Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(3):353-360. doi:10.7150/ijbs.9813 This issue Cite
Research Paper
Gadd45b is a Novel Mediator of Neuronal Apoptosis in Ischemic Stroke
Bin Liu#, Yan-hong Zhang, Ying Jiang, Long-ling Li, Qian Chen, Guo-qian He, Xiao-dan Tan, Chang-qing Li ![]()
Department of Neurology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China
# Current Addresses: Department of Neurology, Shandong Provincial Qianfoshan Hospital, Shandong University, Jinan, China
Received 2014-6-4; Accepted 2015-1-12; Published 2015-2-8
Abstract

Apoptosis plays an essential role in ischemic stroke pathogenesis. Research on the process of neuronal apoptosis in models of ischemic brain injury seems promising. The role of growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) in brain ischemia has not been fully examined to date. This study aims to investigate the function of Gadd45b in ischemia-induced apoptosis. Adult male Sprague-Dawley rats were subjected to brain ischemia by middle cerebral artery occlusion (MCAO). RNA interference (RNAi) system, which is mediated by a lentiviral vector (LV), was stereotaxically injected into the ipsilateral lateral ventricle to knockdown Gadd45b expression. Neurologic scores and infarct volumes were assessed 24 h after reperfusion. Apoptosis-related molecules were studied using immunohistochemistry and Western blot analysis. We found that Gadd45b-RNAi significantly increased infarct volumes and worsened the outcome of transient focal cerebral ischemia. Gadd45b-RNAi also significantly increased neuronal apoptosis as indicated by increased levels of Bax and active caspase-3, and decreased levels of Bcl-2. These results indicate that Gadd45b is a beneficial mediator of neuronal apoptosis.
Keywords: MCAO, Gadd45b, BDNF, Apoptosis
INTRODUCTION
Ischemic stroke is one of the leading causes of death and long-term disability worldwide. Considering the essential role of apoptosis in the pathogenesis of ischemic stroke, a more detailed understanding of the mechanisms involved will have a substantial effect in the optimization and development of treatment strategies. Subsequent to acute ischemic stroke, an early response emerges in the gene expression of key modulators of apoptosis, such as the Bcl-2 family. The release of pro-apoptotic proteins from the mitochondria induces the activation of caspases and other genes that aggravate apoptotic cell death [1,2].
Growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) is originally recognized as an important factor in DNA repair, apoptosis, cell survival, growth arrest, and possibly DNA demethylation. Several studies on non-neuronal cells have suggested Gadd45b as an anti-apoptosis gene [3,4]. Gadd45b was identified as an intrinsic neuroprotective molecule in retinal ganglion cells [5,6]. It's not clear whether Gadd45b was involved in apoptosis induced by ischemic stroke. This study aims to evaluate the effects of Gadd45b downregulation in neuronal apoptosis after cerebral ischemia.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley (SD) male rats (230-300g) were purchased from the Experimental Animal Center of Chongqing Medical University. Rats were group housed (five rats per cage) in quiet in a room maintained at 21-22 °C on a 12/12-h light/dark cycle, and allowed free access to food and water during the study. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Chongqing Medical University and were performed strictly in accordance with the “Guiding Principles for Research Involving Animals and Human Beings”.
A total of 186 rats were included in the study. Data are reported on 104 animals. Eighty-two were excluded from the study due to no neurologic abnormalities (16) or death (66). Rats were randomly assigned to groups: sham (n=14); MCAO (n=30); Lentiviral vector (LV)-control (ischemia and LV-Control, n=30), and LV-shGadd45b (ischemia and LV-shGadd45b, n=30).
Transient MCAO in Rats
To induce transient focal cerebral ischemia in rats, middle cerebral artery occlusion (MCAO) surgery was performed as described previously [7]. Rats were anesthetized using 3.5% chloral hydrate (350 mg/kg, ip) and right common carotid artery (CCA) was surgically exposed by a midline neck incision. Right CCA was ligated distally and external carotid artery (ECA) was ligated proximally to ECA and internal carotid artery (ICA) bifurcation. A 30 mm length of nylon filament (diameter 0.25±0.03mm) was gently inserted into the lumen of ICA from right ECA. Afterwards, reperfusion was established by withdrawal of the nylon filament at 2 h after ischemia. In the sham group, rats underwent the same surgical procedures without occlusion of the right CCA. Rats were then returned to their cages and body temperature were kept at (37±0.5) °C.
