Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(4):361-369. doi:10.7150/ijbs.10858 This issue Cite
Research Paper
PKD1 Mono-Allelic Knockout Is Sufficient to Trigger Renal Cystogenesis in a Mini-Pig Model
Jin He1,2, ![]() , Qiuyan Li1, Suyun Fang1, Ying Guo1, Tongxin Liu1, Jianhua Ye1, Zhengquan Yu1, Ran Zhang1, Yaofeng Zhao1, Xiaoxiang Hu1, Xueyuan Bai3, Xiangmei Chen3, Ning Li1,4,
, Qiuyan Li1, Suyun Fang1, Ying Guo1, Tongxin Liu1, Jianhua Ye1, Zhengquan Yu1, Ran Zhang1, Yaofeng Zhao1, Xiaoxiang Hu1, Xueyuan Bai3, Xiangmei Chen3, Ning Li1,4, ![]()
1. State Key Laboratory for Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing, PR China
2. College of Animal Science and Technology, China Agricultural University, Beijing, PR China
3. Department of Nephrology, State Key Laboratory of Kidney Disease, Chinese PLA General Hospital, Beijing, PR China
4. College of Animal Science and Technology, Yunnan Agricultural University, Kunming, PR China
Received 2014-10-20; Accepted 2014-12-3; Published 2015-2-10
Abstract
PKD1 and PKD2 mutations could lead to autosomal dominant polycystic kidney disease (ADPKD), which afflicts millions of people worldwide. Due to the marked differences in the lifespan, size, anatomy, and physiology from humans, rodent ADPKD models cannot fully mimic the disease. To obtain a large animal model that recapitulates the disease, we constructed a mini-pig model by mono-allelic knockout (KO) of PKD1 using zinc finger nuclease. The mono-allelic KO pigs had lower PKD1 expression than their wild-type littermates at both the transcriptional and translational levels. After approximately six months, renal cysts appeared and grew progressively in the KO pigs. Histological analysis showed that renal cysts were scatteredly distributed in the mutant pig kidneys and were lined by either cuboidal or flattened epithelial cells. Contrast-enhanced computed tomography confirmed that all of the mutant pigs had renal and hepatic cysts, when they were 11-month-old. Immunohistochemical analysis revealed that most of the cysts were derived from the proximal tubules and collecting ducts. Therefore, the PKD1 mono-allelic knockout is sufficient to trigger renal cystogenesis, and this pig model may provide a platform for future study of renal cyst formation.
Keywords: ADPKD, PKD1, ZFN, Disease model
Introduction
Progressively enlarged fluid-filled cysts in kidneys and other organs are the key features of autosomal dominant polycystic kidney disease (ADPKD), which afflicts millions of people worldwide [1]. No effective therapeutic approach, except for renal replacement therapy, is currently available for ADPKD, a disease that ultimately leads to end stage renal disease in approximately half of the affected patients [2, 3]. Genetic studies have demonstrated that mutations in two genes, PKD1 and PKD2, are responsible for ~85% and ~15% of ADPKD cases, respectively [4-6]. In addition, because of the early onset of cystogenesis, patients with PKD1 mutations have significantly more severe disease than those with mutant PKD2 [7]. Although numerous mutations have been found to be associated with ADPKD thus far, no evident mutation hotspots have been identified in either of the two genes, obscuring our understanding of the genetic basis of ADPKD. Therefore, various hypotheses have been proposed to explain the cyst formation, including two-hit, third hit, hypomorphic/hypermorphic effects, and haploinsufficiency, based on which rodent models have been generated to study the etiology of ADPKD [8]. Much information has been obtained using these models thus far, such as the identification of the signaling pathways involved in ADPKD; additionally, drugs have been tested and screened. However, the short lifespan, fast disease progression, dissimilarities in anatomy/physiology to humans, and late/rare cyst formation in mono-allelic PKD1 or PKD2 knockout (KO) rodent models have hindered the utility of rodents as a model for this chronic disease [9, 10].
Pigs, which are highly similar to humans in their anatomy, physiology, and genetics, are becoming a more attractive animal model for biomedical research [11]. A number of genetically modified porcine models have been reported for cystic fibrosis [12], neurodegenerative disorder [13], and xenotransplantation [14]. However, due to the lack of germ-line competent embryonic stem cells, it is very inefficient to produce KO pigs using conventional gene-targeting methods. With the advent of genome editing tools, such as zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), and clustered, regularly interspersed, short panlindromic repeats (CRISPR)/CRISPR-associated (Cas) protein cas9, highly efficient production of KO pigs has become a reality [15-17].
In previous studies, we cloned and characterized the pig PKD1 and PKD2 genes and demonstrated a remarkable similarity between the human and pig orthologs at molecular level [18, 19]. Furthermore, a porcine PKD2 transgenic model was generated, but there were no cystic manifestations by one year of age in that model [20]. To obtain a relatively early-onset disease model and to document whether the mono-allelic KO of PKD1 (PKD1+/-) triggers renal cyst formation, we utilized ZFN technology to generate PKD1+/- Chinese experimental mini-pigs (CEMP), which showed macroscopically visible renal cysts within one year.
