Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and methods
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(1):11-21. doi:10.7150/ijbs.10320 This issue Cite
Research Paper
Molecular Variation and Horizontal Gene Transfer of the Homocysteine Methyltransferase Gene mmuM and its Distribution in Clinical Pathogens
Jianchao Ying1, #, Huifeng Wang1, 2, #, Bokan Bao3, Ying Zhang1, Jinfang Zhang1, Cheng Zhang1, Aifang Li1, Junwan Lu1, 4, Peizhen Li1, Jun Ying1, Qi Liu1, 5, Teng Xu1, Huiguang Yi1, Jinsong Li1, Li Zhou6, Tieli Zhou1, 6, Zuyuan Xu1, Liyan Ni7, ![]() , Qiyu Bao1,
, Qiyu Bao1, ![]()
1. Institute of Biomedical Informatics/Zhejiang Provincial Key Laboratory of Medical Genetics, Wenzhou Medical University, Wenzhou 325035, China
2. National Institute of Biological Sciences, Beijing 102206, China
3. College of Life Science and Agriculture, Cornell University, Ithaca 14850, NY, USA
4. School of Medicine, Lishui College, Lishui 323000, China
5. Institute of Genomic Medicine, Wenzhou Medical University, Wenzhou 325035, China
6. The First Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, China
7. The Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, China
# These authors contributed equally to this work.
Received 2014-8-13; Accepted 2014-10-28; Published 2015-1-1
Abstract

The homocysteine methyltransferase encoded by mmuM is widely distributed among microbial organisms. It is the key enzyme that catalyzes the last step in methionine biosynthesis and plays an important role in the metabolism process. It also enables the microbial organisms to tolerate high concentrations of selenium in the environment. In this research, 533 mmuM gene sequences covering 70 genera of the bacteria were selected from GenBank database. The distribution frequency of mmuM is different in the investigated genera of bacteria. The mapping results of 160 mmuM reference sequences showed that the mmuM genes were found in 7 species of pathogen genomes sequenced in this work. The polymerase chain reaction products of one mmuM genotype (NC_013951 as the reference) were sequenced and the sequencing results confirmed the mapping results. Furthermore, 144 representative sequences were chosen for phylogenetic analysis and some mmuM genes from totally different genera (such as the genes between Escherichia and Klebsiella and between Enterobacter and Kosakonia) shared closer phylogenetic relationship than those from the same genus. Comparative genomic analysis of the mmuM encoding regions on plasmids and bacterial chromosomes showed that pKF3-140 and pIP1206 plasmids shared a 21 kb homology region and a 4.9 kb fragment in this region was in fact originated from the Escherichia coli chromosome. These results further suggested that mmuM gene did go through the gene horizontal transfer among different species or genera of bacteria. High-throughput sequencing combined with comparative genomics analysis would explore distribution and dissemination of the mmuM gene among bacteria and its evolution at a molecular level.
Keywords: comparative genomics, homocysteine methyltransferase gene, horizontal gene transfer, molecular variation
Introduction
Homocysteine methyltransferase (HMT) is a family of enzymes that can catalyze the methylation of homocysteine and convert homocysteine into methionine. HMT plays an important role at the final step of the methionine synthesis. This enzyme is widely distributed among microorganisms, animals and plants, and can be divided into three categories based on methyl group donors including 5-methyltetrahydrofolate: homocysteine S-methyltransferase, betaine-homocysteine methyltransferase and S-methylmethionine: homocysteine methyltransferase.
The 5-methyltetrahydrofolate: homocysteine S-methyltransferase (also known as methionine synthase) can be further divided into two sub-categories based on the catalytic conditions. One is cobalamin-dependent homocysteine S-methyltransferase. As an intermediary methyl carrier, this enzyme catalyzes the methylation of homocysteine with 5-methyltetrahydrofolate as methyl group donor and produces tetrahydrofolic acid and methionine. The activity of this enzyme can be drastically lowered without adequate cobalamin[1]. The other sub-category is cobalamin-independent homocysteine S-methyltransferase. This enzyme catalyzes the direct transfer of the methyl group from 5-methyltetrahydrofolate to homocysteine to produce methionine without any intermediate methyl carrier, and the catalytic process does not require the participation of cobalamin[2]. However, both enzymes require zinc for the activation and binding of homocysteine. The cobalamin-dependent homocysteine S-methyltransferase exists in mammalian tissues and the cobalamin-independent homocysteine S-methyltransferase is in plants, while some microorganisms may have both[3]. The betaine-homocysteine methyltransferase is a cytoplasmic enzyme that is present in mammals and catalyzes the conversions of betaine and homocysteine into dimethyl glycine and methionine, respectively. This reaction also requires the irreversible oxidation of choline[4]. S-methylmethionine: homocysteine methyltransferase encoded by the gene mmuM is currently only found in microorganisms including bacteria and fungus. In the biosynthesis of methionine, this enzyme can use S-methylmethionine as a donor of methyl groups to catalyze methylation of homocysteine. Two molecules of methionine are formed in this process[5].
However, among the three categories of HMT, only S-methylmethionine: homocysteine methyltransferase has been reported to use selenocysteine as a substrate to produce nontoxic selenium compound such as methylselenocysteine[5]. It has high a high similarity in sequence and function to the selenocysteine methyltransferase (SMT) in Se-accumulating plants[6]. This enzyme enables the SMT free microorganisms to enrich selenoproteins and to tolerate high concentrations of environmental selenium, even though such microorganisms do not contain SMT.
Selenium can be absorbed and metabolized through the sulfur assimilation pathway, because both selenium and sulfur elements have similar chemical properties[7-9]. Animals and human beings can obtain selenate or selenocysteine directly from food. In plants, marine algae, yeast and bacteria, selenate and selenite are activated by ATP sulfurylase[10], leading to the eventual selenium assimilation.