Gadd45b-shRNA Injection
LV pLKD.UBC.GFP.U6.shRNA was generated and purchased from Neuron Biotech (Shanghai, China). The prepared rat Gadd45b-shRNA sequence was 5-CGA CAA CGC GGU UCA GAA GUU-3 (sense) and 5-PCU UCU GAA CCG CGU UGU CGUU-3 (antisense). Seven days before MCAO surgery, 4 µl of concentrated viral solution (containing Gadd45b-shRNA) was stereotaxically delivered into the ipsilateral lateral ventricle (coordinates from bregma: AP 1.1 mm, ML 0.8 mm, DV 4.2 mm from the pial surface). The control rats were also injected with the same dose of lentivirus without Gadd45b-shRNA.
Detection of Virus Delivery
Green fluorescent protein (GFP) was observed in brain section under a fluorescence microscope for the detection of virus delivery. To determine the proper therapeutic titer, Gadd45b mRNA and protein expression was examined through real-time PCR and immunohistochemistry. The most appropriate therapeutic titer was used in the following study.
Sample Processing
The rats were killed 24 h after reperfusion, and the brains were quickly removed by neck breaking to collect the cerebral cortex for immunohistochemistry, Western blot analysis, real-time PCR and TTC staining.
RNA Isolation and Real-Time PCR
Animals were anesthetized using 3.5% chloral hydrate (350 mg/kg, ip) 24 h after MCAO. Total RNA from ipsilateral ischemic penumbra was harvested for the assay of Gadd45b and BDNF mRNA levels. The procedure was similar to that in our previous study [8]. The sequence for Gadd45b forward primer was 5ʹ-TGA TCC AAT CGT TCT GCT GC-3ʹ) and the reverse primer was 5ʹ-CGT TTG TGC CTA GAG TCT CTG C-3ʹ. The sequence for BDNF forward primer was 5ʹ-TGT CCG AGG TGG TAG TAC TTC ATC-3ʹ and the reverse primer was 5ʹ-CAT GCA ACC GAA GTA TGA AAT AAC C-3ʹ. The sequence for β-actin forward primer was 5ʹ-CGT TGA CAT CCG TAA AGA CCT C-3ʹ and the reverse primer was 5ʹ-TAG GAG CCA GGG CAG TAA TCT-3ʹ. The real-time PCR protocol was 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Cycle threshold (Ct value) was acquired from the software provided by the manufacturer. A relative quantity was calculated using the 2-ΔΔCT method [9] for each sample.
Immunohistochemistry
Rats were anesthetized using 3.5% chloral hydrate (350 mg/kg, ip) 24 h after MCAO. Paraffin sections were prepared as previously described [8]. In brief, sections (n=5 for each group) were incubated with primary antibodies (rabbit anti-Gadd45b, 1:80; rabbit anti-BDNF, 1:200; rabbit anti-Bax, 1:100; and rabbit anti-Bcl-2, 1:100; all from Santa Cruz, USA) overnight at 4 °C. Afterwards, sections were blocked with normal serum for 30 min at 37 °C. Then, the samples were incubated with goat anti-rabbit IgG for 30 min at 37 °C. Positive activity was revealed with diaminobenzidine. Confocal images were taken with a Nicon microscope. Gadd45b, BDNF, Bax and Bcl-2 expression were quantified using NIH ImageJ software. Immunopositive cells were quantified in sequential fields of 1 mm2 at ipsilateral ischemic penumbra in five animals selected randomly from each group. The number of neurons per field for each sample was the average of 20 consecutive objects in 4 sections/case by an investigator who was blinded to this study.
Western Blot Analysis
Six animals in each group were sacrificed 24 h after reperfusion, and the rat brain tissues were extracted from ipsilateral ischemic penumbra. Western blot analysis was performed as described previously [8]. Anti-Bax (1:200, Santa Cruz, USA), anti-Bcl-2 (1:200, Santa Cruz, USA), anti-BDNF (1:200, Santa Cruz, USA), anti-cleaved caspase-3 (1:500, Cell Signaling Technology), and anti-GAPDH (1:2000; Santa Cruz, USA) were used as primary antibodies. The blots were subjected to gel formatter (BIO-RAD) and quantified through Quantity One analysis. GAPDH was used as an internal loading control.
