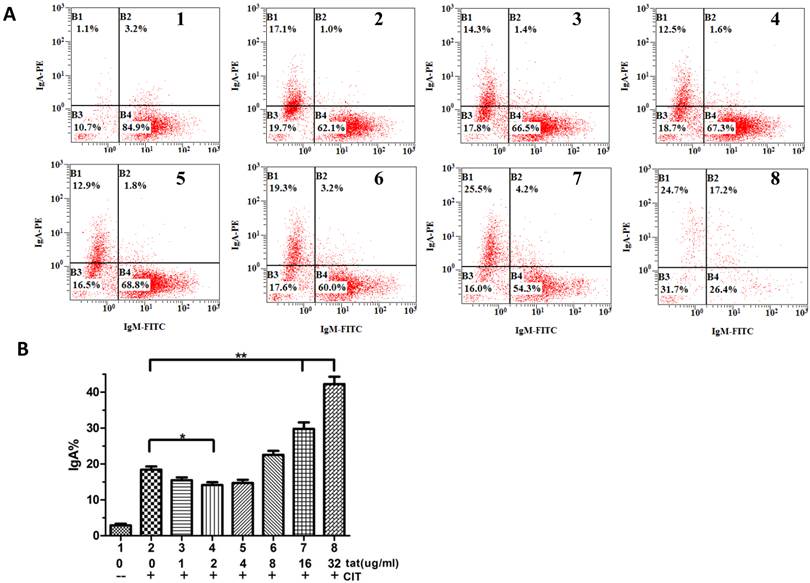
It was reported that DNA-PKcs plays a fundamental role in V(D)J recombination [15] and class switch recombination (CSR) [10]. To determine whether Tat regulates CSR by targeting DNA-PKcs, we used CH12F3 cells capable of switching from IgM to IgA. As shown in Fig. 5A and B, Tat inhibits CSR at low concentrations (≤4 µg/ml) and stimulates CSR at high concentrations (≥8 µg/ml).
HIV-1 Tat regulates class-switch recombination. A, A flow cytometry analysis of CH12F3 cells that were treated with or without CIT and Tat for 3 d and stained with anti-IgA and anti-IgM antibodies. The simultaneous increase of IgA and decrease of IgM expression indicates the effective CSR. B, The statistical analysis of the CSR results from A. Three independent experimental replicates were performed for each dose. B, The statistical analysis of the CSR results from A. *P ≤0.05, **P ≤0.01.
DNA-PKcs modulates HIV transcription with its kinase activity and by forming a complex with CDK9, Cyclin T1 and Tat
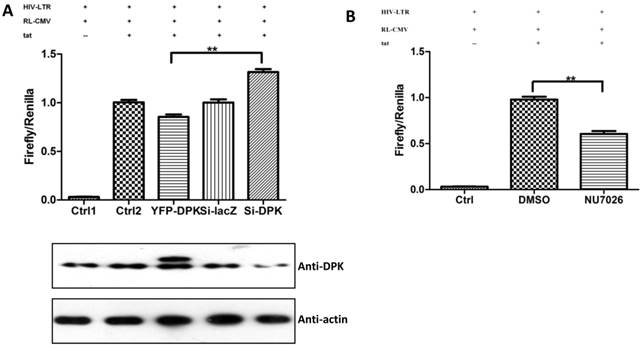
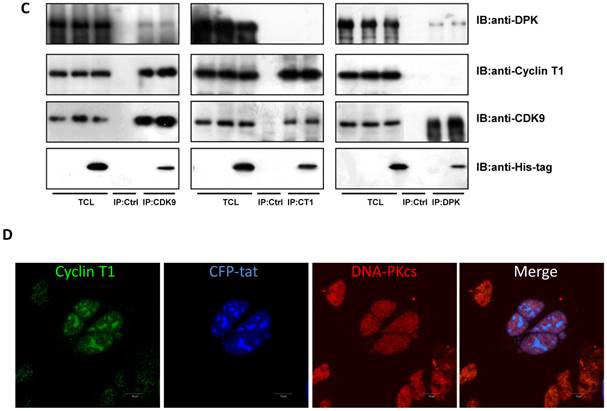
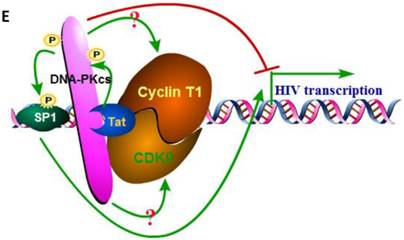
We have proven the interaction between Tat and DNA-PKcs and the regulation of DNA-PKcs by Tat. We next asked if DNA-PKcs modulates HIV-1 transcription by binding to Tat. To verify this hypothesis, we transfected the HIV-LTR reporter RL-CMV together with myc-Tat, YFP-DNA-PKcs, Si-lacZ or Si-DPK plasmids into MT2 cells. As shown in Fig. 6A, the overexpression of DNA-PKcs strongly inhibits HIV transcription, while knockdown of DNA-PKcs greatly stimulates HIV transcription. To further determine if the kinase activity of DNA-PKcs is involved in HIV transcription, we treated MT2 cells transfected with the HIV-LTR reporter plasmid with NU7026, a DNA-PKcs-specific inhibitor. The results demonstrated that inhibition of DNA-PKcs kinase activity drastically suppresses HIV transcription (Fig. 6B). In HIV-1-infected T cells, Tat forms a ternary complex with Cyclin T1 and CDK9 to initiate transcription [1-3]. To further investigate if DNA-PKcs forms a larger complex comprised