One of HMT genes first found in E.coli was once called yagD[5] and was renamed mmuM gene later[6]. mumM is distributed in microbial organisms and mainly encoded in bacterial chromosomes. So far, only a few bacteria have been found to carry mmuM genes on their plasmids[11]. In this work, mmuM gene profiles of clinically isolated pathogenic bacteria have been analyzed through high-throughput sequencing. The sequence diversity and evolution together with the molecular mechanism of horizontal transfer of this gene have also been analyzed.
Results
The distribution of mmuM gene in bacteria
533 mmuM gene sequences covering 70 genera were selected from GenBank and other databases. Among them, three species with the more mmuM sequences were Streptococcus (134), Klebsiella (72) and Lactobacillus (47). Other four genera (Mycobacterium, Bacillus, Escherichia and Xanthomonas) each had more than 20 sequences and sixty genera each had less than 10 sequences (Supplementary Material: Table S1). The frequency of mmuM gene varied from genus to genus. The statistics of 10 genera that had the highest mmuM gene frequencies in the collection showed that Mycobacterium ranked the first (32.22%, 29/90), while Xanthomonas was in the second place (31.58%, 12/38) and the third one was Enterobacter (17.65%, 6/34). Streptococcus had 17.47% (29/166), and Klebsiella and Escherichia only had 4.88% (6/123) and 1.77% (6/339), respectively (Table 1).
The statistics of 10 genera with higher mmuM gene frequencies
| Genus | Genomes with mmuM | Total genomes in database | Gene frequency |
|---|---|---|---|
| Mycobacterium | 29 | 90 | 0.3222 |
| Xanthomonas | 12 | 38 | 0.3158 |
| Enterobacter | 6 | 34 | 0.1765 |
| Streptococcus | 29 | 166 | 0.1747 |
| Leuconostoc | 4 | 37 | 0.1081 |
| Lactobacillus | 21 | 214 | 0.0981 |
| Streptomyces | 6 | 66 | 0.0909 |
| Bacillus | 20 | 283 | 0.0707 |
| Klebsiella | 6 | 123 | 0.0488 |
| Escherichia | 6 | 339 | 0.0177 |
Among 533 mmuM gene sequences collected, most of them were encoded in chromosomes. Only five sequences were located in plasmid genomes. These five mmuM gene-containing plasmids were: pBWB401 (NC_010180) and pMC429 (NC_018689) of Bacillus, pDSHI02 (NC_009956) of Dinoroseobacter, pIP1206 (NC_010558) of Escherichia and pKF3-140 (NC_013951) of Klebsiella. The mmuM gene sequences of pKF3-140 and pIP1206 were exactly the same, while the nucleotide identities of the mmuM genes for pBWB401 and pMC429 was 98.7 %. The mmuM gene sequences of pKF3-140 or pIP1206 were different from those of the other three plasmids pBWB401, pMC429 and pDSHI02 and the nucleotide sequence similarity identities with the three were 51.5%, 51.7% and 50.4%, respectively. The bacterial chromosomes that had mmuM genes with the highest similarities to those mmuM genes on plasmids were analyzed. The mmuM gene encoded in Escherichia coli genome (CP000948) showed an identity of 100% with the mmuM genes of pIP1206 and pKF3-140. The mmuM gene sequence on Halobacillus sp. genome (NZ_ANPF01000013) showed identities of 56.4 % and 56.3 % with those of pBWB401 and pMC429, respectively, while the mmuM gene on Kitasatospora setae genome (AP010968) only showed an identity of 52.3% with that of pDSHI02.
Taken together, mmuM gene is not evenly distributed over bacteria. In this study, many of this gene were collected from genera of Streptococcus, Klebsiella and Lactobacillus; however, genera with higher distribution frequencies of the gene were not them, but Mycobacterium, Xanthomonas and Enterobacter. In addition, the mmuM gene encoded in plasmids provides clues of its dissemination in bacteria through horizontal gene transfer.
Phylogenetic analysis of mmuM gene in bacteria
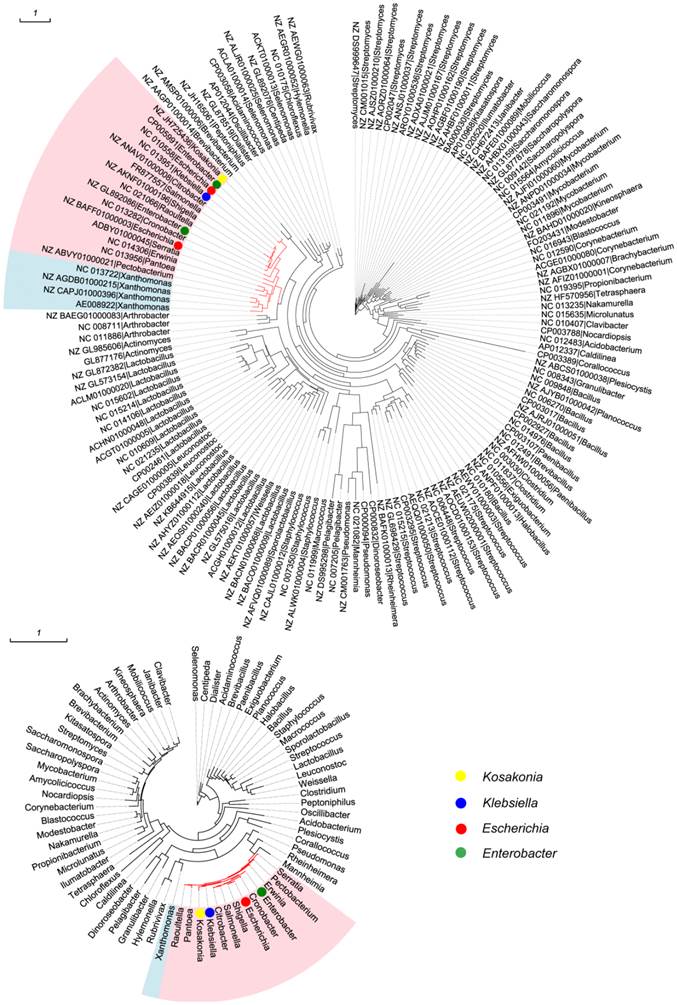
All 533 mmuM genes were clustered based on the amino acid sequence identity with a threshold value of 80% and a total of 144 clusters were obtained (Supplementary Material: Table S1). The sequence with the highest similarity to the consensus sequence in each cluster was selected as the representative sequence used to reconstruct the phylogenetic tree. Meanwhile, 16S rRNA gene sequence in each of 70 genera was selected as a reference from which the mmuM gene was retrieved. The clustering results showed that the genus with the most gene clusters was Lactobacillus which contained 20 mmuM gene clusters, followed by Streptomyces with 13 clusters. The genera with the least gene clusters were Escherichia (2 clusters) and Klebsiella (1 cluster, Supplementary Material: Table S1). The phylogenetic analysis showed that the mmuM genes from the same family (eg Enterobacteriaceae) were located relatively closer in the phylogenetic tree, which was consistent to the locations of 16S rRNA genes in the corresponding 16S rRNA gene phylogenetic tree (Figure 1). It indicated that there was a correlation in evolution between mmuM gene and its host. Generally, the mmuM gene sequences from the same species were closer in the phylogenetic tree. On the contrary, the sequences of NC_010558 and NZ_BAFF01000003 which were both derived from Escherichia were located on two well-separated branches, while the NC_013951 sequence from Klebsiella was on the same branch as NC_010558 of E. coli was. In addition, the mmuM sequences of CP005991 from Enterobacter and NZ_JH725436 from Kosakonia were both located on the same branch. On the 16S rRNA phylogenetic tree, Escherichia and Klebsiella were well separated although both belonged to the same family (Enterobacteriaceae). It suggests that horizontal gene transfer of mmuM probably happened widely among strains, species or genera, even some phylogeneticly remote bacteria.