Neurological Deficits
Neurological deficits were determined 24 h after stroke according to a neurological grading score [10]. The evaluator was blinded to the experimental treatment. Briefly, neurological deficits was scored as follow: 0=no observable deficit, 1=torso flexion to the right side, 2=spontaneous circling to the right side, 3=falling to the right side and 4=no spontaneous movement.
Measurement of Infarct Volume
The rats were euthanized under anesthesia 24 h after reperfusion. The brains were rapidly removed. Coronal sections (n=6 in each group) were cut into 2 mm thick slices and stained with standard 2% TTC for 20 min at 37 °C. The pale-appearing infarcted areas and areas of the uninfarcted hemispheres were digitally analyzed using NIH Image J software. The area of infarction from each slide was added and presented as a percentage of the volume of the uninfarcted hemisphere.
TUNEL Assay
TUNEL staining was done on brain section to detect cell apoptosis according to the manufacturer's protocol of an in situ cell death detection kit (Roche, Basel, Switzerland). TUNEL-positive cells were quantified by individual counts in sequential fields of 1 mm2 at ipsilateral ischemic penumbra in five rats selected randomly from each group. The TUNEL-positive cell number per field for each sample was the average of 20 consecutive objects in 4 sections/case by an investigator who was blinded to the studies.
Statistical Analysis
Data were expressed as means±SD. Differences between groups were compared using one-way analysis of variance (ANOVA). P<0.05 was considered statistically significant. Statistical analysis was performed using SPSS 13.0 for Windows.
Results
Delivery and Silencing Efficiency of shRNA LVs
The titer of LV used was 6.9×108 pfu/ml. The titers of LV were categorized as 1.38×109 pfu/ml (high titer), 6.9×108 (medium titer), and 3.45×108 (low titer). The three titers were able to successfully transfect ipsilateral ischemic penumbra and ipsilateral hippocampus in the rat brains. Delivery efficacy was shown by GFP-positive cells. The transfection efficiency of medium and high titer injections were similar, and better than the efficiency of the low-titer group (Figure 1A).
LV delivery efficacy and inhibition efficiency. Three different titers of LV-shGadd45b were injected 7 d before MCAO through stereotactic surgery. (A) A few GFP-positive cells were localized in the cortex and hippocampus injected with low-titer LVs. A large number of GFP-positive cells were observed in the cortex and hippocampus injected with medium- and high-titer LVs. (B) Gadd45b-shRNA treatment effectively suppressed MCAO-induced Gadd45b expression. Data were expressed as fold of sham. (C) Immunohistochemistry for Gadd45b (original magnification 400×). Images are taken from ipsilateral ischemic penumbra. (D) The bar graph reflects the Gadd45b-positive cells in each group. (*p<0.01, compared with sham group, #p<0.01, compared with LV-shGadd45b group).
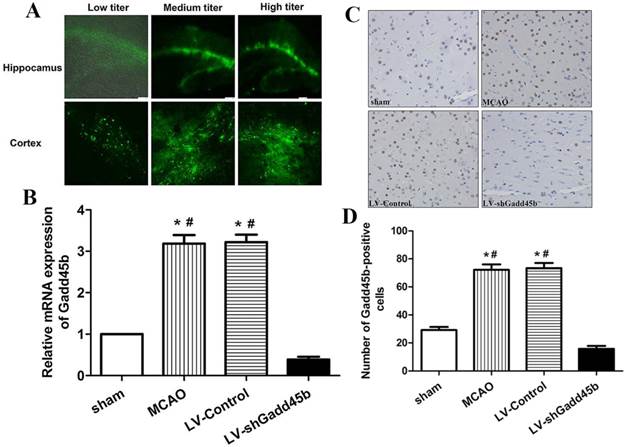
Our previous study has shown that MCAO significantly increased Gadd45b expression in ipsilateral ischemic penumbra from 6 h to 72 h after focal cerebral ischemia [8]. This study showed that Gadd45b-RNAi significantly decreased MCAO-induced Gadd45b mRNA (Figure 1B, p <0.01) and protein (Figure 1C,D, p<0.01) expression in comparison to MCAO and LV-Control groups.