of Tat, Cyclin T1 and CDK9, we performed three immunoprecipitation experiments using anti-DNA-PKcs, anti-Cyclin B1 and anti-CDK9 antibodies in the presence or absence of the Tat protein. As shown in Fig. 6C, DNA-PKcs interacts directly with CDK9 but not Cyclin T1, though Tat does not influence the affinity between DNA-PKcs and CDK9. Although Cyclin T1 co-localizes with DNA-PKcs, there is no direct interaction between the two proteins. These results imply that the DNA-PKcs protein suppresses HIV transcription by interacting with Tat and CDK9 and stimulates HIV transcription via its kinase activity.
DNA-PKcs modulates HIV transcription via its kinase activity and by forming a complex with CDK9, Cyclin T1 and Tat. A, The impact of DNA-PKcs expression levels on HIV transcription. MT2 cells were transfected with or without a DNA-PKcs over-expression plasmid, shRNA plasmid, HIV-LTR, RL-CMV and flag-Tat plasmids, as indicated. B, The plasmids, as indicated, were transfected into MT2 cells and DMSO (control) or NU7026 (DNA-PKcs kinase inhibitor) were added to the cell culture 12 h after transfection. The dual luciferase activity was assayed 48 h after transfection for both A and B. C, After treatment with or without recombinant His-tagged Tat protein (2 µg/ml) for 4 h, the total cell lysates were immunoprecipitated with normal IgG, anti-DNA-PKcs, anti-Cyclin T1 or anti-CDK9 antibody, and a western blot was performed using the indicated antibodies. D, MT2 cells were seeded into six-well plates with coverslips and transfected with a CFP-Tat plasmid and then fixed and stained as described in the Materials and Methods. To increase the adherence of the MT2 cells to the coverslips, the coverslips were pretreated with 0.2% gelatin overnight, prior to seeding. Anti-DNA-PKcs (red), anti-Cyclin T1 (green) and nuclear staining by DAPI were performed. E, A model describing the assembly of the DNA-PKcs/Tat/Cyclin T1/CDK9 complex at the HIV-1 5' LTR and their mutual interaction and regulation. Please see the text for details. Three independent experimental replicates were performed for each group. **P ≤0.01.