Phylogenetic trees. mmuM gene phylogenetic tree (1A) and 16S rRNA gene phylogenetic tree (1B).
Gene mapping of mmuM in nine pathogenic bacteria genomes
To investigate what frequency and coverage of mmuM gene are there in clinical pathogens, we used 90% indentify of amino acid sequences as a threshold to cluster 533 mmuM genes. As a result, 160 clusters were obtained and a reference sequence in each of these clusters was selected for gene mapping. Each reference has the highest similarity to its consensus sequence in each cluster (Supplementary Material: Table S2). The high-throughput sequencing reads of nine pathogens were mapped to 160 mmuM gene reference sequences, respectively. The results showed that only 5 reference sequences had positive mapping results with more than 80% coverage of the full length of reference sequences. Among those five sequences, three were mapped positively in the pooled genome sequences of Staphylococcus aureus and Enterobacter cloacae, respectively. Two were mapped positively in the pooled genome sequences of Klebsiella pneumoniae. For the pooled genome sequences of Acinetobacter baumannii, Salmonella spp. or Enterococcus faecalis, each of them had only one reference sequence mapped positively. No positive mapping result was obtained in the pooled genome sequences of Pseudomonas aeruginosa or Enterococcus faecium (Table 2). It indicated that different pathogen maybe contained different number of the gene mmuM.
The mapping result of nine pooled genome sequences*
| ACGE01000080 | APWG01000155 | CP002824 | NC_007350 | NC_013951 | ||
|---|---|---|---|---|---|---|
| Aba | Coverage | - | 99.89% | - | - | - |
| Depth | - | 33.1190 | - | - | - | |
| Eco | Coverage | - | - | - | - | 100.00% |
| Depth | - | - | - | - | 195.2811 | |
| Efm | Coverage | - | - | - | - | - |
| Depth | - | - | - | - | - | |
| Efa | Coverage | - | - | - | 99.89% | - |
| Depth | - | - | - | 12.0309 | - | |
| Ecl | Coverage | - | 100.00% | 100.00% | - | 99.90% |
| Depth | - | 62.3794 | 360.9861 | - | 77.5100 | |
| Kpn | Coverage | - | 100.00% | - | - | 99.00% |
| Depth | - | 503.4298 | - | - | 4.8193 | |
| Pae | Coverage | - | - | - | - | - |
| Depth | - | - | - | - | - | |
| Sau | Coverage | 97.86% | 99.57% | 91.96% | - | - |
| Depth | 7.3198 | 24.2229 | 2.7867 | - | - | |
| Sal | Coverage | - | - | - | - | 99.90% |
| Depth | - | - | - | - | 21.0843 | |
* only genes with coverage of more than 80% of full length were listed.
Aba: Acinetobacter baumannii, Eco: Escherichia coli, Efa: Enterococcus faecalis, Kpn: Klebsiella pneumoniae, Efm: Enterococcus faecium, Sau: Staphylococcus aureus, Sal: Salmonella spp., Pae: Pseudomonas aeruginosa, Ecl: Enterobacter cloacae.
Among the five reference sequences, NC_013951 from Klebsiella had the highest positive rate. It was identified in the pooled genome sequences of Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae and Salmonella spp. Furthermore, it showed that the gene sequence in Escherichia coli had a higher redundancy of 195.3, while in Klebsiella pneumoniae, it only had a redundancy of 4.8 (Table 2).
Verification of mmuM gene
To verify presence of the gene mmuM in the pathogen genomes, polymerase chain reaction (PCR) was performed using reference NC_013951 as the target gene against several genera of bacteria and the amplified products were verified by sequencing. The upstream primer sequence of mmuM for PCR amplification was 5'-CGGAATTCTTGCGTTGTGCTATGGTGCT (mmuM-F), and the downstream primer sequence of mmuM was 5'-CCAAGCTTTCAGCTTCGCGCTTTTAACG (mmuM-R). The sequencing results of the PCR products confirmed the mapping results that all four bacteria (Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae and Salmonella spp.) with positive mapping results harbored mmuM genes. 29 sequences of PCR products from more than 600 strains were completely consistent with the reference sequence of NC_013951. Among these 29 sequenced PCR products, 13 were amplified from 150 strains of E. coli (13/150), 13 from 185 strains of Enterobacter cloacae (13/185), 2 from 200 strains of Klebsiella pneumoniae (2/200) and 1 from 89 strains of Salmonella spp. (1/89). It confirmed the mapping results above that all four bacteria contained the mmuM Gene (NC_013951) and that the mmuM gene in pooled samples of E. coli and Enterobacter cloacae has a higher redundancy than in those of Klebsiella pneumoniae and Salmonella.
