Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Acknowledgements
Conflicts of interest
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):879-891. doi:10.7150/ijbs.12101 This issue Cite
Research Paper
Magnolia Bioactive Constituent 4-O-Methylhonokiol Prevents the Impairment of Cardiac Insulin Signaling and the Cardiac Pathogenesis in High-Fat Diet-Induced Obese Mice
Zhiguo Zhang1, 2, 3, Jing Chen3, Shanshan Zhou1, 2, 3, Shudong Wang1, 2, 3, Xiaohong Cai2, Daniel J. Conklin4, Ki-Soo Kim5, Ki Ho Kim6, Yi Tan2, 3, Yang Zheng1, ![]() , Young Heui Kim7,
, Young Heui Kim7, ![]() , Lu Cai2, 3
, Lu Cai2, 3
1. Department of Cardiology at the First Hospital of Jilin University, Changchun, 130021, China
2. The Chinese-American Research Institute for Diabetic Complications and the Second Affiliated Hospital, Wenzhou Medical University, Wenzhou 325035, China
3. Kosair Children's Hospital Research Institute, Department of Pediatrics of University of Louisville, Louisville, KY 40202
4. Diabetes and Obesity Center, University of Louisville, Louisville, KY 40202
5. Bioland Biotec HaiMen Co., Ltd, Linjiang New District, Haomen, 226100, China
6. KHBios, 505 Venture Center, 194-41, Osongsaengmyeong 1, Osong, Cheongju, Chungbuk 363-951, Republic of Korea
7. Bioland R&D Center, 59 Songjeongni 2-gil, Byeongcheon, Dongnam, Cheonan, Chungnam 330-863, Republic of Korea
Received 2015-3-11; Accepted 2015-5-13; Published 2015-6-5
Abstract

In obesity, cardiac insulin resistance is a putative cause of cardiac hypertrophy and dysfunction. In our previous study, we observed that Magnolia extract BL153 attenuated high-fat-diet (HFD)-induced cardiac pathogenic changes. In this study, we further investigated the protective effects of the BL153 bioactive constituent, 4-O-methylhonokiol (MH), against HFD-induced cardiac pathogenesis and its possible mechanisms. C57BL/6J mice were fed a normal diet or a HFD with gavage administration of vehicle, BL153, or MH (low or high dose) daily for 24 weeks. Treatment with MH attenuated HFD-induced obesity, as evidenced by body weight gain, and cardiac pathogenesis, as assessed by the heart weight and echocardiography. Mechanistically, MH treatment significantly reduced HFD-induced impairment of cardiac insulin signaling by preferentially augmenting Akt2 signaling. MH also inhibited cardiac expression of the inflammatory factors tumor necrosis factor-α and plasminogen activator inhibitor-1 and increased the phosphorylation of nuclear factor erythroid-derived 2-like 2 (Nrf2) as well as the expression of a Nrf2 downstream target gene heme oxygenase-1. The increased Nrf2 signaling was associated with decreased oxidative stress and damage, as reflected by lowered malondialdehyde and 3-nitrotyrosine levels. Furthermore, MH reduced HFD-induced cardiac lipid accumulation along with lowering expression of cardiac fatty acid translocase/CD36 protein. These results suggest that MH, a bioactive constituent of Magnolia, prevents HFD-induced cardiac pathogenesis by attenuating the impairment of cardiac insulin signaling, perhaps via activation of Nrf2 and Akt2 signaling to attenuate CD36-mediated lipid accumulation and lipotoxicity.
Keywords: Nrf2, obesity, lipid accumulation, lipotoxicity, cardiac insulin signaling, 4-O-methylhonokiol
Introduction
Obesity is a major risk for the development of type 2 diabetes mellitus and cardiovascular disease [1, 2]. Over-nutrition manifests itself as hyperlipidemia, which is believed to be a major factor for the development of insulin resistance and lipotoxicity in humans and animals [3, 4]. It was reported that lipid overload leads to an accumulation of intramyocellular lipid-derived metabolites, which coincide with insulin resistance [5]. Previous studies have demonstrated that the lipid accumulation of metabolites such as long chain acyl CoAs, diacylglycerols, ceramide and/or triglycerides (TGs) results from an impaired ability of the mitochondria to oxidize fatty acids [6, 7]. These lipid metabolites activate a number of different serine kinases that negatively modulate the insulin response [8]. Although statins are used to treat hyperlipidemia, they increase the incidence of diabetes [9]. These findings emphasize the need for novel medications that prevent obesity and diabetes and the associated cardiac disease.
Natural products with anti-obesity and anti-diabetes effects yet lacking toxicity are attractive for the prevention of and therapy against the growing epidemic [10]. The stem bark of Magnolia officinalis has been used as a traditional medicine to treat diabetes in China and Japan. Honokiol (HON), magnolol (MAG), and 4-O-methylhonokiol (MH) are considered major bioactive constituents of Magnolia stem bark [11]. Recent studies revealed that MAG and HON, bioactive constituents of Magnolia, ameliorated body fat accumulation and insulin resistance in high-fat diet (HFD)-fed mice [12]. Moreover, MAG decreases fasting blood glucose and plasma insulin levels in a type 2 diabetic model without affecting body weight gain [13] or glucose uptake in adipocytes [14]. In our previous study, we observed that BL153, a Magnolia officinalis extract, partially attenuated heart damage including lipid accumulation, inflammation, oxidative stress, and apoptosis in a HFD-induced obese mouse model [15]. Because Magnolia officinalis also contains other ingredients except for the bioactive component MH, [16, 17], we investigated whether the pure bioactive component play the role in the prevention by Magnolia officinalis extract of HFD-metabolic syndrome. Our pilot study demonstrated that MH prevented HFD-induced obesity and systemic insulin resistance in C57BL/6J mice [18]. Therefore, we further determined whether MH can also effectively prevent obesity-induced heart damage and impaired cardiac insulin signaling.