Discussion
Although the DNA-PKcs promoter was identified 17 years ago [19, 20], there has been very little research concerning its regulation at the transcriptional level [24]. The reason is most likely ascribed to its location on the chromosome and its relatively small promoter when compared with other eukaryotic genes. PRKDC and MCM4, which are neighbors on chromosome 8, are transcribed divergently and share a small common promoter. There is no TATA or CAAT box in the promoter region, a common feature of housekeeping genes. Bioinformatics analysis has demonstrated that there are a few indicators that PRKDC is regulated at the transcriptional level. Recent research showed that estrogen receptor-α binds to the -220 region of the PRKDC promoter to activate its transcription [24], which was the first demonstration that DNA-PKcs can be modulated at the transcriptional level. Previous research has shown the core promoter region to reside between -106 to -3, with respect to ATG. However, our study further shortened the core region to -63 to -1.
Unlike other transactivators, which only bind to DNA, HIV-1 Tat can bind to HIV proviral DNA [25], host cell DNA [26] and viral RNA LTRs and affect transcription. In the HIV 5'-LTR region, there are multiple SP1 binding sites. However, whether Tat regulates transcription by interacting with SP1 remains controversial [21, 22]. Interestingly, comparison between the PRKDC core promoter region and the HIV-1 5' LTR revealed that they share a high homology (>45%). Reporter and EMSA assays clearly demonstrated that Tat represses PRKDC by directly binding to its core promoter, most likely by binding to its SP1 binding sites. Coincidently, our previous work found that Tat stimulates Cyclin B1 expression by targeting a region comprised of SP1 binding sites [17]. This implies that targeting SP1 binding sites may be a commonly used mechanism by Tat to modulate gene expression, although its up- or down-regulation depends on other endogenous transcription factors.
As one of the most important regulatory proteins encoded by HIV-1, Tat interacts with hundreds of host-cell proteins to modulate their function [27, 28]. However, only a few interacting partners of DNA-PKcs were reported and confirmed, including Ku [8], c-Abl [29], Lyn tyrosine kinase[30] and KIP [31], due to its large size. Our research further categorized each partner to its own interacting protein list. The Tat-interacting region of DNA-PKcs, which is comprised of a FAT domain (3023-3470aa), is also targeted by the Ku70/Ku80 heterodimer (3002-3850aa) [29]. The canonical theory states that DNA-PKcs is activated by DNA breaks, via an unclear mechanism, during NHEJ, CSR or V(D)J recombination [6, 8, 9, 32, 33]. When a DNA DSB occurs, Ku70/Ku80 is recruited to the DSB sites, which further recruits DNA-PKcs. DNA-PKcs is then activated by the DNA ends. In vitro, DNA-PKcs has an intrinsic affinity for DNA. In low salt buffers (<100 mM NaCl), it is activated by DNA even in the absence of Ku or any other regulatory subunits [34]. Interestingly, recent research revealed that DNA-PKcs was activated by HIV-1 viral integration to initiate apoptosis [35]. However, our results demonstrated a new mechanism showing that DNA-PKcs can be activated by Tat in the absence of DNA. This provides a very useful tool for investigating the mechanism of DNA-PKcs activation. Nevertheless, we do have observed the dual effect of Tat on DNA-PKcs, and both are the direct action. We considered that these are two independent events produced by Tat, but concordantly facilitate the HIV-1 transcription and infection. On one hand, maintaining of the kinase activity within a certain low level of DNA-PKcs could be necessary and sufficient for HIV-1 transcription (Figure 6A & B). On the other hand, a deceased DNA-PKcs expression (but not complete depletion) caused by Tat may play a role in the immunopathogenesis processing of HIV-1 infection but without influence on its transcription.