Comparative analysis of the structure of the plasmids with mmuM genes
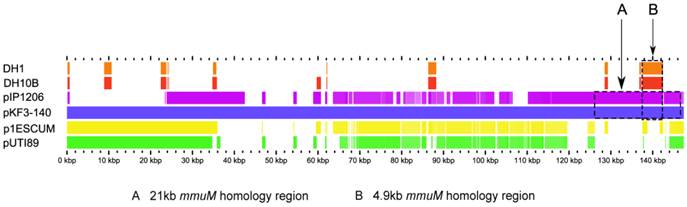
As mentioned above, of 533 mmuM gene sequences selected from the database, only 5 genes were encoded on plasmids. The structures of the plasmids with mmuM genes were analyzed carefully. Comparative genomics study showed that, although pKF3-140 plasmid had the highest similarity with E. coli plasmids p1ESCUM and pUTI89 (with gene identities of 98% and 92%, respectively)[11], there was no mmuM gene in these two plasmids. The plasmid pIP1206[12], however, only showed a gene identities of 49 % with plasmid pKF3-140, but the mmuM gene on pIP1206 showed the highest similarity with that on pKF3-140. As mentioned above, mmuM gene sequences encoded by pIP1206 and pKF3-140 are identical (with an identity of 100%). Furthermore, the 21 kb flanking regions of the mmuM genes on both plasmids showed a nucleotide sequence identity of 99.3 % (Figure 2, 3).
Comparison of mmuM genes on plasmids and chromosomes. The comparative map was created with the genomes of four plasmids (pKF3-140, pIP1206, pESCUM and pUTI89) and two chromosomes (DH1 for E. coli DH1 and DH10B for E. coli K12 DH10B). The nucleotide sequence of pKF3-140 was used as a reference and compared with those of five query genomes. The bars within the five slots of query sequences showed that the regions hit on the reference sequence with a higher degree of similarity (70% or higher). Empty regions on the query slots indicated the parts without similar hits between the reference sequence and the query sequences. Arrow A shows the 21 kb homology region and arrow B shows the 4.9 kb homology region.
Genome structure comparison of pKF3-140 with pIP1206. Corresponding blocks from these two plasmids were shown according to the gene contents or sequence similarities. Blocks below the line in the plasmid pIP1206 genome indicated that the sequence in the block was in a reverse direction compared to the corresponding region in the plasmid pKF3-140. Density and height of colored lines in the box illustrated the gene density and their similarity. The denser and higher lines represented more genes and higher similarity between the sequences. Regions outside blocks were lack of detectable homology between the two plasmid genomes.
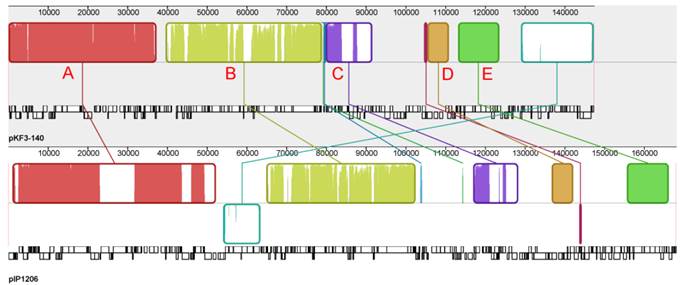
The whole plasmid genome sequences of pKF3-140 and pIP1206 were compared with each other to further analyze the evolutionary relationship between these two bacteria. It showed that pIP1206 was about 20 kb longer than pKF3-140. There were a large number of similar regions between these two plasmids. Most of the regions were in the same orientation. Each plasmid could be divided into five blocks based on the gene functions. Block A contained more genes than the other four blocks. The genes in this region were concerned with various functions, such as the restriction modification system, replication, transfer leading regions and metabolic functions (for example, S-methylmethionine metabolism). Block B included genes related to conjugation. Block C had genes mainly for replication. Block D had genes associated with drug resistance, while block E contained the genes related to the transport system. It was interesting to mention that an about 21 kb fragment (126.2-146.8 kb) harboring mmuM gene in block A of pKF3-140 showed a high similarity of 99.3% to the corresponding region in the plasmid pIP1206 (159.5-139.0 kb, Figure 3). The other four blocks also showed sequence similarities in their homolog genes, which suggested that these two plasmids might have the same backbone origin.
Comparative genomics analysis of the mmuM-encoding regions between the plasmid pKF3-140 and E. coli genomes
The common 21 kb mmuM-encoding regions of both plasmid pKF3-140 and pIP1206 were used as a reference in searching similar fragments from all prokaryotic genome sequences available in GenBank database. The searching results revealed that the 4.9 kb mmuM-encoding regions of the plasmids (137560-142459 of pKF3-140 and 148065-143166 of pIP1206) were matched to six E. coli genomes (such as E. coli str. K12 substr. DH10B genome 247283-253259, Figure 2, 4). These six E. coli chromosomes included E. coli K12 strains DH10B (CP000948.1), W3110 (AP009048.1), MG1655 (U00096.2), DH1 (ME8569, AP012030.1) and DH1 (CP001637.1) and chromosome minutes 4-6 (U70214.1). The first five strains have complete genome sequences available in the database.
Comparison of the mmuM gene regions on pKF3-140, pIP1206 and E. coli DH10B. The homologue genes are marked with same color and lined together, respectively, while non-homologous genes were left blank.