To further explore the preventive efficacy of MH in HFD-induced cardiac damage, the effects of MH at 0.5 mg/kg were compared with BL153 used at a near equivalent reference dose of 5 mg/kg, which has been shown to reduce HFD-induced cardiac damage [15]. Furthermore, the protective mechanisms of BL153 (and MH) remain unclear. In this study, we investigated both the efficacy and mechanisms of BL153- and MH-mediated protection against diet-induced cardiac insulin resistance and the associated lipotoxicity.
Materials and methods
Magnolia extract (BL153) and its bioactive constituent MH
BL153 and MH were provided by Bioland Co., Ltd. (Chungnam, Korea). A voucher specimen was deposited at the Herbarium of Chungbuk National University, Chungbuk, Korea (voucher specimen #: CNBU2009006). Briefly, the air-dried bark of Magnolia officinalis was extracted twice with 95% ethanol. The ethanol extract of BL153 was analyzed by HPLC to ensure that it contained 10.2% 4-O-methylhonokiol (Lot No. MBLEH-052140). This fraction was purified by silica gel chromatography to obtain pure MH (purity >95.45%). The BL153 and MH were delivered by gavage in 0.5% ethanol in deionized water (vehicle) as described previously [18].
Animal model
All experiments involving animals were carried out in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Louisville Institutional Animal Care and Use Committee.
Eight-week-old male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME), housed in the University of Louisville Research Resources Center under a constant temperature of 22 °C with a 12-h light/dark cycle, and fed standard rodent chow and tap water to acclimate for 1 week after delivery. Eighty mice were randomly assigned to the following 8 groups (n=10): normal diet (ND, 10 kcal% fat; 3.85 kcal/g, D12450B, Research Diets Inc.) or HFD (60 kcal% fat; 5.24 kcal/g, D12492, Research Diets Inc.) and simultaneous daily gavage-treated vehicle (0.5% ethanol), BL153 (5 mg/kg), low-dose MH (L-MH, 0.5 mg/kg, equal to 5 mg/kg BL153), or high-dose MH (H-MH, 1.0 mg/kg) for 24 weeks. The gavage volume was 1% of the mouse body weight and was adjusted weekly based on the change in body weight.
Non-invasive blood pressure (BP) and echocardiography
Both BP recording and echocardiography were performed at the 24th week of the experiment. BP was measured with a non-invasive tail cuff Plethysmograph system (CODA6TM, Kent Scientific, Torrington, CT). Mice were restrained in a plastic tube for 5 min prior to starting BP measurement on a temperature-control platform. Tails were exposed for positioning of tail-cuff inflation/deflation and a pressure sensor. BP was recorded after mice were trained at least 3 days. Given that tail-cuff measurement using a volume pressure recording system can only accurately measure systolic BP and underestimates diastolic BP compared with direct telemetry measurement [19], only systolic BP values are reported here.
Transthoracic echocardiography was measured with a Vevo 770 ultrasound system (Visualsonics, Toronto, Canada). Mice were anesthetized with intraperitoneal injection of 1.2% Avertin (Sigma, St. Louis, MO) and placed in the supine position on a temperature-controlled platform. The chest hair was removed with a depilatory to reduce ultrasound attenuation. The images were recorded in parasternal long-axis and short-axis views. The left ventricular posterior wall (LVPW) and interventricular septum (IVS) were measured in parasternal short axis M-mode images. At the same time, the ejection fraction (EF), fractional shortening (FS), and LV mass were calculated using Vevo770 software. The data were averaged over 10 cardiac cycles [20].
Sample collection and preparation
After a 6-h fasting 10 mice in each group were randomly divided into two subgroups for intraperitoneal injection of human insulin (Humalin R; Eli Lily, Indianapolis, IN) at 1.5 U/kg body weight or phosphate-buffered saline 15 min prior to euthanasia. Hearts were isolated, weighed, and collected for RNA extraction, protein preparation, and cryosectioning (OCT-embedded). Mouse tibia length was measured with digital micrometer for calculation of heart hypertrophy index.
Oil Red O staining for lipid accumulation
Oil Red O staining was performed to examine lipid accumulation in hearts after 24 weeks of HFD feeding [21]. After cryostat-sectioning, heart tissue slides were fixed in 10% neutral buffered formalin for 5 min., briefly washed with running tap water, immersed in 60% isopropanol for a few seconds, and then stained with Oil Red O working solution (saturated Oil red O isopropanol solution was diluted as 4:6 into 60% isopropanol, Sigma-Aldrich, St. Louis, MO) for 10 min at room temperature. Slides were washed with 60% isopropanol and counterstained with hematoxylin (DAKO, Carpinteria, CA) for 45 s, and images were captured (Nikone Eclipse E600 microscope, Melville, NY). The area of Oil red O staining was analyzed in nine fields of view for each heart. The area value is the size of the selection within the set threshold, and the integrated optical density, a measurement parameter in Image Pro Plus 6.0 software (Media Cybernetics, Bethesda, MA), used to quantitatively analyze the area of Oil Red O staining.
Determination of lipid peroxidation
Lipid peroxidation was assessed using a thiobarbituric acid (TBA) reactive substances (TBARS) assay according to the formation of malondialdehyde (MDA) during acid hydrolysis of the lipid peroxide compound. The reaction mixtures included 20 μl 8.1% sodium dodecyl sulfate, 50 μl supernatant of homogenate samples, 210 μl 0.571% TBA, and 150 μl 20% acetic acid solution (pH 3.5). All samples were assayed in duplicate. The mixtures were incubated at 90 ºC for 1 h and cooled on ice. Then 100 μl distilled water was added, and the mixtures were centrifuged at 4000 rpm for 15 min. The absorbance of the supernatant was recorded at 540 nm. The TBARS level is presented as nmol/mg protein [22].