Gadd45b-RNAi increased cerebral infarction and worsened neurological scores
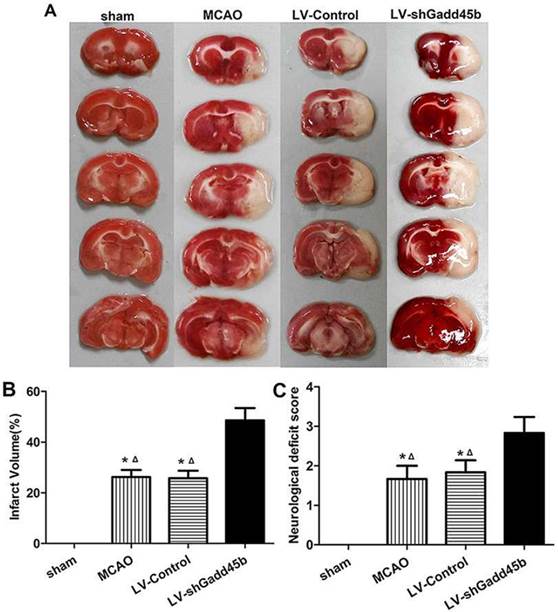
Infarct volume was measured 24 h after reperfusion. No infarction was observed in the sham group, whereas extensive lesion was developed in the striatum and lateral cortex in the LV-Control and MCAO groups. Gadd45b-RNAi significantly increased infarct volume in comparison to rats in LV-Control and MCAO groups (Figures 2A, B, p<0.05). The effect of Gadd45b-RNAi in neurological deficits was also investigated. No neurological deficiency was observed in the sham group. The rats exhibited focal neurological deficiency following MCAO with failure to fully extend the forepaw. Treatment with Gadd45b-RNAi showed a significant increase in neurological score (Figure 2C, p<0.05), indicating that Gadd45b-RNAi can worsen neurological deficits.
Gadd45b-RNAi treatment increased infarct size and worsened the neurological behavior at 24 h after reperfusion. (A) TTC staining of the brain. (B) The bar graph reflects the infarct volume in each group. (C) Determination of neurological deficits. (*p<0.01, compared with sham group, △p<0.05, compared with LV-shGadd45b group).
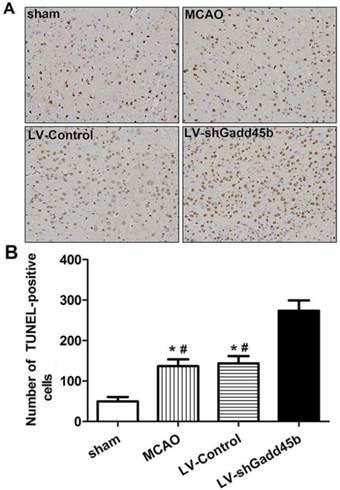
Gadd45b-RNAi increased neuronal apoptosis after cerebral ischemia
The apoptotic activity shown by TUNEL was a common method for detecting DNA fragmentation that results from apoptotic signaling cascades. A small number of TUNEL-positive cells were found in the sham-operated animals. In samples collected from the MCAO and LV-Control group, a large number and density of TUNEL-positive staining was found and further enhanced by Gadd45-RNAi treatment (Figure 3, p<0.01).
Gadd45b-RNAi treatment increased cell apoptosis in cerebral ischemia. (A) TUNEL assay after cerebral ischemia (original magnification 400×). (B) The bar graph reflects the TUNEL-positive cell number in ipsilateral ischemic penumbra in each group. (*p<0.01, compared with sham group, # p<0.01, compared with LV-shGadd45b group).