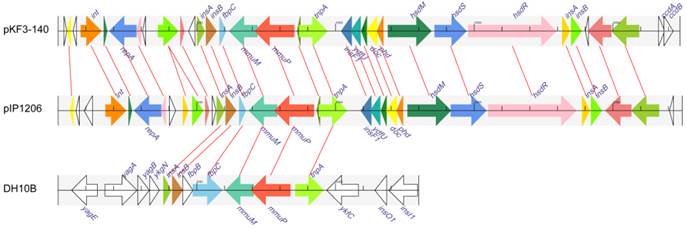
However, the regions in E. coli genomes that demonstrated similarity to 4.9 kb regions of the plasmids were about 1 kb longer. Taking E. coli str. K12 substr DH10B as an example, its mmuM-encoding region was 5977 bp (247283-253259) in length and 1077 bp (251414-252490 ) longer than the corresponding regions of the plasmids ( Figure 4 ), although the common 4.9 kb showed a high similarity with an identity of 99.9% and contained only five different nucleotides. The common 4.9 kb sequence encoded six genes (or gene fragments) including insA, insB, fbpC, mmuM, mmuP and insH-1. The plasmids encoded a truncated fbpC while E. coli str. K12 substr. DH10B encoded not only a complete fbpC gene, but also a complete fbpB gene more (Figure 4). Further analysis showed that this 4.9 kb mmuM-encoding region comprised a transposon. The left side of the region was an insertion sequence 1 (IS1) with 23 bp flanking imperfect inverted repeats (IRs). The transposase of IS1 was encoded by insA and insB genes. The right side of the region was an insertion sequence 5 (IS5) with 16 bp flanking IRs. The transposase of IS5 was encoded by the insH-1 gene. In the middle of the region, there were other genes including a partial fbpC (or fbpB-fbpC in E. coli genomes), mmuM and mmuP genes. The fbpB and fbpC genes were related to Fe (3+) ion absorption. By the way, on the both sides of the 4.9 kb region in these two plasmids, there were 8 bp imperfect direct repeats (DRs). Therefore, the 4.9 kb region in plasmids (or 6.0 kb in E. coli) was a transposon which consisted of IS1, truncated fbpC (or fbpB-fbpC), mmuM, mmuP and IS5. As previously reported, under certain conditions, the transposon could transpose between chromosomes and plasmids through a series of processes and could also make a horizontal gene transfer among different species or different genera of bacteria.
Discussion
HMT is known as the key enzyme that catalyzes the last step in methionine biosynthesis. However, S-methylmethionine: homocysteine methyltransferase also contribute to excretion and detoxification of selenium in organisms.
Selenium is an essential trace element for growth and metabolism of animals, plants and microbial organisms. It plays a key role in certain cell processes in forms of selenocysteine and selenoenzymes[13]. If Se accumulation reaches a higher concentration as selenocysteine in organisms, Se will be non-specifically incorporated into other proteins, leading to toxicity effects[8]. Organisms usually do have selenium excretion and detoxification mechanism, which could prevent the toxicity resulted from the accumulation of selenocysteine or selenide. In eukaryotes[14] and prokaryotes[15], there are two pathways for selenium detoxification. One common way is methylation of selenocysteine or selenomethionine by methyltransferase and the other pathway is to reduce selenite into elemental selenium[16]. In the non-Se-accumulating plants, excessive amounts of selenocysteine can be converted into selenomethionine through the sulfur assimilation pathway. The resulted selenomethionine is methylated with methyltransferase, and eventually volatilized in the form of dimethylselenide[10, 17]. In Se-accumulating plants, selenocysteine is converted into dimethyldiselenide with the help of SMT which catalyzes methyl transfer from S-methylmethionine to selenocysteine[8]. In animals, the inorganic selenium ions can be methylated to dimethylselenium and trimethylselenonium which are eventually excreted via respiration or urination[18]. Some microorganisms also have the ability to detoxify and excrete selenium. For example, Pseudomonas syringae harbors trimethyl purine methylase gene (tmp) and can convert selenate, selenite and selenocysteine into dimethylselenide and dimethyl diselenide[19]. Thiopurine methyltransferase[20] and homologs of calichaemicin methyltransferase[21] found in Pseudomonas can also convert selenite and selenocysteine into dimethylselenide and dimethyl diselenide, respectively. S-methylmethionine: homocysteine methyltransferase found on pKF3-140 plasmid of Klebsiella pneumoniae KF3 was reported to enable the host E. coli to tolerate high concentrations of selenite[11]. This enzyme catalyzes the methylation of selenocysteine to form MeSeCys and the function of the enzyme is similar to that of SMT found in Se-accumulating plants.
The gene mmuM is widely distributed in varieties of bacteria, including both Gram-positive and Gram-negative bacilli, cocci, and mycobacteria. Of the 70 selected genera in this study, the genomes of Mycobacterium and Xanthomonas have especially high frequencies of mmuM gene. The gene mmuM in different species shows different conservative profiles. A comparative analysis of mmuM genes within each genus with a cut-off identity of 80% similarity of amino acid sequences revealed that all 71 mmuM genes in Klebsiella could be clustered into one cluster (71/1). 27 genes of Escherichia could be divided into two clusters (27/2), while mmuM genes from certain genera showed more varieties, for example, 134 mmuM genes of Streptococcus were clustered into 11 clusters (134/11) and 47 mmuM genes of Lactobacillus were clustered into 20 clusters (47/20). It indicated that mmuM genes in Klebsiella and Escherichia were more conservative than those of Streptococcus and Lactobacillus. Further analysis of the mmuM polymorphism by mapping all the pooled sequencing reads of nine pathogens' genomes using the sequence of NC_013951 as the reference showed that none of the single nucleotide polymorphism (SNP) locus could be identified. In addition, sequencing of 29 PCR products of mmuM genes from Escherichia coli, Klebsiella pneumoniae, Salmonella spp. and Enterobacter cloacae, showed the same result as that from gene mapping. All these results further confirmed the conservation of the mmuM gene (NC_013951 as a reference).
The phylogenetic tree of mmuM genes showed that some mmuM genes might have closer phylogenetic relationship with those from different genera (such as mmuM genes between Escherichia and Klebsiella, and between Enterobacter and Kosakonia) than those in the same genus. It suggested that the mmuM gene in the same genus might have different origins while those in the different genera might have a common ancestor. The mmuM genes in Klebsiella are so highly conserved that they might have evolved from one common ancestor. The mmuM genes in Escherichia might have at least two origins, with one origin very close to that in Klebsiella.