Western blotting assays
Western blotting assays were performed as described previously [21]. The primary antibodies included those against phospho-nuclear factor erythroid 2 related factor 2 (p-Nrf2), tumor necrosis factor-alpha (TNF-α), peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC-1α), sirtuin 1 (Sirt1), and fatty acid translocase/cluster of differentiation 36 (CD36), all of which were obtained from Abcam (Cambridge, MA). Total Nrf2 (t-Nrf2), hexokinase II (HK II), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). AMP-activated protein kinase α (AMPKα), phospho-AMPKα (Thr172), phospho-Akt (Ser473, p-Akt), total Akt (t-Akt), phospho-Akt2 (Ser474, p-Akt2), and total Akt2 (t-Akt2) were all obtained from Cell Signaling Technology (Danvers, MA), and 3-nitrotyrosine (3-NT) was purchased from Millipore (Billerica, MA). Quantitative densitometry was performed on identified bands using Image Quant 5.2 software (GE Healthcare Bio-Sciences AB) [23, 24].
RNA isolation and real-time RT-PCR
The mRNA levels of heme oxygenase 1 (HO-1) in the heart were quantified by real-time RT-PCR. Briefly, total RNA was extracted from heart tissues using Trizol reagent (RNA STAT60 Tel-Test; Ambion, Austin, TX). RNA concentration and purity were quantified using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE). One microgram of total RNA was used for first strand cDNA synthesis using iScript cDNA Synthesis system (Master Cycler, Eppendorf, NY). For real-time PCR, primers (mouse HO-1: Mm00516005_m1; mouse GAPDH Mm99999915_g1) were purchased from Applied Biosystems (Foster City, CA). The amplification reactions were carried out in triplicate 20-μl reactions that contained 10 μl TaqMan Universal PCR Master Mix (Applied Biosystems), 1 μl primers, 3 μl cDNA, and 6 μl DD H2O, in the ABI 7300 Real-Time PCR system (Life Technologies Corporation, Carlsbad, CA) with an initial hold step at 50 °C for 2 min, followed by 95 °C for 10 min, and then 50 cycles of a two-step procedure (92 °C for 15 s and 60 °C for 1 min). The comparative cycle time (Ct) method was used to determine fold differences between samples and determine the amount of target, normalized to an endogenous reference GAPDH and relative to a calibrator (2-ΔΔCt) [22].
Statistical analysis
Data are presented as means ± standard deviation (SD). Comparisons were performed by two-way analysis of variance for the different groups, followed by post-hoc pair-wise repetitive comparisons with Tukey's test using Origin 8.6 Lab data analysis and graphing software (Origin Lab Corporation, Northampton, MA). Statistical significance was considered if P <0.05.
Results
Effects of MH on HFD-induced obesity, heart weight, BP, and cardiac function in mice
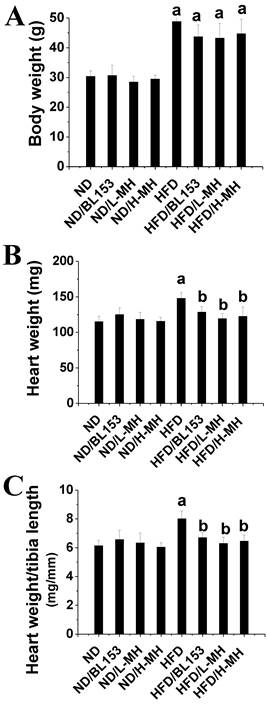
Body weight was measured as the index of obesity. HFD feeding significantly increased body weight compared with ND feeding. MH and BL153 treatment only slightly inhibited the body weight increase (Fig. 1A) compared to that in the HFD group. No body weight change was observed in ND-fed mice treated with MH or BL153 (Fig. 1A). Heart weights and tibia lengths were measured, and no differences in heart weight (Fig. 1B), tibia length, or the ratio of heart weight to tibia length (Fig. 1C) were observed among the ND groups. However, treatment with either MH or BL153 significantly decreased HFD-induced increases in heart weight and the heart weight to tibia length ratio.
Effects of MH on HFD-induced mouse body and heart weights. Eight-week-old male C57BL/6J mice were fed either a ND (10% kcal as fat) or a HFD (60% kcal as fat) with vehicle (0.5% ethanol), BL-153 (5 mg/kg body weight), or MH (0.5 or 1 mg/kg) for 24 weeks. (A) Body weight; (B) heart weight; (C) heart weight to tibia length. Data are presented as means ± SD (n=10). a, p< 0.05 vs. ND; b, p< 0.05 vs. HFD.
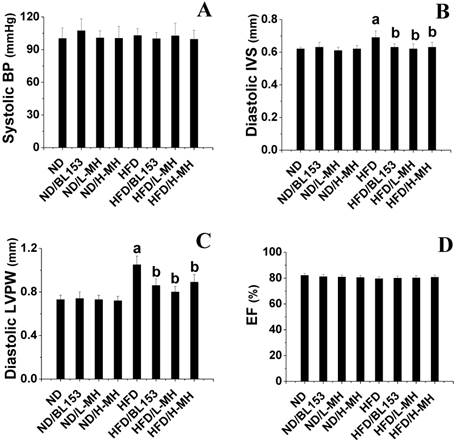
Although neither the ND nor HFD led to an obvious change in systolic BP (Fig. 2A), echocardiography examination indicated that HFD feeding induced cardiac hypertrophy as indicated by increased wall thickness including diastolic IVS and diastolic LVPW compared with those in ND-fed mice (Fig. 2B,C). MH and BL153 treatments significantly attenuated cardiac hypertrophy induced by HFD feeding (Fig. 2B,C). No change in overall myocardial systolic function was detected (as per EF; Fig. 2D) between ND- and HFD-fed mice. Treatment with MH and BL153 did not affect myocardial systolic function or systolic BP in ND- and HFD-fed mice (Fig. 2D).