Materials and methods
ZFN
ZFN was designed by Sigma-Aldrich. Different amounts of ZFN were used for optimization of transfection. Twenty-four hours after transfection, the fibroblasts were subjected to DNA extraction (Qiagen). Then the ZFN-F (TAATTACAGGGGCCAAGCAG) and ZFN-R (GAGGCAGGGAAGACGTTGT) primers were designed to sequence the target site, and ZFN-mediated gene editing efficiency was calculated according to one hundred TA-clone sequencing results. The optimized transfection conditions resulted in high gene editing efficiency and low cell toxicity. Based on the standard, transfection with 0.5 μg of each ZFN plasmid was used for subsequent study.
Chinese experimental mini-pig
Chinese experimental mini-pig (CEMP) was established by full-sib inbreeding and negative selection of Xiang pigs [21]. CEMP, which has a life-span of 16-18 years, reaches sexual maturity at 4-6 months old. The sow can deliver 7~8 piglets per litter with a gestation period of 114 days. The neonatal pigs weigh about 0.4-0.5 kg, and then grow to 5 kg at 2-month-old. When they are 6-month-old, the body weight is about 20 kg. The adult pigs weigh approximately 40-50 kg with a height of 45-50 cm at one-year-old. The kidney, liver, heart, and stomach weight-to-body weight ratio of CEMP are similar to human being's, which makes it an intriguing model for biomedical research [21].
Generation of cloned pigs
CEMP embryonic fibroblasts were obtained from an E30 day male embryo and were cultured in DMEM (Sigma) with 10% fetal bovine serum (Gibco) at 37ºC. A neomycin-resistant cassette was derived from the pL452 plasmid by EcoRI and BamHI digestion. A total of 1.5 μg DNA (0.5 μg for each ZFN construct and 0.5 μg of neomycin-resistant cassette) was transfected into 1×106 fibroblasts. Subsequent procedures, including the screening of the cells, colony formation, transfer, and conservation were similar to a previous report [20]. Genotyping of the 72 surviving colonies was performed using the ZFN-F/R primers. Potential off-target sites were sequenced using eight pairs of primers (listed in Supplementary Material: Table S1) flanking these sites. After ruling out off-target events, four fibroblast colonies (Nos. 60, 63, 77, and 96) were selected for somatic cell nuclear transfer [22]. Thirteen surrogate sows each received an average of 384 reconstructed embryos. After approximately 114 days, 20 piglets were delivered naturally from five sows. Genotyping was performed immediately using the ZFN-F/R primers and DNA extracted from ear biopsies. The cloned pigs were fed the same food and water.
PKD1 expression analysis
Quantitative real-time PCR (qRT-PCR) was employed to detect PKD1 expression in WT and PKD1+/- pigs. RNA was extracted from ear-biopsies and kidneys of the cloned pigs by RNeasy (Qiagen) and RNase-free DNase (Qiagen). The primers for PKD1 3' part were 3'-Q-F (CATGTGGCTCCTCTCAAGCA) and 3'-Q-R (GCTTCCAGCAGGACCTTGAGT) targeting exons 36 & 37, for 5' part were 5'-Q-F (ACGTCGGGCTCCTAGAGAA) and 5'-Q-R (TTCCCGCTCAGGTTTATTTC) targeting exons 2 & 3, while those for the internal control, GAPDH, were GAPDH-F (ATCACCATCTTCCAGGAGCGA) and GADPH-R (AGCCTTCTCCATGGTCGTGAA). One microgram of RNA from these tissues was reverse-transcribed into cDNA using oligo dT or random primer in a volume of 25 μL. Then, qRT-PCR reactions were performed in triplicate.
Membrane protein was extracted from the kidneys using the Membrane and Cytosol Protein Extraction Kit (Beyotime, Nantong, China). Eighty micrograms of membrane protein per lane was separated, transferred, and blotted according to a previously established protocol [18]. The antibodies used to detect PC1 were 7e12 (gift from Dr. Christopher Ward) with 1:500 dilution. For internal control, the mouse monoclonal anti-Na+, K+-ATPase antibody (Abcam) was utilized at a 1:1,000 dilution.
Histological analysis
Kidneys from cloned pigs were harvested, minced, and fixed in 10% neutral buffered formalin solution. Then, the samples were dehydrated and embedded in paraffin. For HE analysis, 5-μm-thick sections were stained with hematoxylin and eosin. Antibodies for α-SMA (1:100, Abcam, Shanghai, China), Lrp2 (1:100, Abcam), THP (1:100, Santa Cruz), Calbindin-D-28K (1:100, Sigma-Aldrich), and biotinylated-DBA (1:200, Vector Laboratories, CA) were used for immunohistochemistry by the ABC method (Vector Laboratories). Cyst index, which was calculated using ImageJ (NIH, Bethesda, MD) as the area of lumen divided by the section area, was performed on the WT kidney sections and the non-cystic regions of mutant kidneys.