Horizontal gene transfer (HGT) is a common biological process by which organisms acquire foreign genes or DNA fragments to across species boundaries. By rapidly introducing newly evolved donor genes into the host genomes and by avoiding the slow steps of ab initio gene creation, HGT therefore accelerates genome innovation and evolution[22, 23]. Some genes with special functions, such as genes for antibiotics resistance and genes for extreme environment, would be spread among organisms by HGT[24, 25]. HGT generally consists of two steps. One is a transfer process and the other is integration of foreign DNA into the host genome. The pathways for DNA transfer mainly include transformation, conjugation and transduction. HGT in bacteria is often involved with a number of mobile DNA elements such as the insertion sequence (IS), the transposon (Tn), the integron and some phages. Foreign DNA can be integrated into host cells through homologous recombination, illegitimate recombination, site-specific recombination, and the reconstruction of plasmids[26].
It demonstrated in this work that the mmuM gene also experienced the horizontal gene transfer to spread itself among bacteria. For example, the phylogenetic analysis showed that, although genetic relationship of most mmuM genes in the same genus or between closely related genera are closer, there exist higher homology genes among different genera than those belong to the same genus, such as the genetic relationship of the mmuM gene from an Escherichia strain (NC_010558) was closer to one from a Klebsiella strain (NC_013951) than that in another Escherichia strain (NZ_BAFF01000003). Similarly, the mmuM gene from Enterobacter (CP005991) shared the highest homology to that from Kosakonia (NZ_JH725436).
In addition, our study showed that the sequence similarity between plasmids pIP1206 and pKF3-140 was lower than that between pKF3-140 and p1ESCUM, or between pKF3-140 and pUTI89[11]. However, pKF3-140 and pIP1206 shared a 21 kb homologous region which encodes a mmuM gene and a number of mobile DNA elements such as insertion sequences, transposon and integron, which indicated that the 21 kb homologous region was a transferable region. It could be formed by a horizontal gene transfer. Analysis of the entire prokaryotic genomes and plasmid sequences in the NCBI database showed that a 4.9 kb fragment of the 21 kb homologous region in these two plasmids had homologous sequences in certain E. coli chromosomes, and the 4.9 kb fragment was identified to be a transposon. Therefore, this finding supported the hypothesis that 4.9 kb mmuM gene containing transposon was derived from the E. coli chromosome (such as E. coli str. K12 substr. DH10B genome, etc.) by HGT. Further HGT occurred between plasmids of Escherichia coli and Klebsiella pneumonia. As a result, the 21 kb mmuM gene containing regions were observed.
The comparative genomic analyses of mmuM-encoding plasmids and bacterial chromosomes have showed that mmuM gene could transfer between chromosome and plasmid genomes or among plasmid genomes. This mmuM gene migration among bacteria could not only enhance the methionine synthesis capability, but also enable the bacteria to tolerate a high concentration of selenium. It is also an evidence that bacteria evolved to adapt to extreme environment. Structure analysis of mmuM gene related sequences and the gene distribution would help to understand the molecular mechanism of dissemination and evolution of the mmuM gene.
Materials and methods
Collection and processing of mmuM gene sequences
mmuM gene sequences were obtained from NCBI Nucleotide database using mmuM as the key word. The resulted sequences were filtered and only sequences from bacteria were kept. The CDS of the mmuM gene was retrieved if the search result was a whole genome of a bacterium. Finally, 533 complete mmuM CDSs from 70 genera of bacteria were selected until June, 2014. The sequence retrieving, statistics analysis and other bioinformatics tools used in this study were completed with Python and Biopython scripts [27].
Clustering and phylogenetic analysis of mmuM gene sequences
The amino acid sequences of mmuM genes from the same genus were clustered using a cut-off threshold of 80% identity using CD-hit[28]. The 16S rRNA sequences were downloaded from NCBI Nucleotide database. Multiple sequence alignments were performed using MAFFT[29]. Phylogenetic trees were reconstructed by maximum likelihood method and the resulted trees were tested with bootstrap values of 100 replicates using PhyML3.0[30]. The best-fitting models of the amino acids and nucleotide substitutions were selected using Prottest3[31] and Modeltest3.7[32], respectively. Visualizing and annotating of phylogenetic trees were made using EvolView[33].
Collection of clinical pathogens and Hiseq 2000 DNA Sequencer sequencing
Nine pathogens, each with about 200 strains isolated from the clinical samples in the First Affiliated Hospital of Wenzhou Medical University, China, over the years 2009-2011, were collected for sequencing. They were Acinetobacter baumannii (Aba), Escherichia coli (Eco), Enterococcus faecalis (Efa), Klebsiella pneumoniae (Kpn), Enterococcus faecium (Efm), Staphylococcus aureus (Sau), Salmonella spp. (Sal), Pseudomonas aeruginosa (Pae) and Enterobacter cloacae (Ecl). Bacteria were isolated and identified according to National Guide to Clinical Laboratory Procedures, and each strain was confirmed by the Vitek-60 microorganism auto-analysis system (bioMe´rieux Corporate, France). A single clone of all 9 genera of bacteria was cultured in 5 ml of liquid broth over night at 37℃, the cultures of the same species were mixed together. The genomic DNAs of the pooled bacteria were extracted and sequenced with Hiseq-2000 DNA Sequencer in Beijing Genomics Institute. The average depth of the sequencing of each bacterium was calculated (Table 3).