Effects of MH on HFD-induced BP change and cardiac hypertrophy in mice. Systolic BP was measured by non-invasive tail cuff Plethysmograph system (A). Cardiac wall thickness was measured by echocardiography including diastolic interventricular septum (IVS, B) and diastolic left ventricular posterior wall (LVPW, C). At the same time, the ejection fraction (EF, D) was calculated using Vevo770 software. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD.
Effects of MH on cardiac inflammation and oxidative stress in obese mice
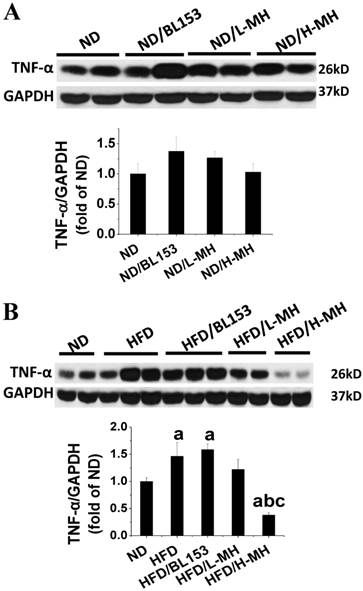
Cardiac inflammation and oxidative stress are important contributors to the development of cardiac hypertrophy [25]. In present study, the inflammatory marker TNF-α in cardiac tissue was measured by Western blotting. In ND-fed mice, MH did not affect the expression of TNF-α (Fig. 3A). HFD feeding significantly elevated cardiac TNF-α expression; yet these markers were significantly diminished by H-MH treatment (Fig. 3B).
Effects of MH on HFD-induced inflammation in heart tissue. The expression of inflammation marker TNF-α was measured in ND-fed mice (A) and HFD-fed mice (B) by western blotting and quantified by densitometric analysis. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD; c, p< 0.05 vs. HFD/BL153.
Animal studies have shown that HFD-induced obesity increases oxidative stress [15]. Here we found that HFD feeding significantly increased oxidative stress in the heart tissue as demonstrated by increased levels of MDA (TBARS assay, Fig. 4A) and 3-NT (Fig. 4C). Both MH and BL153 significantly reduced the endogenous (basal) and HFD-induced levels of MDA (Fig. 4A) and 3-NT (Fig. 4B,C).
Effects of MH on HFD-induced oxidative stress and damage in heart tissue. (A) Lipid peroxidation in heart tissue was measured by a thiobarbituric acid reactive substances (TBARS) assay (TBARS was equivalent to MDA). (B, C) The levels of reactive nitrogen stress were measured by 3-nitrotyosine (3-NT) in ND-fed mice (B) and HFD-fed mice (C) with western blotting. (D, E) The levels of phosphorylated p-Nrf2 were measured in ND-fed mice (D) and HFD-fed mice (E) by western blotting. (F) mRNA expression of HO-1, as the target gene of p-Nrf2, was measured by real-time RT-PCR. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD
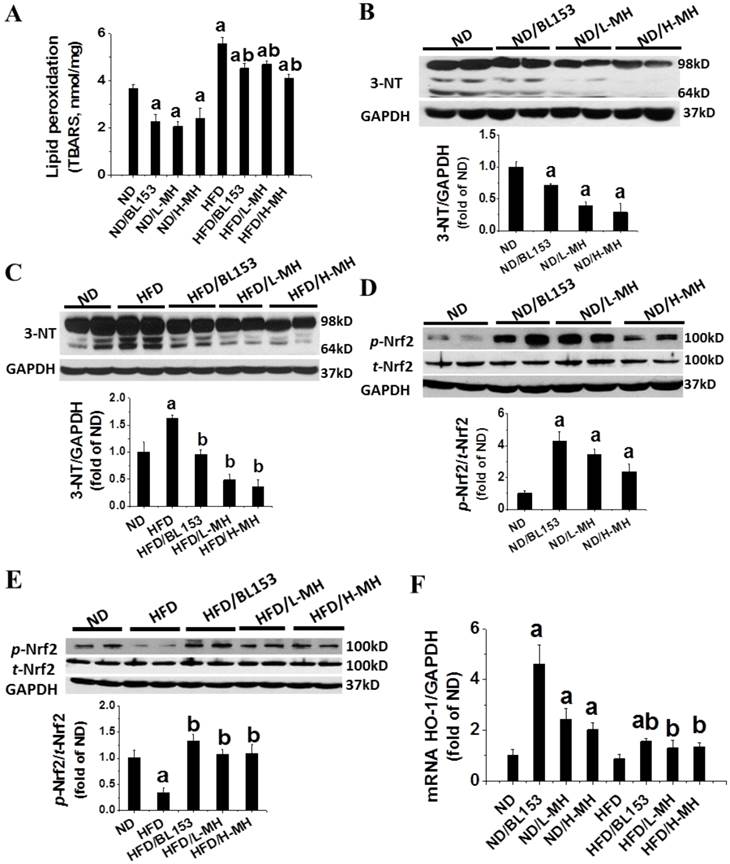
In ND-fed mice, both MH and BL153 significantly increased the levels of Nrf2 phosphorylation (Fig. 4D) and HO-1 mRNA expression (Fig. 4F). HFD feeding was found to significantly down-regulate Nrf2 phosphorylation (Fig. 4E). MH or BL153 could significantly upregulate Nrf2 phosphorylation and HO-1 mRNA expression in both ND- and HFD-fed mice (Fig. 4F).