Contrast enhanced computed tomography
After anesthetization, pigs were injected with Ultravist (Bayer) as contrast medium. Then, CT was performed with a multi-splice spiral CT instrument (Lightspeed Plus, GE), with the following parameters: 120 kV, 500 ms, and 1.5 mm splice thickness. The adjusted renal volume was presented as the mean value of the left and right kidneys normalized to the body weight. CT was performed in the radiology department of either Zhuozhou Chinese Medical Hospital or PLA General Hospital. The renal/hepatic cyst identification and 3D reconstruction were conducted by experienced radiologists from the Department of Radiology, PLA General Hospital.
Statistical analysis
Data are presented as either mean ± SEM or mean ± SD. The unpaired student's t test was used to compare the difference between the WT pigs and each PKD1+/- genotype. The paired student's t test was employed to determine changes in the cyst diameter. P<0.05 was considered statistically significant.
Ethical Statement
All of the procedures were conducted according to the guidelines developed by the China Council on Animal Care and Protocol and were approved by China Agricultural University (No. SKLAB-2012-04-03).
Results and discussion
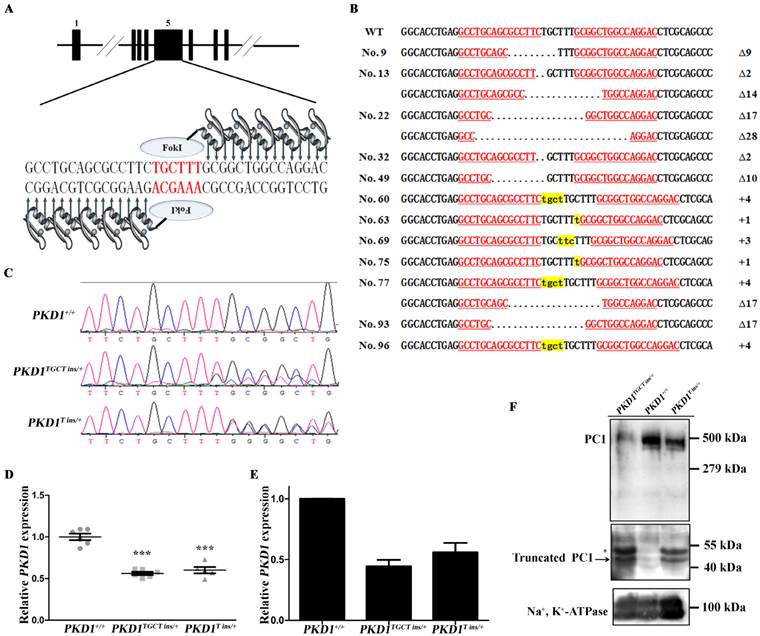
ZFN was designed to target exon 5 of PKD1, where ZFN-induced nucleotide insertions and deletions (indels) are expected to cause frame-shifting, and disrupt normal gene expression (Fig. 1A). After optimization of the transfection conditions (Supplementary Material: Table S2), ZFN plasmids, together with a neomycin-resistant cassette, were then delivered into CEMP fibroblasts obtained from a male embryo (Supplementary Material: Fig. S1A). After seven days of selection with G418, 72 colonies survived. PCR was performed to examine the genotypes of these cells, and 12 colonies (16.7%) were found to harbor mutant PKD1 alleles, of which three colonies contained compound heterozygous KO events (4.2%) (Fig. 1B). Our results showed a higher efficiency than previous reports, which generated heterozygous and homozygous KO fibroblasts with efficiencies of 4.2% and 1%, respectively [15, 16]. To select colonies without off-target modifications of the genome, eight most likely potential off-target sites, each containing six mismatched nucleotides to the ZFN target, were sequenced for all 12 mutant colonies. These potential off-target sites were screened based on the released pig genome sequence [23]. No mutations were identified in the off-target sites, proving high specificity of the ZFN used in our experiment (Supplementary Material: Table S1). Mono-allelic KO colonies Nos. 60, 63, and 96 (PKD1+/-) and a compound heterozygous allele-harboring colony No. 77 (PKD1-/-) were then selected as donors for somatic cell nuclear transfer (SCNT). A total of 4,987 reconstructed embryos were transferred to 13 surrogate sows, and 20 healthy piglets were delivered naturally from five recipients (Supplementary Material: Table S3). Sequencing was conducted to confirm the genotypes of these cloned pigs, of which seven piglets had a TGCT insertion allele (PKD1TGCT ins/+; c.642_643insTGCT, p.Phe215Lysfs*197), while six piglets had a T insertion allele (PKD1T ins/+; c.642_643insT, p.Ala216Cysfs*195) (Fig. 1C). The two frame-shifted alleles with the appearance of TGA at 1,447-1,449 bp or 1,450-1,452 bp of the PKD1 mRNA (GenBank ID: NM_001246202) encoding two 2-residue-difference truncated polypeptides (~42 kDa) that only preserved signal peptide and LRR domains of Polycystin-1 (PC1) (Supplementary Material: Fig. S1B). Moreover, no piglets derived from colony No. 77 were born in our experiment, perhaps because colony No. 77 was incapable of SCNT, or the PKD1-/- resulted in embryonic lethality. As in mouse models that disrupted either PKD1 or PKD2, prenatal and perinatal deaths were common phenomena, which demonstrates that both genes are required for normal placental development [24]. Because PKD1 is ubiquitously expressed in various organs [18], qRT-PCR was performed using RNA from ear biopsies of these piglets. As shown in Fig. 1D and Supplementary Material: Fig. S1C, PKD1+/- (PKD1TGCT ins/+ and PKD1T ins/+) pigs showed a nearly 40% reduction in PKD1 expression, proving that the majority of the mutant alleles cannot produce full length mRNA. To confirm decreased PKD1 expression in PKD1+/- pig kidneys, one neonatal pig (48h) with each genotype was randomly chosen and sacrificed. RNA and membrane protein extracted from the kidneys were subjected to qRT-PCR and Western blot analysis, respectively. As shown in Figs. 1E and F and Supplementary Material: Fig. S1D, PKD1 expression in KO pigs were lower than in PKD1+/+ pig at both the transcriptional and translational levels. Additionally, we detected additional bands with similar molecular weight to the truncated PC1 in PKD1+/- pigs (Fig. 1F), which meant that mutant alleles could generate truncated PC1 to some extent. At sexual maturity, some of the PKD1+/- pigs were mated with WT sows, and a total of 64 offspring were secured, of which 35 piglets inherited the mutant alleles (Supplementary Material: Table S4). This result demonstrated that the mutant alleles could be passed through germline transmission.
Generation and molecular characterization of PKD1+/- pigs. (A) Schematic representation of the ZFN targeting site of PKD1. The red letters indicate the ZFN cleavage site, and the black letters are the ZFN binding sequence. (B) The genotypes of the 12 mutant colonies, of which Nos. 13, 22, and 77 were compound heterozygous colonies. Δ and + indicate the deletion and insertion of nucleotides, respectively, compared to the WT allele. The underlined red letters represents the ZFN binding sequence. The lowercase letters highlighted in yellow denote the inserted nucleotides, and dots mean the deleted nucleotides. (C) Representative direct sequencing results of the WT, TGCT insertion and T insertion alleles, respectively. (D) qRT-PCR analysis of PKD1 expression (mean ± SEM) in neonatal pigs, showing that PKD1TGCT ins/+ (n=6) and PKD1T ins/+ (n=5) pigs had reduced expression compared to WT pigs (n=6, *** P<0.001). (E) qRT-PCR of PKD1 expression in neonatal kidneys showing a reduction of renal PKD1 in PKD1+/- pigs. Data obtained from three replicated experiments are presented as the mean ± SD. Primers amplifying 3' part of PKD1 were used in (D) and (E). (F) The PC1 levels of these cloned pigs were measured by Western blotting of membrane protein extracted from neonatal kidneys using 7e12 antibody. The full-length of PC1 is shown in the upper panel in all pigs (exposure time: 10 min). The arrow in the middle panel indicates the truncated PC1 produced by mutant PKD1 alleles, while the asterisk indicates an ~50 kDa band that might be the glycosylated form of the truncated PC1 (exposure time: 30 min). The PKD1+/+ pig does not have the truncated PC1 fragment. Na+, K+-ATPase was used as an internal control (exposure time: 5 min). The three panels are from three different blots, but they are running simultaneously with the same load of protein.