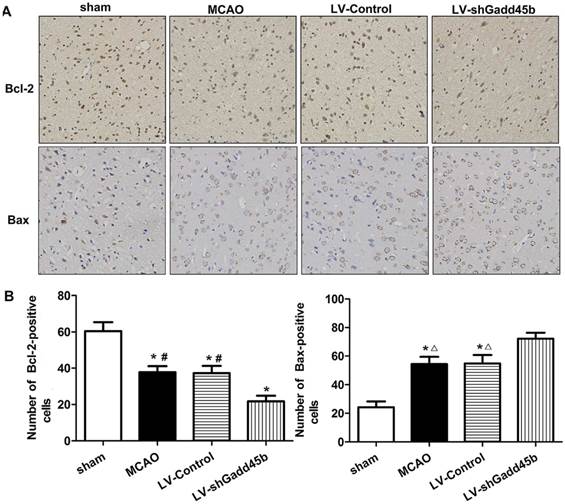
Gadd45b-RNAi upregulated the apoptotic and downregulated anti-apototic proteins
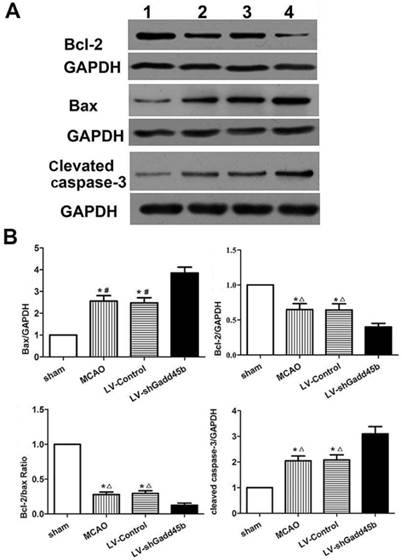
Considering the importance of Bcl-2, Bax, and cleaved caspase-3 in the regulation of neuronal apoptosis, the effects of Gadd45b-RNAi on these protein levels were determined. Immunochemistry analysis of the ipsilateral ischemic penumbra showed a strong upregulation of Bax (p<0.01, Figure 4A,B) following MCAO as well as in the LV-Control group, and it was enhanced by Gadd45b-RNAi (p<0.05). Immunochemistry analysis of the ipsilateral ischemic penumbra showed a strong downregulation of Bcl-2 (p<0.01, Figure 4A, B) following MCAO as well as in the LV-Control group, but it was markedly inhibited by Gadd45b-RNAi (p<0.05). Western blot analysis of the ipsilateral ischemic penumbra in each group showed that the expression levels of Bax (Figure 5A, B) and cleaved caspase-3 ( Figure 5A,B) were significantly increased in the MCAO and LV-Control groups compared to those of the sham group (P<0.05). Gadd45b-RNAi enhanced the levels of cleaved caspase-3 and Bax proteins (P<0.05). The protein levels of Bcl-2 were significantly decreased in the MCAO and LV-Control groups compared to those of the sham group (P<0.05), and Gadd45b-RNAi further decreased the levels of Bcl-2 protein (P<0.05).
Immunohistochemical analysis of Bax and Bcl-2 expression in stroke rats. (A) Immunohistochemistry for Bax and Bcl-2 (original magnification 400×). Images are taken from ipsilateral ischemic penumbra. (B) The bar graph reflects the Bcl-2 and Bax-positive cells in each group. (*p<0.01, compared with sham group, #p<0.01, compared with LV-shGadd45b group, △p<0.05, compared with LV-shGadd45b group).
Protein levels of cleaved caspase-3, Bax, and Bcl-2 in stroke rats. (A) Western blot assay of cleaved caspase-3, Bax, and Bcl-2 expression. 1: sham group; 2: MCAO group; 3: LV-Control group; 4: LV-shGadd45b group. (B) The bar graph reflects the cleaved caspase-3, Bax, and Bcl-2 protein expressions in each group. Data were expressed as fold of sham. (*p<0.01, compared with sham group, #p<0.01, compared with LV-shGadd45b group, △p<0.05, compared with LV-shGadd45b group).
Gadd45b-RNAi decreased expression of the neuro-protective agent BDNF
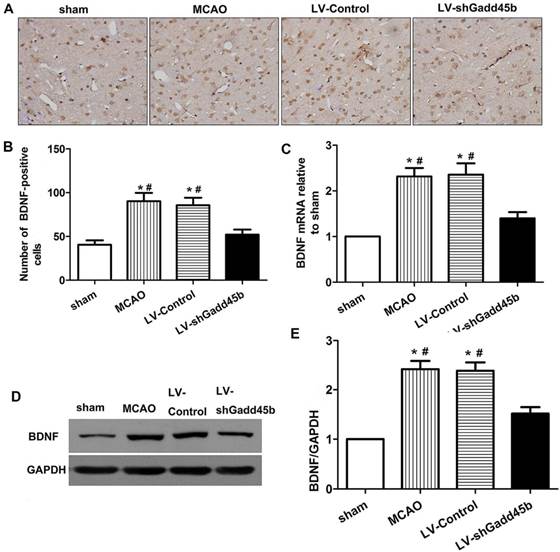
BDNF activation was shown to upregulate Bcl-2, downregulate Bax, inhibit caspase-3 activation and subsequent apoptosis following cerebral ischemia [11,12]. Gadd45b was shown to regulate BDNF expression in brain [13]. To further evaluate the mechanism of Gadd45b in apoptosis, we investigate the effect of Gadd45b-RNAi on BDNF expression. The changes in BDNF protein and mRNA levels were studied through immunohistochemistry, real-time PCR, and Western blot analysis 24 h after focal cerebral ischemia. The brain BDNF levels are shown in Figure 6. In ipsilateral ischemic penumbra, immunohistochemical studies showed that expression of BDNF-positive cells (p<0.01, Figure 6A,B) increased after cerebral ischemia. Gadd45b-RNAi decreased BDNF-positive cells in comparison to MCAO and LV-Control groups (p<0.01). The BDNF protein levels, determined by Western blot analysis, reflected the immunohistochemical results in the same region (p<0.01, Figure 6D,E). Moreover, in ipsilateral ischemic penumbra, the mRNA levels of BDNF were consistent with the protein levels in each group (p<0.01, Figure 6C).