As mentioned above, DNA-PKcs plays a critical role in V(D)J recombination [15, 36, 37], but its role in CSR remains controversial [6, 10-12, 38]. In developing lymphocytes, the Ig- and T-cell-receptor variable-region exons are assembled from germline variable (V), diversity (D) and joining (J) gene segments. In this process, RAG endonuclease introduces DSBs between V-, D- and J-coding sequences, and the following events are accomplished by members of the NHEJ pathway, including DNA-PKcs. Unlike its role in V(D)J recombination, there is current debate as to the role of DNA-PKcs in CSR. Bosma and associates reported that DNA-PKcs is not required for Ig CSR, by using Ig-transgenic SCID mice that lacked either DNA-PKcs kinase activity or the DNA-PKcs protein [11]. However, other groups have reported that DNA-PKcs plays an indispensable role in CSR[6, 10, 12], which more closely resembles our own results. As mentioned in the Results section, Tat paradoxically down-regulates DNA-PKcs at the transcriptional level but up-regulates its kinase activity. Interestingly, Tat also shows opposing effects on CSR. At low concentrations, Tat suppresses CSR, while at high concentrations, Tat activates CSR. This can be explained by stating that at low Tat concentrations, the transcriptional-suppression effect of Tat dominates over its kinase-activation effect, which has a reverse effect at high Tat concentrations. However, other possibilities cannot be ruled out. For example, Tat likely regulates CSR by targeting other NHEJ effectors, including Ku70, Ku80, Artemis, XRCC4, Lig4 and XLF.
Tat has been reported to suppress the immune system, which is closely related to the effect of Tat on cellular gene expression. Tat up-regulates IL-10 and inhibits gp120-specific T-cell responses in an IL-10-dependent manner [39]. In addition, Tat inhibits interferon-induced nitric oxide synthase activity in macrophages, which renders the host vulnerable to infection [40]. The inhibitory effects of Tat on MHC I expression also suggests another mechanism for the induction of immune suppression by Tat [41, 42]. Tat was reported recently to activate T-cell by induction IL-17[43]. Tat may also play an important role in CD4+ T-cell apoptosis induced by the up-regulation of Cyclin B1 [17, 44, 45]. Our results provide new evidence that Tat impairs humoral immunity by targeting DNA-PKcs, which plays a vital role in V(D)J and class-switch recombination. However, the effect of Tat on V(D)J recombination and its detailed mechanism in modulating CSR still requires further investigation.
In the prevailing model of HIV transcription, Tat binds to an RNA stem-loop structure called TAR, located at the 5' end of all initiated viral transcripts. After binding to TAR, Tat recruits the P-TEFb complex, which is composed of cyclin T2 and CDK9 [2, 46]. The P-TEFb complex phosphorylates serine residues in the C-terminal domain of RNA polymerase II and potentiates the latter to synthesize full-length HIV transcripts [3, 47, 48]. In addition to transcription factors, SP1, CREB, eIF2α and NF-κB have been reported to be triggered by Tat. Tat promotes SP1 phosphorylation via an unknown mechanism, and thereby, SP1 binds to the LTRs to up-regulate the HIV promoter.
On the other hand, DNA-PKcs has been studied in HIV-1 life cycle, but its role in virus integration remained controversial [49-51]. Interestingly, HTLV-1 Tax protein, homology of HIV-1 Tat, also modulates DNA-PKcs by direct binding [52]. Our results showed that DNA-PKcs forms a large complex with Tat, cyclin T1, and CDK1, and the over-expression of DNA-PKcs will inhibit the transcription of HIV. A reasonable explanation is that DNA-PKcs acts as a scaffold in this complex and inhibits cyclin T1 or CDK9. However, the inhibitory effect is not due to its kinase activity because in vitro phosphorylation assays were unable to prove that cyclin T1 or CDK9 could be phosphorylated by DNA-PKcs (data not shown). In addition, the kinase activity of DNA-PKcs is essential for HIV transcription, as we demonstrated that the DNA-PKcs-specific kinase inhibitor NU7026 could drastically decrease HIV transcription. Moreover, SP1 was reported to be phosphorylated by DNA-PKcs [53]. Based upon these results, we speculate that Tat activates DNA-PKcs, which phosphorylates SP1, and then SP1 stimulates HIV transcription. Recently, the activation of DNA-PKcs was reported to involve in killing of CD4+ T cells during HIV-1 integration, and inhibition of DNA-PKcs could abolish the cell death during HIV-1 infection [35]. Taking together, the inhibitors or intervention measures directing toward DNA-PKcs may provide additional strategy and valuable candidates for the search and development of novel drugs of anti-HIV-1 therapeutics.