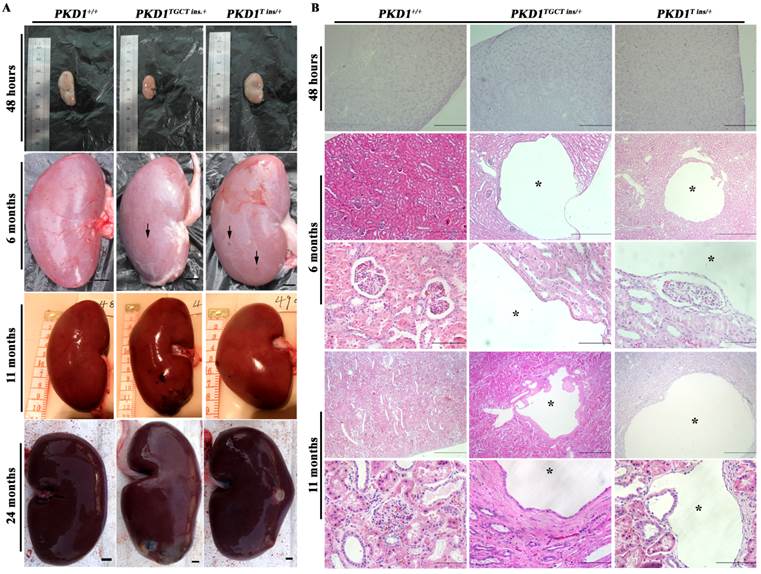
No macroscopic or microscopic cysts had developed in the PKD1+/- pig kidneys by 48 hours after birth, suggesting that half the amount of normal PC1 might be sufficient for renal development (Fig. 2). To investigate whether there are pathological changes in 6-month-old PKD1+/- pig kidneys, one pig from each of the three groups was sacrificed, and the kidneys were collected and fixed. Some small but macroscopically visible fluid-filled cysts began to emerge on the surfaces of the PKD1+/- pig kidneys (Fig. 2A). The largest cysts at this stage were ~5 mm in diameter. At 11 months old, the cysts were growing larger and the number increased, though the cysts were still scattered throughout the PKD1+/- kidneys. In the cystic kidneys, both flattened and cuboidal epithelial cells were found in the lining of the renal cysts (Fig. 2B and Supplementary Material: Fig. S2A). At 24 months, these cysts began to slightly deform the normal kidney shape (Fig. 2A). Further analysis also confirmed the appearance of renal cysts in F1 PKD1+/- piglets by as early as 3 months after birth, suggesting the stable inheritance of PKD1 mutant alleles (Supplementary Material: Fig. S2A). Additionally, we measured the cyst index of the non-cystic regions of mutant kidneys. Although we could find at least 50 cysts/microcysts in each KO pig kidneys, the cyst index did not show any signs of tubule dilation (20.0% ± 0.8%) compared with WT kidney sections (19.5% ± 2.0%) (Supplementary Material: Fig. S2B). This result suggested that the disease was in the early-stage without affecting the whole kidney.
PKD1+/- pigs show the typical ADPKD phenotype. (A) The gross morphology of the kidneys collected at different ages exhibits macroscopic cysts (arrow) on the surface of the PKD1+/- pig kidneys (scale bar =1 cm). (B) Hematoxylin and eosin analysis of the kidneys reveals typical renal cysts (*) lined by either flattened or cuboidal epithelia in the PKD1+/- pigs (scale bar for neonatal kidneys is 1 mm; for upper panel of 6-month-old kidneys, the bar is 500 μm; for the lower panel of 6-month-old kidneys, it is 100 μm; for the upper panel of 11-month-old kidneys, it is 1 mm; for the lower panel of 11-month-old kidneys, it is 100 μm). For each stage, at least 30 renal sections or 20 cysts were analyzed.
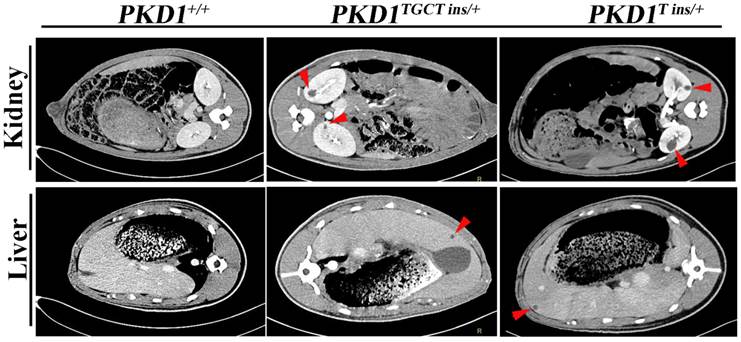
As an accurate approach for detecting renal cysts, contrast-enhanced computed tomography (CT) was employed to examine the 11-month-old pigs. To our knowledge, there is no CT diagnostic criterion for ADPKD; thus, we adopted an ultrasonographic standard to examine these pigs [25]. In this criterion, the presence of at least 3 cysts in one or two kidneys from patients younger than 30 years of age would confirm the diagnosis of ADPKD. Because contrast-enhanced CT can detect a cyst of at least 1.5 mm, we set the threshold to 5 mm for counting cysts to minimize the false-positive diagnosis. We found that many different-sized cysts were present in the bilateral kidneys of all tested PKD1+/- pigs (Fig. 3 and Supplementary Material: Table S5). Means of nine and five cysts in bilateral kidneys of the 11-month-old pigs had larger diameters than 5 mm in the PKD1TGCT ins/+ and PKD1T ins/+ pigs, respectively. The largest diameter was 28.3 mm in a PKD1TGCT ins/+ pig, while a PKD1T ins/+ pig had a 19.3 mm cyst. In addition to renal cysts, liver cysts, observed in approximately 80% of ADPKD cases [26], were found in all PKD1+/- pigs (Fig. 3).