Gadd45b-RNAi treatment suppressed BDNF expression 24 h after MCAO. (A) Immunohistochemistry for BDNF (original magnification 400×). Images are taken from ipsilateral ischemic penumbra. (B) The bar graph reflects the BDNF-positive cells in each group. (C) The bar graph reflects the BDNF mRNA expression in each group. Data were expressed as fold of sham. (D) Western blot assay of the BDNF expression in each group. (E) The bar graph reflects the BDNF protein expressions in each group. Data were expressed as fold of sham. (*p<0.01, compared with sham group, #p<0.05, compared with LV-shGadd45b group).
DISSCUSSION
The aim of this study was to determine the role of Gadd45b on apoptosis during focal cerebral ischemia. In the present study, a previously unknown, intrinsic, anti-apoptotic molecule Gadd45b in ischemic brain was elucidated. Research on enhancing the neuroprotective function of Gadd45b may provide effective therapeutic strategies for preventing the death of the neurons in stroke.
Apoptosis plays an essential role in the acute and chronic phases of ischemic stroke [14,15]. Many studies have shown that inhibition of apoptosis has a beneficial effect in acute ischemic stroke process. The genes responsible for ischemic stroke-induced neuronal cell death have not been fully identified, although various studies have sought to elucidate the molecular basis of acute ischemic stroke. Our results in this study demonstrated that Gadd45b is essential for ischemic stroke-induced neuronal apoptotic death based on the following evidence. First, our previous study showed that mRNA and protein levels of Gadd45b were significantly increased by transient cerebral ischemia in ischemic brain [8]. Second, ischemic stroke-induced neuronal death was remarkably aggravated by the down-regulation of Gadd45b.
Gadd45 is a highly conserved three-gene family consisting of Gadd45a, Gadd45b, and Gadd45g. Gadd45 has been identified as a stress-response gene to physiological or environmental conditions, and is originally induced by genotoxic agents [16]. Gadd45a has been demonstrated as a pro-apoptosis gene in neuronal injuries [17,18] and Gadd45b has been implicated as an anti-apoptosis factor. The present research showed that Gadd45b inhibits neuronal apoptosis after cerebral ischemia. Gadd45b is significantly involved in preventing RGC death [5,6], which is consistent with our findings regarding its anti-apoptotic role in neurons. Gadd45b also inhibits apoptosis in other cell types, such as NIH3T3 and INS-1Eb cells [19,20]. By contrast, Gadd45b was found to induce apoptotic death in cardiomyocytes [21], murine hepatocytes [22,23], and other cell types [24]. Accordingly, the function of Gadd45b in apoptosis appears to be cell type specific. Apoptosis occurs after acute ischemic stroke, and is regulated by the apoptotic and anti-apoptotic proteins of the Bcl-2 family. Pro-survival Bcl-2 proteins include Bcl-2 and Bcl-xL, as well as pro-apoptotic proteins including Bax, Bad, and Bak [25,26]. The relative amounts of active anti- and pro-apoptotic Bcl-2 family proteins determine the resistance of the cells to apoptosis. Increased Bcl-2 expression with a concurrent decrease of Bax mainly functions in protecting the brain against apoptosis induced by acute ischemic stroke [27]. Caspase-3 is also a well-identified pro-apoptotic executioner that is involved in apoptosis [28]. Several groups have provided evidence that activation of caspase-3 is involved in neurons undergoing ischemic cell death [29]. As demonstrated in the present study, Gadd45b down regulation increased brain damage following ischemic stroke. The increase in brain damage is associated with increased apoptosis as indicated by increased levels of Bax and active caspase-3, and decreased levels of Bcl-2.