In conclusion, our study shows the dual effects of HIV-1 Tat on DNA-PKcs, i.e. depressing its transcription and activating its kinase activity. The interaction of HIV-1 Tat and DNA-PKcs affects the class switch recombination (CSR) in a Tat concentration-dependent manner. DNA-PKcs can form a large complex comprised of Cyclin T1, CDK9 and Tat via direct interacting with CDK9 and Tat. Our results provide new mechanistic clues indicating the influence of Tat protein on the host humoral immunity.
Acknowledgements
This work was supported by the China National Science Foundation grant (Grant#81071361, # 81272998, # 81372925, # 81172119). We thank Dr. Ruichuan Chen (Xiamen University, China) for providing the HIV-LTR plasmid and Dr. David Chen (Southwestern Medical Center, UT, USA) for providing the pEYFP-DNA-PKcs plasmid. We also thank Dr.Hua-Dong Pei for providing the cell line and materials for class switch recombination assay. We thank Dr.Ya Wang for proofreading the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T. et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622-32
2. Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y. et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633-44
3. Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747-51 doi:10.1038/nature09131nature09131 [pii]
4. Zhang SM, Song M, Yang TY, Fan R, Liu XD, Zhou PK. HIV-1 Tat impairs cell cycle control by targeting the Tip60, Plk1 and cyclin B1 ternary complex. Cell Cycle. 2012;11:1217-34 doi:10.4161/cc.11.6.1966419664 [pii]
5. Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118-21 doi:10.1038/nature08648nature08648 [pii]
6. Rooney S, Alt FW, Sekiguchi J, Manis JP. Artemis-independent functions of DNA-dependent protein kinase in Ig heavy chain class switch recombination and development. Proc Natl Acad Sci U S A. 2005;102:2471-5 doi:0409857102 [pii]10.1073/pnas.0409857102
7. Reynolds P, Anderson JA, Harper JV, Hill MA, Botchway SW, Parker AW. et al. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012;40:10821-31 doi:10.1093/nar/gks879gks879 [pii]
8. Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131-42 doi:0092-8674(93)90057-W [pii]
9. Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J. et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333-8 doi:10.1101/gad.1015202
10. Franco S, Murphy MM, Li G, Borjeson T, Boboila C, Alt FW. DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. J Exp Med. 2008;205:557-64 doi:10.1084/jem.20080044jem.20080044 [pii]
11. Bosma GC, Kim J, Urich T, Fath DM, Cotticelli MG, Ruetsch NR. et al. DNA-dependent protein kinase activity is not required for immunoglobulin class switching. J Exp Med. 2002;196:1483-95
12. Kiefer K, Oshinsky J, Kim J, Nakajima PB, Bosma GC, Bosma MJ. The catalytic subunit of DNA-protein kinase (DNA-PKcs) is not required for Ig class-switch recombination. Proc Natl Acad Sci U S A. 2007;104:2843-8 doi:0611359104 [pii]10.1073/pnas.0611359104
13. Gapud EJ, Sleckman BP. Unique and redundant functions of ATM and DNA-PKcs during V(D)J recombination. Cell Cycle. 2011;10:1928-35 doi:16011 [pii]
14. Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, Brush JW. et al. Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci U S A. 2011;108:2028-33 doi:10.1073/pnas.10192931081019293108 [pii]
15. Oksenych V, Kumar V, Liu X, Guo C, Schwer B, Zha S. et al. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci U S A. 2013;110:2234-9 doi:10.1073/pnas.1222573110
16. Sun Y, Huang YC, Xu QZ, Wang HP, Bai B, Sui JL. et al. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. Int J Radiat Oncol Biol Phys. 2006;65:842-50 doi:10.1016/j.ijrobp.2006.02.040