The information of high-throughput sequencing of all nine species of pathogens
| Bacteria (accession No.) | Reference genome size(Mb) | Number of read | Strains sequenced | Depth |
|---|---|---|---|---|
| Aba (CP000521) | 3.98 | 377056430 | 200 | 47.41 |
| Eco (NC_000913) | 4.64 | 200565354 | 200 | 21.60 |
| Efm (NC_017960) | 2.70 | 187637758 | 231 | 30.11 |
| Efa (NC_004668) | 3.22 | 150936992 | 240 | 19.54 |
| Ecl (NC_014121) | 5.31 | 294298898 | 238 | 23.27 |
| Kpn (FO834906) | 5.44 | 182032468 | 240 | 13.95 |
| Pae (CP000438) | 6.54 | 140478390 | 239 | 8.99 |
| Sau (NC_002951) | 2.81 | 58304508 | 233 | 8.91 |
| Sal (AE014613) | 4.79 | 132858534 | 201 | 13.79 |
Aba: Acinetobacter baumannii, Eco: Escherichia coli, Efa: Enterococcus faecalis, Kpn: Klebsiella pneumoniae, Efm: Enterococcus faecium, Sau: Staphylococcus aureus, Sal: Salmonella spp., Pae: Pseudomonas aeruginosa, Ecl: Enterobacter cloacae.
Mapping of the bacterial genomes
When reconstructing phylogenetic tree of the gene mmuM above, clustering was performed for the genes from the same genus. In order to have fewer references to reconstruct a phylogenetic tree, we used cut-off value of 80% identity. However, we selected cut-off value of 90% identity to get more references in gene mapping which can help us to map more sequence reads from pooled pathogen genomes. 533 MmuM amino acid sequences were clustered together at 90% sequence identity using CD-hit[28] and a total of 160 clusters were obtained. The nucleotide sequence of the mmuM gene with the highest similarity to the consensus protein sequence in each cluster was chosen as the reference sequence for gene mapping using SOAPaligner/soap2[34] (Supplementary Material: Table S2). The high-throughput sequencing reads of all nine bacteria were mapped to 160 reference sequences of the mmuM genes, respectively. The coverage and the redundancy were calculated according to the mapping result for each reference sequence. In this work, a coverage of 80% or higher of the full length of the reference sequence was considered as a positive result.
Detection of mmuM gene by PCR and sequencing
Based on the mapping result, primers were designed using Primer Premier 5[35] to amplify the full length of mmuM gene. All strains of each species with positive mmuM gene mapping results were screened three times by PCR and the PCR products were confirmed by sequencing. Vector NTI[36] was used to assemble sequencing products and to examine the sequencing quality. Multiple sequence alignments were performed using MEGA5.2[37] and CLUSTAL W[38] to identify SNPs.
Analysis of the origin of the mmuM gene
The plasmid and chromosome genome sequences used in this study for extensively comparative analysis were downloaded from NCBI GenBank database (http://www.ncbi.nlm.nih.gov). The accession numbers of the related plasmid and the E. coli chromosome sequences were pKF3-140 (FJ876827/ NC_013951), pIP1206 (AM886293), p1ESCUM (CU928148), pUTI89 (CP000244) and E. coli str. K12 substr. DH10B (CP000948). Open reading frames (ORFs) were predicted and annotated using Glimmer3[39] and BASys[40], respectively. Insertion sequences were predicted by using IS Finder[41]. Nucleotide sequence was compared against the nucleotide collection database in NCBI using BlastN[42]. Figure 2 was generated using GView server[43]. Mauve2.3.1[44] was used to perform comparative plasmid genome alignment. Figure 4 was drawn using GenomeDiagram[27, 45]. Orthologous groups of genes from pKF3-140, pIP1206 and DH10B were identified using BLAST+[46].
Supplementary Material
Tables S1 - S2.
Abbreviations
HGT: Horizontal gene transfer; HMT: homocysteine methyltransferase; SMT: selenocysteine methyltransferase.
Acknowledgements
This work is supported by the Natural Science Foundation of Zhejiang Province, China (LY14C060005); the Science and Technology Foundation of National Health and Family Planning Commission of China (WKJ2012-2-032); the National Natural Science Foundation of China (81401702, 81171614, 31100917); Technology Innovation Team of Zhejiang Province, China (2010R50048-13); Science and Technology Foundation of Wenzhou City, China (H20100011) and the Science and Technology Foundation of Zhejiang Province, China (2008C23074).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Abdel-Azeim S, Li X, Chung LW, Morokuma K. Zinc-Homocysteine Binding in Cobalamin-Dependent Methionine Synthase and its Role in the Substrate Activation: DFT, ONIOM, and QM/MM Molecular Dynamics Studies. Journal of computational chemistry. 2011;32:3154-67 doi: 10.1002/Jcc.21895
2. Pejchal R, Ludwig ML. Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS biology. 2005;3:e31. doi:10.1371/journal.pbio.0030031
3. Zhang Z, Tian C, Zhou S, Wang W, Guo Y, Xia J. et al. Mechanism-based design, synthesis and biological studies of N(5)-substituted tetrahydrofolate analogs as inhibitors of cobalamin-dependent methionine synthase and potential anticancer agents. European journal of medicinal chemistry. 2012;58:228-36 doi:10.1016/j.ejmech.2012.09.027
4. Feng Q, Kalari K, Fridley BL, Jenkins G, Ji Y, Abo R. et al. Betaine-homocysteine methyltransferase: human liver genotype-phenotype correlation. Molecular genetics and metabolism. 2011;102:126-33 doi:10.1016/j.ymgme.2010.10.010
5. Neuhierl B, Thanbichler M, Lottspeich F, Bock A. A family of S-methylmethionine-dependent thiol/selenol methyltransferases. Role in selenium tolerance and evolutionary relation. The Journal of biological chemistry. 1999;274:5407-14
6. Thanbichler M, Neuhierl B, Bock A. S-methylmethionine metabolism in Escherichia coli. J Bacteriol. 1999;181:662-5
7. Terry N, Zayed AM, De Souza MP, Tarun AS. Selenium in Higher Plants. Annual review of plant physiology and plant molecular biology. 2000;51:401-32 doi:10.1146/annurev.arplant.51.1.401
8. Ellis DR, Salt DE. Plants, selenium and human health. Current opinion in plant biology. 2003;6:273-9
9. Sors TG, Ellis DR, Salt DE. Selenium uptake, translocation, assimilation and metabolic fate in plants. Photosynthesis research. 2005;86:373-89 doi:10.1007/s11120-005-5222-9