Contrast enhanced computed tomography was performed to diagnose the renal and hepatic cysts in PKD1+/- pigs at 11-month-old. Red arrowheads show the typical cysts in the kidneys and livers.
To confirm that these cysts could grow gradually, CT was performed on 18-month-old pigs. The detected renal cysts were larger than those from 11-month-old pigs by 1.27 ± 0.30 mm (P=0.0002) (Supplementary Material: Fig. S1E). In current studies, total kidney volume (TKV) is recognized as a more accurate marker for polycystic kidney disease, as changes in this measurement mainly reflect alterations in the cyst numbers and volumes [27]. Thus, in our study, 3D reconstruction and calculation of kidney volumes were performed, but no TKV difference was observed in the PKD1+/- pigs at this stage compared with the WT littermates (Supplementary Material: Fig. S1F). The result meant that these mono-allelic KO pigs were in the early stage of disease progression without significant renal enlargement.
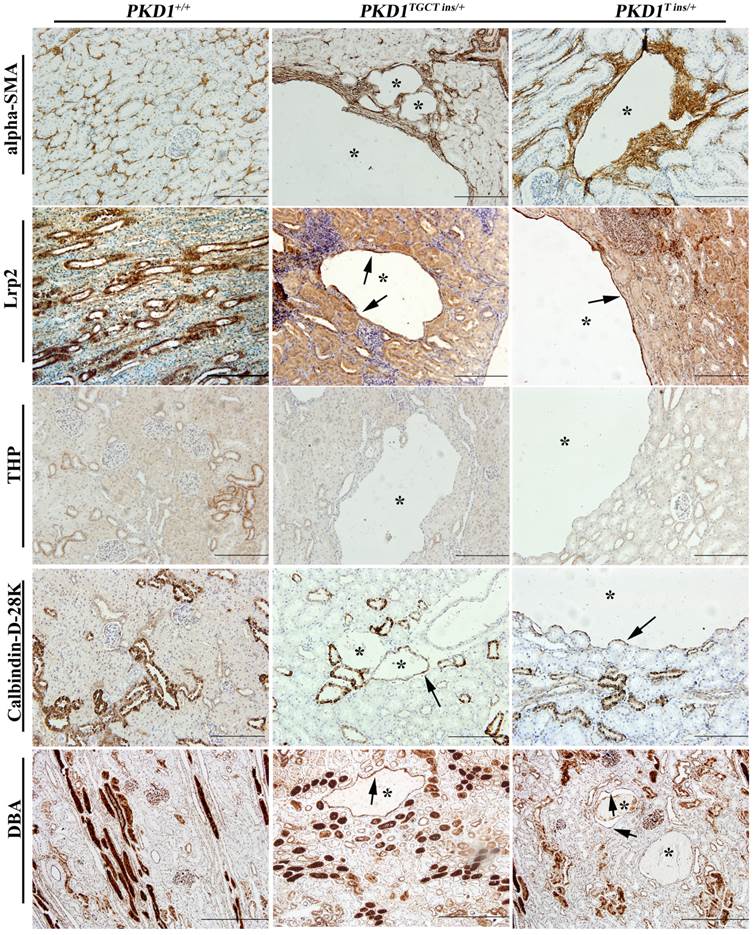
As human renal cysts can arise from all nephron segments and collecting ducts, segment-specific markers for nephrons and collecting ducts were used to identify the origin of the cysts in our experiment. All the markers except for THP, which stains thick ascending limbs of loop of Henle (LH) and distal tubules (DT), were positively stained (Fig. 4), indicating that the cysts derived mainly from proximal tubules (PT) and collecting ducts (CD). Our results are similar to the recent report of Hopp et al, who demonstrated in a mouse ADPKD model that cysts originating from the LH and DT were consistently <1% [28]. Another ADPKD characteristic like renal fibrosis, which is associated with disease progression, can also be observed in the PKD1+/- pigs [29]. Early-stage human ADPKD often have focal staining of α-SMA, similar to our result shown in Fig. 4, where α-SMA mainly surrounded the renal cysts suggesting the increased number of interstitial myofibroblasts. These cells are typical feature of fibrosis, and can participate in the deposition of extracellular matrix [29].
Immunohistochemical characterization of renal cysts in PKD1+/- pigs. The upper panel shows that α-SMA staining fibrosis surrounding renal cysts (*) in the PKD1+/- pigs. The origin of renal cysts was determined using segment-specific markers/antibodies for proximal tubules (Lrp2), thick ascending limbs of loop of Henle/distal convoluted tubules (Tamm-horsefall protein, THP), distal convoluted tubules/cortical collecting ducts (Calbindin-D-28K), and collecting ducts (Dolichos Biflorus agglutinin, DBA). Cysts were positively stained by all of these antibodies/markers (arrow) except for THP. Each tubular segment specific marker was used to stain at least 30 cysts in each PKD1+/- kidneys. Scale bars in the images are 200 μm, except for DBA staining, which are 500 μm.