17. Zhang SM, Sun Y, Fan R, Xu QZ, Liu XD, Zhang X. et al. HIV-1 Tat regulates cyclin B1 by promoting both expression and degradation. FASEB J. 2010;24:495-503 doi:10.1096/fj.09-143925fj.09-143925 [pii]
18. Pei H, Wu X, Liu T, Yu K, Jelinek DF, Lou Z. The histone methyltransferase MMSET regulates class switch recombination. J Immunol. 2013;190:756-63 doi:10.4049/jimmunol.1201811jimmunol.1201811 [pii]
19. Fujimoto M, Matsumoto N, Tsujita T, Tomita H, Kondo S, Miyake N. et al. Characterization of the promoter region, first ten exons and nine intron-exon boundaries of the DNA-dependent protein kinase catalytic subunit gene, DNA-PKcs (XRCC7). DNA Res. 1997;4:151-4
20. Connelly MA, Zhang H, Kieleczawa J, Anderson CW. The promoters for human DNA-PKcs (PRKDC) and MCM4: divergently transcribed genes located at chromosome 8 band q11. Genomics. 1998;47:71-83 doi:10.1006/geno.1997.5076
21. Loregian A, Bortolozzo K, Boso S, Sapino B, Betti M, Biasolo MA. et al. The Sp1 transcription factor does not directly interact with the HIV-1 Tat protein. J Cell Physiol. 2003;196:251-7 doi:10.1002/jcp.10271
22. Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, Fan H. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol. 1993;67:6224-33
23. Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K. et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709-15 doi:M408827200 [pii]10.1074/jbc.M408827200
24. Medunjanin S, Weinert S, Poitz D, Schmeisser A, Strasser RH, Braun-Dullaeus RC. Transcriptional activation of DNA-dependent protein kinase catalytic subunit gene expression by oestrogen receptor-alpha. EMBO Rep. 2010;11:208-13 doi:10.1038/embor.2009.279embor2009279 [pii]
25. El Kharroubi A, Piras G, Zensen R, Martin MA. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol Cell Biol. 1998;18:2535-44
26. Pessler F, Cron RQ. Reciprocal regulation of the nuclear factor of activated T cells and HIV-1. Genes Immun. 2004;5:158-67 doi:10.1038/sj.gene.63640476364047 [pii]
27. Li JC, Yim HC, Lau AS. Role of HIV-1 Tat in AIDS pathogenesis: its effects on cytokine dysregulation and contributions to the pathogenesis of opportunistic infection. AIDS. 2010;24:1609-23 doi:10.1097/QAD.0b013e32833ac6a000002030-201007170-00001 [pii]
28. Easley R, Van Duyne R, Coley W, Guendel I, Dadgar S, Kehn-Hall K. et al. Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim Biophys Acta. 2010;1799:275-85 doi:10.1016/j.bbagrm.2009.08.008S1874-9399(09)00102-3 [pii]
29. Jin S, Kharbanda S, Mayer B, Kufe D, Weaver DT. Binding of Ku and c-Abl at the kinase homology region of DNA-dependent protein kinase catalytic subunit. J Biol Chem. 1997;272:24763-6
30. Kumar S, Pandey P, Bharti A, Jin S, Weichselbaum R, Weaver D. et al. Regulation of DNA-dependent protein kinase by the Lyn tyrosine kinase. J Biol Chem. 1998;273:25654-8
31. Wu X, Lieber MR. Interaction between DNA-dependent protein kinase and a novel protein, KIP. Mutat Res. 1997;385:13-20 doi:S0921-8777(97)00035-9 [pii]
32. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781-94 doi:S0092867402006712 [pii]
33. Meek K, Lees-Miller SP, Modesti M. N-terminal constraint activates the catalytic subunit of the DNA-dependent protein kinase in the absence of DNA or Ku. Nucleic Acids Res. 2012;40:2964-73 doi:10.1093/nar/gkr1211gkr1211 [pii]
34. Hammarsten O, Chu G. DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci U S A. 1998;95:525-30
35. Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature. 2013;498:376-9 doi:10.1038/nature12274nature12274 [pii]