10. Mehdi Y, Hornick JL, Istasse L, Dufrasne I. Selenium in the environment, metabolism and involvement in body functions. Molecules. 2013;18:3292-311 doi:10.3390/molecules18033292
11. Bai J, Liu Q, Yang Y, Wang J, Li J, Li P. et al. Insights into the evolution of gene organization and multidrug resistance from Klebsiella pneumoniae plasmid pKF3-140. Gene. 2013;519:60-6 doi:10.1016/j.gene.2013.01.050
12. Perichon B, Bogaerts P, Lambert T, Frangeul L, Courvalin P, Galimand M. Sequence of conjugative plasmid pIP1206 mediating resistance to aminoglycosides by 16S rRNA methylation and to hydrophilic fluoroquinolones by efflux. Antimicrobial agents and chemotherapy. 2008;52:2581-92 doi:10.1128/AAC.01540-07
13. Stolz JF, Basu P, Santini JM, Oremland RS. Arsenic and selenium in microbial metabolism. Annual review of microbiology. 2006;60:107-30 doi:10.1146/annurev.micro.60.080805.142053
14. Hassoun BS, Palmer IS, Dwivedi C. Selenium detoxification by methylation. Research communications in molecular pathology and pharmacology. 1995;90:133-42
15. Heider J, Bock A. Selenium metabolism in micro-organisms. Advances in microbial physiology. 1993;35:71-109
16. Stolz JF, Basu P, Oremland RS. Microbial transformation of elements: the case of arsenic and selenium. International microbiology: the official journal of the Spanish Society for Microbiology. 2002;5:201-7 doi:10.1007/s10123-002-0091-y
17. Tagmount A, Berken A, Terry N. An essential role of s-adenosyl-L-methionine:L-methionine s-methyltransferase in selenium volatilization by plants. Methylation of selenomethionine to selenium-methyl-L-selenium- methionine, the precursor of volatile selenium. Plant physiology. 2002;130:847-56 doi:10.1104/pp.001693
18. Schrauzer GN. Selenomethionine: a review of its nutritional significance, metabolism and toxicity. The Journal of nutrition. 2000;130:1653-6
19. Ranjard L, Prigent-Combaret C, Nazaret S, Cournoyer B. Methylation of inorganic and organic selenium by the bacterial thiopurine methyltransferase. J Bacteriol. 2002;184:3146-9
20. Ranjard L, Nazaret S, Cournoyer B. Freshwater bacteria can methylate selenium through the thiopurine methyltransferase pathway. Applied and environmental microbiology. 2003;69:3784-90
21. Ranjard L, Prigent-Combaret C, Favre-Bonte S, Monnez C, Nazaret S, Cournoyer B. Characterization of a novel selenium methyltransferase from freshwater bacteria showing strong similarities with the calicheamicin methyltransferase. Biochimica et biophysica acta. 2004;1679:80-5 doi:10.1016/j.bbaexp.2004.05.001
22. Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: the complexity hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3801-6
23. Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Molecular biology and evolution. 2003;20:1598-602 doi:10.1093/molbev/msg154
24. Gootz TD. The global problem of antibiotic resistance. Critical reviews in immunology. 2010;30:79-93
25. Hanage WP, Fraser C, Tang J, Connor TR, Corander J. Hyper-recombination, diversity, and antibiotic resistance in pneumococcus. Science. 2009;324:1454-7 doi:10.1126/science.1171908
26. Brigulla M, Wackernagel W. Molecular aspects of gene transfer and foreign DNA acquisition in prokaryotes with regard to safety issues. Applied microbiology and biotechnology. 2010;86:1027-41 doi:10.1007/s00253-010-2489-3
27. Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422-3 doi:10.1093/bioinformatics/btp163
28. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658-9 doi:10.1093/bioinformatics/btl158
29. Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular biology and evolution. 2013;30:772-80 doi: 10.1093/molbev/mst010
30. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst Biol. 2010;59:307-21 doi: 10.1093/sysbio/syq010
31. Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164-5 doi: 10.1093/bioinformatics/btr088
32. Posada D. Using MODELTEST and PAUP* to select a model of nucleotide substitution. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al]. 2003 Chapter 6: Unit 6 5. doi:10.1002/0471250953.bi0605s00
33. Zhang H, Gao S, Lercher MJ, Hu S, Chen WH. EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic acids research. 2012;40:W569-72 doi:10.1093/nar/gks576
34. Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966-7 doi:10.1093/bioinformatics/btp336
35. Singh VK, Mangalam AK, Dwivedi S, Naik S. Primer premier: program for design of degenerate primers from a protein sequence. BioTechniques. 1998;24:318-9
36. Lu G, Moriyama EN. Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform. 2004;5:378-88
37. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular biology and evolution. 2011;28:2731-9 doi: 10.1093/molbev/msr121
38. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research. 1994;22:4673-80
39. Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic acids research. 1999;27:4636-41
40. Van Domselaar GH, Stothard P, Shrivastava S, Cruz JA, Guo AC, Dong XL. et al. BASys: a web server for automated bacterial genome annotation. Nucleic acids research. 2005;33:W455-W9 doi: 10.1093/Nar/Gki593
41. Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic acids research. 2006;34:D32-6 doi:10.1093/nar/gkj014
42. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic acids research. 2008;36:W5-9 doi:10.1093/nar/gkn201
43. Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. 2010;26:3125-6 doi:10.1093/bioinformatics/btq588
44. Darling AE, Mau B, Perna NT. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PloS one. 2010 doi: 10.1371/journal.pone.0011147
45. Pritchard L, White JA, Birch PR, Toth IK. GenomeDiagram: a python package for the visualization of large-scale genomic data. Bioinformatics. 2006;22:616-7 doi:10.1093/bioinformatics/btk021
46. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. et al. BLAST+: architecture and applications. Bmc Bioinformatics. 2009;10:421. doi:10.1186/1471-2105-10-421
Author contact
![]() Corresponding author: Qiyu Bao: baoqycn; Liyan Ni: nily2001com
Corresponding author: Qiyu Bao: baoqycn; Liyan Ni: nily2001com