As young human patients only have rare cysts, which is reflected by the diagnostic criterion [25], our PKD1+/- pigs, which is comparable to human adolescence, shows some aspects of early-stage human ADPKD in terms of sporadic renal and hepatic cysts. Until now, mouse models, which have provided us with essential knowledge of ADPKD, are still the predominant animal models used in ADPKD research [8]. However, no effective therapies have developed thus far from these models, e.g., some drugs, which slow down disease progression in ADPKD mice, failed in preclinical trials [30, 31]. The possible reason is likely due to the differences in anatomy, physiology, metabolism and genetics between mouse and human. Over the mouse models, pigs show many advantages, which may either supplement our understanding of ADPKD from a different angle or act as a bridge between clinical trial and basic research. First, porcine kidney has a comparable size and similar multipapillate structure to human kidney, making it an ideal model for histological study and sample [32]. Second, ADPKD is a systemic disease with many extrarenal manifestations [1], such as cardiovascular abnormalities, intracranial aneurysm and hypertension, some of which can be better studied using pigs than mice.
Third, compared to mono-allelic PKD1 or PKD2 KO mice, which showed late-onset cystogenesis or no cyst formation [9, 10, 33, 34], our PKD1+/- pigs have a relatively early and progressed cystogenesis. Finally, as a chronic disease, our CEMP pigs, which have a lifespan of at least 16 years, could be readily used for longitudinal studies of ADPKD. Additionally, the PKD1+/- pigs could also be used as a platform to facilitate the construction of other related models. As we showed that PKD1 mutant alleles could be passed to offspring by germline transmission (Supplementary Material: Table S4), PKD1+/- embryonic fibroblasts can be established after the mating of PKD1+/- and PKD1+/+ pigs. With modifications to the normal PKD1 allele in fibroblasts by introducing a hypomorphic mutation, a faster progressing disease model than PKD1+/- could be obtained; or by knockout of PKD2, a pig with the PKD1+/-; PKD2+/- genotype can be produced to elucidate the trans-heterozygosity hypothesis.
Conclusion
Our experiment demonstrated that mono-allelic KO of PKD1 could trigger renal and hepatic cysts formation. This model may be served as an alternative model for ADPKD because of the observed early-onset of cystogenesis, combined with relatively slow disease progression, mimicking the chronic nature of human ADPKD. Therefore, the model we developed could be used either to study the molecular mechanism of cystogenesis, or as a basis to construct other fast-progressed disease models.
Supplementary Material
Tables S1-S5, Figures S1-S2.
Abbreviations
ADPKD, autosomal dominant polycystic kidney disease; CEMP, Chinese Experimental mini-pig; CRISPR/Cas9, clustered, regularly interspersed, short panlindromic repeats (CRISPR)/CRISPR-associated (Cas) protein cas9; CT, computed tomography; DT, distal tubule; indel, insertion and deletion; KO, knockout; LH, loops of Henle; PC1, polycystin-1; PKD1, polycystic kidney disease 1 gene; PKD2, polycystic kidney disease 2 gene; PT, proximal tubule; SCNT, somatic cell nuclear transfer; TALEN, transcription activator like effector nuclease; TKV, total kidney volume; WT, wild type; ZFN, zinc finger nuclease.
Acknowledgements
This work was supported by National Basic Research Program of China (No. 2011CB944103) and National Natural Science Foundation of China (No. 81370840). H.J. was also sponsored by the Chinese Universities Scientific Fund (No. 2013BH002). We thank Dr. Tian Shuping (Chinese PLA General Hospital) for the CT analysis.
Competing Interest
A patent application on the PKD1TGCT ins/+ and PKD1T ins/+ pigs has been submitted by N. L and J. H. as the inventors.