Effects of MH on cardiac insulin signaling in obese mice
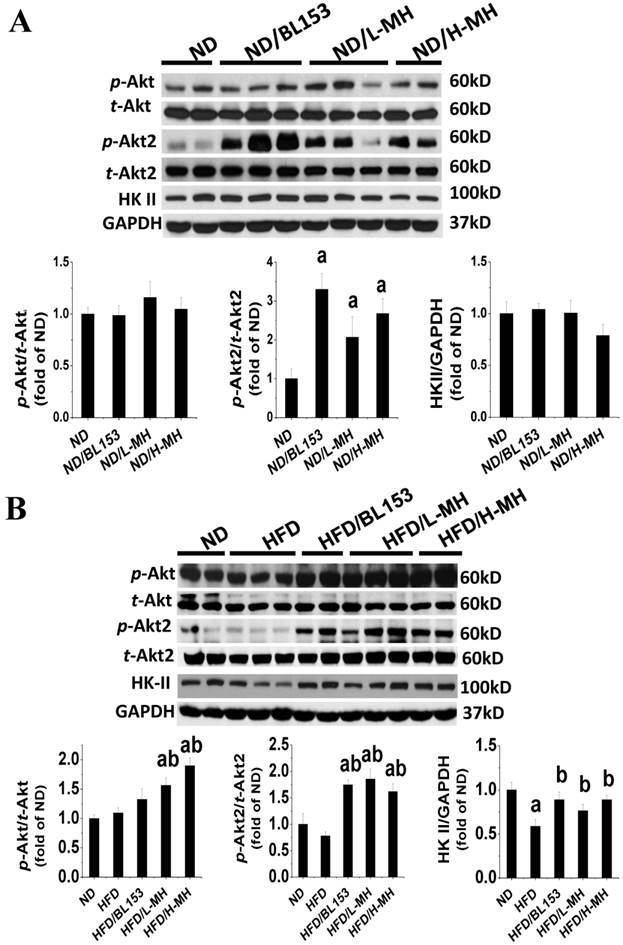
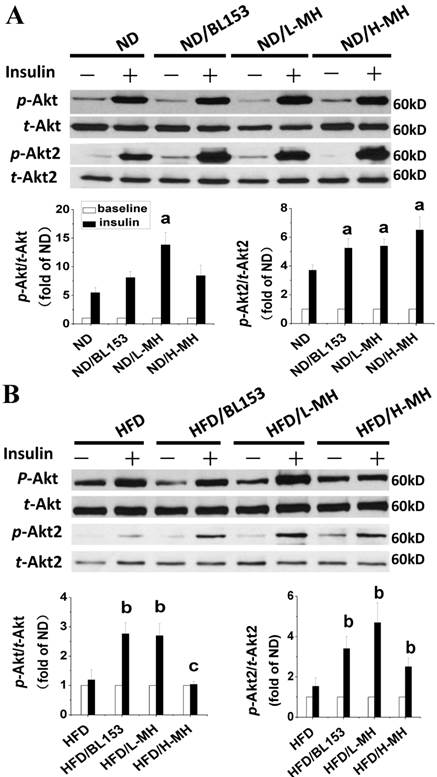
Cardiac lipid accumulation reportedly causes insulin resistance in HFD-fed obese mice [26]. Thus, we examined whether MH and BL153 affected in vivo insulin targeting of Akt, which is required for insulin-stimulated glucose uptake, and HK II, a glycolysis regulator. Consistent with the results of a previous study [26], HFD feeding impaired cardiac insulin signaling, as demonstrated by downregulation of Akt2 phosphorylation and a significant reduction of HK II expression (Fig. 5B). In ND-fed mice, both MH and BL153 significantly increased insulin-induced Akt2 activation, as reflected by the increased ratio of phosphorylated Akt2 to total Akt2 expression (Fig. 5A). Compared to HFD feeding alone, treatment of HFD-fed mice with MH significantly increased the ratio of phosphorylated Akt to total Akt expression, and both MH and BL153 significantly increased Akt2 activation and HK II expression (Fig. 5B). Insulin treatment in vivo (15 min) significantly increased phosphorylation levels of Akt and Akt2 in ND-fed mice (Fig. 6A) but not in HFD-fed mice (Fig. 6B). Treatment with MH or BL153 dramatically boosted insulin-induced phosphorylation levels of both Akt and Akt2 except for Akt phosphorylation upon treatment with H-MH in HFD-fed mice.
Effects of MH on cardiac glucose metabolism. Protein expression of p-Akt, p-Akt2, and HK II in ND-fed mice (A) and HFD-fed mice was measured by western blot analysis. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD.
Effects of MH on cardiac glucose metabolism after insulin treatment for 15 min. The data for insulin treatment (black column) are presented as the fold change relative to baseline (without insulin treatment, white column). Protein expression of p-Akt, p-Akt2, and HK II in ND-fed mice (A) and HFD-fed mice (B) was measured by western blot analysis. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND+Insulin; b, p< 0.05 vs. HFD+Insulin; c, p<0.05 vs. HFD/BL153+Insulin.