Many animal studies have documented the anti-apoptotic capability of BDNF in experimental ischemic stroke. BDNF reduces neuronal death after transient forebrain ischemia in vivo [30,31]. Intravenous BDNF counterregulates the expression pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins after focal cerebral ischemia [11]. BDNF also inhibits caspase-3 activation and subsequent apoptosis following hypoxic-ischemic injury [12]. Previous studies have clearly showed that activation of BDNF-associated signaling pathways inhibits neuronal apoptotic cell death.
Gadd45b reduces brain apoptosis, but the mechanism of Gadd45b in apoptosis is not clear. As is shown in this study, Gadd45b may regulates apoptosis by regulating BDNF and downstream regulatory apoptotic proteins in ischemic stroke. First, Gadd45b promoteregulatess DNA demethylation of the regulatory regions of BDNF. Gadd45b knockout mice show a decrease in BDNF gene expression. Neurons lacking Gadd45b fail to demethylate DNA, thereby failing to activate BDNF by previous studies [13,32] and our recent research [33]. Second, many previous studies have clearly showed that activating BDNF-associated apoptotic proteins inhibits neuronal apoptotic cell death. Thirdly, this study indicated that Gadd45b-RNAi treatment obviously down regulated BDNF expression and subsequent apoptosis after ischemic brain injury.
Decreased BDNF levels may related to the decreased demethylation of BDNF by Gadd45b down regulation. Despite all this, the role of BDNF in Gadd45b-mediated apoptosis was not clear. Therefore, it would be interesting to show whether the down regulation of BDNF affect the role of Gadd45b in neuronal apoptotic cell death.
In summary, this study indntify Gadd45b as a critical mediator of cerebral ischemia-induced neuronal apoptotic death. Our findings suggest that treatment strategies targeting Gadd45b could be used to inhibit neuronal apoptotic cell death after acute ischemic stroke.
Abbreviations
Gadd45b, Growth Arrest and DNA-Damage-inducible protein 45 beta; MCAO, middle cerebral artery occlusion; RNAi, RNA interference; LV, lentiviral vectors; CCA, common carotid artery; ECA, external carotid artery; ICA, internal carotid artery; GFP, Greenfluorescent protein; TTC, triphenyltetrazolium chloride; BDNF, brain-derived neurotrophic factor.
Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 81271306) and the Medical Science Research Project of Chongqing Municipal Health Bureau (2012-1-037).
Author Contributions
Conceived and designed the experiments: BL CL. Performed the experiments: BL YZ GH. Analyzed the data: YJ. Contributed reagents/materials/analysis tools: LL XT. Wrote the manuscript: BL.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331-9
2. Joza N, Susin SA, Daugas E, Stanford WL, Cho SK. et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;401:549-554
3. Yamamoto Y, Negishi M. The antiapoptotic factor growth arrest and DNA-damage-inducible 45 beta regulates the nuclear receptor constitutive active/androstane receptor-mediated transcription. Drug Metab Dispos. 2008;36:1189-93
4. Yamamoto Y, Moore R, Flavell RA, Lu B, Negishi M. Nuclear Receptor CAR Represses TNFα-Induced Cell Death by Interacting with the Anti-Apoptotic GADD45B. PLoS One. 2010;5:e10121
5. Liu B, Suyeoka G, Papa S, Franzoso G, Neufeld AH. Growth arrest and DNA damage protein45b (Gadd45b) protects retinal ganglion cells from injuries. Neurobiol Dis. 2009;33:104-10
6. Liu B, Sun X, Suyeoka G, Garcia JG, Leiderman YI. TGFβ signaling induces expression of Gadd45b in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2013;54:1061-9
7. Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616-1622
8. Liu B, Li J, Li L, Yu L, Li C. Electrical stimulation of cerebellar fastigial nucleus promotes the expression of growth arrest and DNA damage inducible gene β and motor function recovery in cerebral ischemia/reperfusion rats. Neurosci Lett. 2012;520:110-114
9. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-408