References
1. Gabow PA. Autosomal dominant polycystic kidney disease--more than a renal disease. Am J Kidney Dis. 1990;16:403-13
2. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149-68 doi:10.1038/ki.2009.128
3. Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151-64 doi:10.1056/NEJMra022161
4. Kimberling WJ, Kumar S, Gabow PA, Kenyon JB, Connolly CJ, Somlo S. Autosomal dominant polycystic kidney disease: localization of the second gene to chromosome 4q13-q23. Genomics. 1993;18:467-72
5. Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ. et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339-42
6. Pignatelli PM, Pound SE, Carothers AD, Macnicol AM, Allan PL, Watson ML. et al. Multipoint mapping of adult onset polycystic kidney disease (PKD1) on chromosome 16. J Med Genet. 1992;29:638-41
7. Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB. et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013-9 doi:10.1681/ASN.2006080835
8. Wilson PD. Mouse models of polycystic kidney disease. Curr Top Dev Biol. 2008;84:311-50 doi:10.1016/S0070-2153(08)00606-6
9. Lu W, Fan X, Basora N, Babakhanlou H, Law T, Rifai N. et al. Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet. 1999;21:160-1 doi:10.1038/5944
10. Chang MY, Parker E, Ibrahim S, Shortland JR, Nahas ME, Haylor JL. et al. Haploinsufficiency of Pkd2 is associated with increased tubular cell proliferation and interstitial fibrosis in two murine Pkd2 models. Nephrol Dial Transplant. 2006;21:2078-84 doi:10.1093/ndt/gfl150
11. Aigner B, Renner S, Kessler B, Klymiuk N, Kurome M, Wunsch A. et al. Transgenic pigs as models for translational biomedical research. J Mol Med (Berl). 2010;88:653-64 doi:10.1007/s00109-010-0610-9
12. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ. et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837-41 doi:10.1126/science.1163600
13. Yang D, Wang CE, Zhao B, Li W, Ouyang Z, Liu Z. et al. Expression of Huntington's disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum Mol Genet. 2010;19:3983-94 doi:10.1093/hmg/ddq313
14. Dai Y, Vaught TD, Boone J, Chen SH, Phelps CJ, Ball S. et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol. 2002;20:251-5 doi:10.1038/nbt0302-251
15. Yang D, Yang H, Li W, Zhao B, Ouyang Z, Liu Z. et al. Generation of PPARgamma mono-allelic knockout pigs via zinc-finger nucleases and nuclear transfer cloning. Cell Res. 2011;21:979-82 doi:10.1038/cr.2011.70
16. Hauschild J, Petersen B, Santiago Y, Queisser AL, Carnwath JW, Lucas-Hahn A. et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci U S A. 2011;108:12013-7 doi:10.1073/pnas.1106422108
17. Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M. et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A. 2012;109:17382-7 doi:10.1073/pnas.1211446109
18. He J, Wang Q, Ye J, Hu X, Li N. Identification of porcine polycystic kidney disease 1 (PKD1) gene: molecular cloning, expression profile, and implication in disease model. Gene. 2011;490:37-46 doi:10.1016/j.gene.2011.08.027
19. Wang Q, Yin H, He J, Ye J, Ding F, Wang S. et al. cDNA cloning of porcine PKD2 gene and RNA interference in LLC-PK1 cells. Gene. 2011;476:38-45 doi:10.1016/j.gene.2011.01.017
20. He J, Ye J, Li Q, Feng Y, Bai X, Chen X. et al. Construction of a transgenic pig model overexpressing polycystic kidney disease 2 (PKD2) gene. Transgenic Res. 2013;22:861-7 doi:10.1007/s11248-012-9686-z
21. Yu Shu-min, Wang Chuan-wu, Zhao De-ming, Zhang Qing-cai, De-zhi P. Raising and pathogen purification of chinese experimental mini-pig. Laboratory Animal Sciences and Management. 2003;20:44-6
22. Wei H, Li Q, Li J, Li Y, Dai Y, Ma Y. et al. Effect of leptin on oocyte maturation and subsequent pregnancy rate of cloned embryos reconstructed by somatic cell nuclear transfer in pigs. Progress in Natural Science. 2008;18:1583-7 doi:10.1016/j.pnsc.2008.05.018
23. Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF. et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491:393-8 doi:10.1038/nature11622
24. Garcia-Gonzalez MA, Outeda P, Zhou Q, Zhou F, Menezes LF, Qian F. et al. Pkd1 and Pkd2 are required for normal placental development. PLoS One. 2010 5. doi:10.1371/journal.pone.0012821
25. Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E. et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205-12 doi:10.1681/ASN.2008050507
26. Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM. et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64-9 doi:10.2215/CJN.00080605
27. Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148-57 doi:10.2215/CJN.00330705
28. Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG. et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257-73 doi:10.1172/JCI64313
29. Norman J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim Biophys Acta. 2011;1812:1327-36 doi:10.1016/j.bbadis.2011.06.012
30. Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C. et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830-40 doi:10.1056/NEJMoa1003491
31. Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J. et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820-9 doi:10.1056/NEJMoa0907419
32. Ibrahim Z, Busch J, Awwad M, Wagner R, Wells K, Cooper DK. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation. 2006;13:488-99 doi:10.1111/j.1399-3089.2006.00346.x
33. Ahrabi AK, Terryn S, Valenti G, Caron N, Serradeil-Le Gal C, Raufaste D. et al. PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol. 2007;18:1740-53 doi:10.1681/ASN.2006010052
34. Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM. et al. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol. 2009;20:2389-402 doi:10.1681/ASN.2008040435
Author contact
![]() Corresponding author: hejin47edu.cn; ninglicaulabcom
Corresponding author: hejin47edu.cn; ninglicaulabcom