36. Lee BS, Gapud EJ, Zhang S, Dorsett Y, Bredemeyer A, George R. et al. Functional intersection of ATM and DNA-dependent protein kinase catalytic subunit in coding end joining during V(D)J recombination. Mol Cell Biol. 2013;33:3568-79 doi:10.1128/MCB.00308-13MCB.00308-13 [pii]
37. Malu S, De Ioannes P, Kozlov M, Greene M, Francis D, Hanna M. et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J Exp Med. 2012;209:955-63 doi:10.1084/jem.20111437jem.20111437 [pii]
38. Manis JP, Dudley D, Kaylor L, Alt FW. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 2002;16:607-17 doi:S1074761302003060 [pii]
39. Gupta S, Boppana R, Mishra GC, Saha B, Mitra D. HIV-1 Tat suppresses gp120-specific T cell response in IL-10-dependent manner. J Immunol. 2008;180:79-88 doi:180/1/79 [pii]
40. Barton CH, Biggs TE, Mee TR, Mann DA. The human immunodeficiency virus type 1 regulatory protein Tat inhibits interferon-induced iNos activity in a murine macrophage cell line. J Gen Virol. 1996;77( Pt 8):1643-7
41. Howcroft TK, Strebel K, Martin MA, Singer DS. Repression of MHC class I gene promoter activity by two-exon Tat of HIV. Science. 1993;260:1320-2
42. Matsui M, Warburton RJ, Cogswell PC, Baldwin AS Jr, Frelinger JA. Effects of HIV-1 Tat on expression of HLA class I molecules. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;11:233-40
43. Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R. et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A. 2013;110:13588-93 doi:10.1073/pnas.13086731101308673110 [pii]
44. Piedimonte G, Corsi D, Paiardini M, Cannavo G, Ientile R, Picerno I. et al. Unscheduled cyclin B expression and p34 cdc2 activation in T lymphocytes from HIV-infected patients. AIDS. 1999;13:1159-64
45. Cannavo G, Paiardini M, Galati D, Cervasi B, Montroni M, De Vico G. et al. Abnormal intracellular kinetics of cell-cycle-dependent proteins in lymphocytes from patients infected with human immunodeficiency virus: a novel biologic link between immune activation, accelerated T-cell turnover, and high levels of apoptosis. Blood. 2001;97:1756-64
46. Chen R, Liu M, Zhang K, Zhou Q. Isolation and functional characterization of P-TEFb-associated factors that control general and HIV-1 transcriptional elongation. Methods. 2011;53:85-90 doi:10.1016/j.ymeth.2010.04.005S1046-2023(10)00114-3 [pii]
47. Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681-91 doi:10.1093/emboj/17.13.3681
48. Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR. et al. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013;9:e1003338. doi:10.1371/journal.ppat.1003338PPATHOGENS-D-12-02022 [pii]
49. Daniel R, Katz RA, Skalka AM. A role for DNA-PK in retroviral DNA integration. Science. 1999;284:644-7
50. Baekelandt V, Claeys A, Cherepanov P, De Clercq E, De Strooper B, Nuttin B. et al. DNA-Dependent protein kinase is not required for efficient lentivirus integration. J Virol. 2000;74:11278-85
51. Ariumi Y, Turelli P, Masutani M, Trono D. DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus type 1 integration. J Virol. 2005;79:2973-8 doi:79/5/2973 [pii]10.1128/JVI.79.5.2973-2978.2005
52. Durkin SS, Guo X, Fryrear KA, Mihaylova VT, Gupta SK, Belgnaoui SM. et al. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J Biol Chem. 2008;283:36311-20 doi:10.1074/jbc.M804931200M804931200 [pii]
53. Jackson SP, MacDonald JJ, Lees-Miller S, Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell. 1990;63:155-65 doi:0092-8674(90)90296-Q [pii]
Author contact
![]() Corresponding author: Shi-Meng Zhang, E-mail: zhangshimengcom; Ping-Kun Zhou; Email: zhoupkac.cn.
Corresponding author: Shi-Meng Zhang, E-mail: zhangshimengcom; Ping-Kun Zhou; Email: zhoupkac.cn.