Effects of MH on cardiac lipid accumulation in obese mice
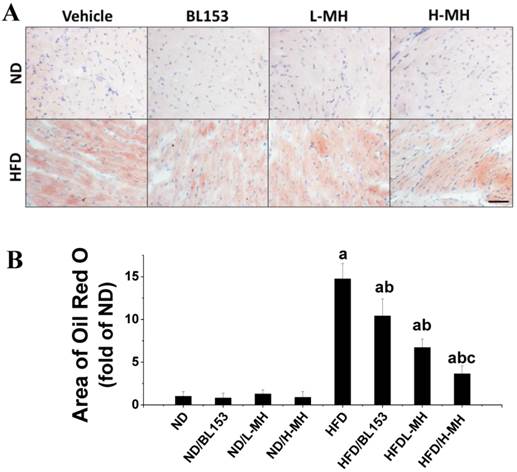
Lipid accumulation in cardiomyocytes is a marker of lipotoxicity and a feature of HFD-induced obesity. Histological detection by Oil Red O staining revealed that both MH and BL153 treatment significantly inhibited HFD-induced lipid accumulation in the heart. A quantitative analysis further showed that the area of Oil Red O staining in hearts of HFD-fed mice was almost 15 times higher than that present in ND-fed mice. This effect of the HFD was significantly reduced with either MH or BL153 treatment, and MH treatment results in a dose-dependent decrease in lipid accumulation that was more effective than that observed with BL153 treatment (Fig. 7). No lipid accumulation was observed in the ND-fed groups.
Effects of MH on HFD-induced lipid accumulation in heart tissue. Cardiac lipid accumulation was detected by Oil Red O staining (20×, bar=50 µm) of cryosections (A). The area of Oil Red O staining was quantified as a fold difference from that in sections from ND-fed mice (B). Data are presented as means ± SD (n=10). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD; c, p< 0.05 vs. HFD/BL153.
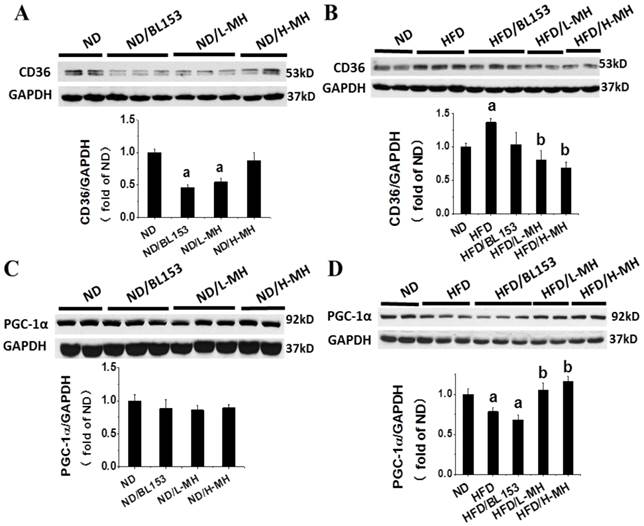
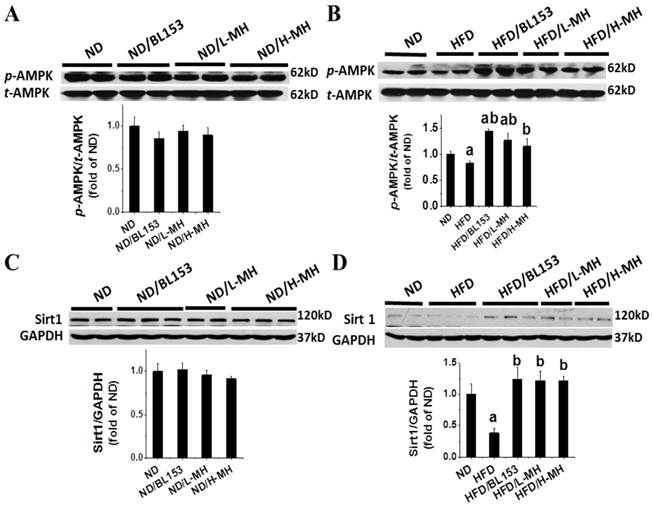
To understand how MH regulates lipid metabolism in the heart, we assessed the expression of fatty acid uptake protein (CD36) and controlling lipidolysis protein, PGC-1α. In ND-fed mice low-dose MH and BL153, but not high-dose MH, significantly reduced CD36 expression (Fig. 8A). However, MH and BL153 did not affect PGC-1α expression in ND-fed mice (Fig. 8C). HFD feeding significantly increased the expression of CD36 and simultaneously decreased the expression of PGC-1α compared to levels in ND-fed mice. Treatment of HFD-fed mice with MH, but not BL153, significantly reduced the effect of HFD on CD36 and PGC-1α expression (Fig. 8B,D). To better understand the influence of diet on PGC1-1α, we further investigated the upstream role of AMPK via NAD+-dependent type III deacetylase Sirt1 activity [27], because activation of both AMPK and Sirt1 can increase the expression of PGC-1α [27, 28]. We found that MH treatments did not affect the expression of either phospho-AMPK (i.e., activity of AMPK) or Sirt in ND-fed mice (Fig. 9A,C). HFD feeding decreased the expression of both phospho-AMPK and Sirt1 compared to levels in ND-fed mice (Fig. 9B,D), and both targets were significantly restored by treatment with either BL153 or MH, suggesting a potential pathway to reverse the effects of a HFD.
Effects of MH on cardiac lipid metabolism in HFD-fed mice. Protein expression of CD36 and PGC-1α was measured in ND-fed mice (A,C) and HFD-fed mice (B,D) by western blot analysis. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD.
Effects of MH on cardiac AMPK and Sirt1 expression in HFD-induced obese mice. AMPK and Sirt1 were measured in ND-fed mice (A,C) and HFD-fed mice (B,D) by western blot analysis. Data are presented as means ± SD (n=5). a, p<0.05 vs. ND; b, p< 0.05 vs. HFD.