10. Hunter AJ, Hatcher J, Virley D, Nelson P, Irving E, Hadingham SJ. et al. Functional assessments in mice and rats after focal stroke. Neuropharmacology. 2010;39:806-816
11. Schäbitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M. et al. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212-7
12. Han BH, D'Costa A, Back SA, Parsadanian M, Patel Set. et al. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2007;7:38-53
13. Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML. et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074-1077
14. Legos JJ, Barone FC. Update on pharmacological strategies for stroke: Prevention, acute intervention and regeneration. Curr Opin Investig Drugs. 2003;4:847-858
15. Mitsios N, Gaffney J, Krupinski J, Mathias R, Wang Q. et al. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem Biophys. 2007;47:73-86
16. Liebermann DA, Hoffman B. Gadd45 in the response of hematopoietic cells to genotoxic stress. Blood Cells Mol Dis. 2007;39:344-347
17. Befort K, Karchewski L, Lanoue C, Woolf CJ. Selective up-regulation of the growth arrest DNA damage-inducible gene Gadd45 alpha in sensory and motor neurons after peripheral nerve injury. Eur J Neurosci. 2003;18:911-922
18. Uberti D, Meli E, Memo M. Expression of cell-cycle-related proteins and excitoxicity. Amino Acids. 2002;23:27-30
19. Larsen CM, Dossing MG, Papa S, Franzoso G, Billestrup N. et al. Growth arrest- and DNA-damage-inducible 45b gene inhibits c-Jun N-terminal kinase and extracellular signal-regulated kinase and decreases IL-1beta-induced apoptosis in insulin-producing INS-1E cells. Diabetologia. 2006;49:980-989
20. Engelmann A, Speidel D, Bornkamm GW, Deppert W, Stocking C. Gadd45b is a pro-survival factor associated with stress-resistant tumors. Oncogene. 2008;27:1429-1438
21. Kim MY, Seo EJ, Lee DH, Kim EJ, Kim HS. et al. Gadd45beta is a novel mediator of cardiomyocyte apoptosis induced by ischaemia/hypoxia. Cardiovasc Res. 2010;87(1):119-26
22. Cho HJ, Park SM, Hwang EM, Baek KE, Kim IK. et al. Gadd45b mediates Fas-induced apoptosis by enhancing the interaction between p38 and retinoblastoma tumor suppressor. J Biol Chem. 2010;285:25500-5
23. Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B. et al. Transforming growth factor-b-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278:43001-43007
24. Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521-30
25. Lazou A, Iliodromitis EK, Cieslak D, Voskarides K, Mousikos S. et al. Ischemic but not mechanical preconditioning attenuates ischemia/reperfusion induced myocardial apoptosis in anaesthetized rabbits: the role of Bcl-2 family proteins and ERK1/2. Apoptosis. 2006;11:2195-2204
26. Mayer B, Oberbauer R. Mitochondrial regulation of apoptosis. News Physiol Sci. 2003;18:89-94
27. Rybnikova E, Sitnik N, Gluschenko T, Tjulkova E, Samoilov MO. The preconditioning modified neuronal expression of apoptosis-related proteins of Bcl-2 superfamily following severe hypobaric hypoxia in rats. Brain Res. 2006;1089:195-202
28. Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269-290
29. Namura S, Zhu J, Fink K, Endres M, Srinivasan A. et al. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischaemia. J Neurosci. 1998;18:3659-3668
30. Beck T, Lindholm D, Castren E, Wree A. BDNF protects against ischemic cell damage in rat hippocampus. J Cereb Blood Flow Metab. 1994;14:689-692
31. Tsukahara T, Yonekawa Y, Tanaka K, Ohara O, Wantanabe S. et al. The role of BDNF in transient forebrain ischemia in the rat brain. Neurosurgery. 1994;34:323-330
32. Gavin DP, Sharma RP, Chase KA, Matrisciano F, Dong E. et al. Growtharrest and DNA-damage-inducible, beta (GADD45b)-mediated DNA demethylation in major psychosis. Neuropsychopharmacology. 2012;37:531-42
33. Liu B, Li LL, Tan XD, Zhang YH, Jiang Y. et al. Gadd45b Mediates Axonal Plasticity and Subsequent Functional Recovery After Experimental Stroke in Rats. Mol Neurobiol. 2014 Oct 17
Author contact
![]() Corresponding author: E-mail:licq9217edu.cn
Corresponding author: E-mail:licq9217edu.cn