Discussion
In the present study, we explored the preventive effects of MH, an active constituent of BL153, on cardiac insulin resistance and associated pathogenic changes including structural remodeling, inflammation and oxidative stress induced by HFD feeding. The preventive effect of L-MH treatment is not significantly different from that of BL153 treatment in terms of cardiac hypertrophy, lipid accumulation, inflammation, and oxidative stress [15, 18]. In current study, HFD feeding increased heart weight and cardiac wall thickness (IVS and LVPW) and slightly decreased cardiac function (decreased EF), as measured by echocardiographic examination. MH and BL153 treatments attenuated HFD feeding-induced cardiac hypertrophy. These findings are in a line with those of previous reports, indicating that obesity increased cardiac lipid accumulation, which was associated with insulin resistance, cardiac hypertrophy, and cardiac dysfunction in humans [29] and animal models [30]. Several studies have proved that chronic HFD feeding promotes ectopic fatty acid (FA) storage as TGs in cardiomyocytes, leading to an induction of cardiac insulin resistance in rodents, even though the heart obviously is not a normal site for lipid storage [6, 7, 15, 31]. Cardiac insulin resistance often causes cardiac hypertrophy as a consequence [32]. Overloaded TG-induced impairment of the insulin/Akt pathway is thought to be a causative factor in diabetic cardiomyopathy [33]. In our study, HFD feeding did not affect the levels of phospho-Akt and phospho-Akt2 compared to those levels in ND-fed mice. In HFD-fed mice, however, the increases in phospho-Akt and phospho-Akt2 levels after insulin-treatment for 15 min were significantly lower than those in ND-fed mice. The expression of HK II, a rate-limiting enzyme in glucose metabolism, is reduced in obesity and diabetes [21, 34]. In our current study, HKII expression was significantly reduced in HFD-fed mice, which indicates that HFD feeding impairs cardiac insulin signaling and glycolysis.
Although it has been known that Magnolia bioactive constituent (HON, MAG, and MH) can ameliorate systemic insulin resistance induced by HFD feeding [12, 18], the molecular mechanisms by which Magnolia improves HFD-induced systemic insulin resistance, particularly in the heart, are still unclear. It was reported that HON and MAG stimulate glucose uptake by activating Akt phosphorylation, a key locus in the insulin signaling pathway, in L6 myotubes [35]. There are three isoforms in the Akt family [36]. Akt1 is involved in cellular survival pathways that support physiological growth and function, and Akt3 may play an important role in the prevention of neurological disorders [36]. Also, Akt2 plays a key role in the insulin signaling pathway [37]. Insulin resistance is observed in mice with an Akt2 gene deletion [37] but not in mice with an Akt1 gene deletion [38]. One novel finding of our present study is that another bioactive constituent of Magnolia, MH, significantly upregulated the activity of Akt (predominantly the Akt2 isoform) in both ND- and HFD-fed mice. To further explore Akt function, we treated ND- and HFD-fed mice with insulin in vivo and then measured the phosphorylation level of Akt2. HFD feeding prevented the insulin-induced increase in Akt2 phosphorylation, which was significantly upregulated in ND-fed mice. However, treatment with MH both preserved in HFD-fed mice the phosphorylation level of Akt2 and further enhanced it in ND-fed mice as well. These results suggest that MH restores the impaired insulin signaling caused by HFD feeding probably through upregulation of phosphorylation of Akt2.
It is known that cardiac insulin resistance is associated with lipid accumulation [5]. A previous study reported that reduction of intracellular lipid levels by decreasing FA uptake of cells and increasing FA oxidation reversed insulin resistance [39]. Therefore, another novel finding in the present study is that MH inhibits the expression of CD36 that can enhance FA uptake in cardiomyocytes. This finding suggests that MH may be an inhibitor of CD36, such that MH attenuates cardiac lipid accumulation by inhibiting the expression of CD36. On the other hand, an investigation of FA oxidation pathway has revealed that the activity and expression of the critical FA oxidator, PGC-1α, was reduced, leading to mitochondrial dysfunction in patients with insulin resistance and type 2 diabetes [40]. Consistently, we showed here that the expression of PGC-1α is markedly decreased in the heart of HFD-fed mice, an effect that is significantly restored by MH. In the PGC-1α pathway, AMPK is an important sensor that regulates lipid metabolism [41]. Activated AMPK upregulates the cellular NAD+/NADH ratio, which in turn activates Sirt1 (an NAD+-dependent type III deacetylase) [42]. Sirt1 can deacetylate PGC-1α and increase its activity [43]. Taken together, our results suggest that MH may enhance mitochondrial function and oxidative capacity by restoring AMPK and Sirt1 activities and their downstream target, PGC-1α. Although it has been reported that MH is a PPARγ agonist [44] that plays an important role in hypolipidemic activity [45], the expression of PPARγ in the heart is relatively low and PPARγ probably also does not play the key role in cardiac metabolism regulation [46]. Therefore, in our study we did not measure the levels of PPARγ.
Increased oxidative stress in obesity and diabetes is associated with the development of insulin resistance [47, 48]. It has been reported that HFD-induced obesity induces cardiac oxidative stress by increasing reactive oxygen species and nicotinamide adenine dinucleotide phosphate oxidase levels in the cytosol [49, 50]. Consistent with the results of these studies, in our study, HFD-fed mice also exhibited increased cardiac oxidative stress, as shown by increased MDA and 3-NT levels (Fig. 4A and 4C). MH dramatically prevented the induction of oxidative and/or nitrosative stress under HFD feeding conditions in a dose-dependent manner. We further investigated how MH inhibits cardiac oxidative stress and found that the activity of Nrf2, as reflected indirectly by its target gene HO-1 at the mRNA level, was upregulated in the MH treatment group. Nrf2 is crucial in the regulation of cellular defenses against oxidative stress [51]. In the nucleus, phospho-Nrf2 forms heterodimers that bind to antioxidant-responsive element (ARE) and then trigger basal expression and induction of detoxification enzymes/proteins such as HO-1 [52]. Therefore, these results suggest that through upregulation of Nrf2, MH may ameliorate oxidative stress to additionally help preserve cardiac insulin signaling in HFD-fed mice.
Regarding the mechanism by which MH activates Nrf2, it has been shown recently that Nrf2 transcriptional activity can be upregulated by insulin signaling. Specifically, activated Akt can enhance Nrf2 activity by prevention of Nrf2 nuclear exclusion and degradation in cytosol [53]. In our study, we found that MH treatment not only upregulated the phosphorylation of Akt but also increased Nrf2 activation. Therefore, we propose that MH enhances Nrf2 activity probably by upregulation of Akt signaling.
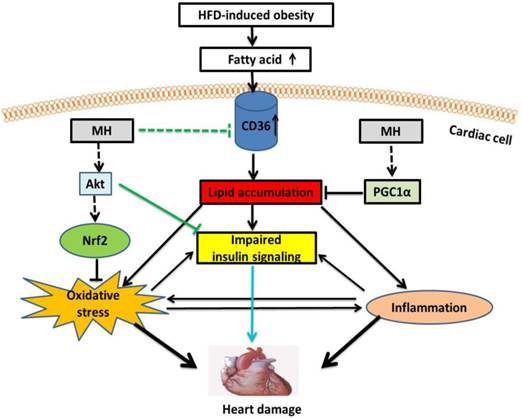
In summary, here we demonstrated that MH as the key bioactive component in Magnolia officinalis extract play the key role in the prevention by Magnolia officinalis extract of HFD-metabolic syndrome and its cardiovascular damage. Our working hypothesis is presented in Fig. 10, illustrating the potential mechanisms by which MH restores the impaired cardiac insulin signaling in HFD-induced obese mice. First, MH may upregulate Akt (predominantly Akt2 isoform) activity to improve insulin signaling. Second, MH may attenuate intracellular lipid accumulation by decreasing FA uptake through inhibition of CD36 expression and increasing FA oxidation through AMPK-Sirt1-PGC-1α pathway, and subsequently improve insulin signaling. Lastly, MH may attenuate oxidative stress-impaired insulin signaling by activating an Akt-Nrf2 pathway. Compared with the selected reference (BL153), MH also showed a similar effect on cardiac hypertrophy and insulin signaling, but because MH is a further purified constituent, it potentially should have much less severe side effects/toxicity compared with BL153. Taken together, these findings indicate that the bioactive constituent MH from Magnolia officinalis extract represents a new therapeutic candidate for cardiac hypertrophy induced by cardiac insulin resistance in obese/pre-diabetic patients. Due to the purity of MH compared to previously used BL153, it may have more potential to be considered as an alternative approach alone or combined with other routinely used medication to preventing the cardiovascular disorders in the obesity individuals.
A proposed mechanism for the effects of MH on cardiac insulin resistance in obese mice. MH might upregulate Akt (mainly Akt2) activity to ameliorate obesity-impaired cardiac insulin signaling. On the other hand, MH might attenuate oxidative stress-induced insulin resistance by activating the Akt-Nrf2 pathway. Lastly, MH might inhibit expression of CD36, which decreases FA uptake, and preserve the FA oxidation pathway (AMPK-Sirt1-PGC-1α), subsequently reducing intracellular lipid accumulation and finally reversing insulin signaling.
Acknowledgements
This study was supported in part by Chungbuk Technopark Grant Bio-International Collaborating Research Project, funded by Chungbuk Province, Republic of Korea, a collaborative project between University of Louisville and Bioland Co. Lid for “Screening Anti-diabetes and/or Obesity Naturally Extracted Compounds”, funded by Bioland Co., Ltd. (10-0826 Bioland), a Start-Up Fund for Chinese-American Research Institute for Diabetic Complications at Wenzhou Medical University (to YT & LC), a Research Development Fund of Wenzhou Medical University (QTJ 13007 to YT), a rsearch grant from the National Natural Science Foundation of China (No. 81370318 to ZY), a research project of the Diabetes and Obesity Center, funded by the National Institute of General Medical Sciences (GM103492, to DC), and projects funded by Natural Science Foundation of Zhejiang Province (Y2110277) and Zhejiang Province Science and Technology (2013C33174).
Conflicts of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
References
1. Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am. 2011;95:875-892
2. Abbasi F, Brown BW Jr, Lamendola C, McLaughlin T, Reaven GM. Relationship between obesity, insulin resistance, and coronary heart disease risk. J Am Coll Cardiol. 2002;40:937-943
3. Calder PC, Ahluwalia N, Brouns F, Buetler T, Clement K, Cunningham K. et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br J Nutr. 2011;106(Suppl 3):S5-78
4. Heidemann C, Scheidt-Nave C, Richter A, Mensink GB. Dietary patterns are associated with cardiometabolic risk factors in a representative study population of German adults. Br J Nutr. 2011;106:1253-1262
5. Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507-520
6. Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC. et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4:465-474
7. Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J. et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A. 2007;104:17075-17080
8. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005-2011
9. Chogtu B, Magazine R, Bairy KL. Statin use and risk of diabetes mellitus. World J Diabetes. 2015;6:352-357
10. Li WL, Zheng HC, Bukuru J, De Kimpe N. Natural medicines used in the traditional Chinese medical system for therapy of diabetes mellitus. J Ethnopharmacol. 2004;92:1-21
11. Maruyama Y, Kuribara H. Overview of the pharmacological features of honokiol. Cns Drug Reviews. 2000;6:35-44
12. Kim YJ, Choi MS, Cha BY, Woo JT, Park YB, Kim SR. et al. Long-term supplementation of honokiol and magnolol ameliorates body fat accumulation, insulin resistance, and adipose inflammation in high-fat fed mice. Mol Nutr Food Res. 2013;57:1988-1998
13. Sohn EJ, Kim CS, Kim YS, Jung DH, Jang DS, Lee YM. et al. Effects of magnolol (5,5'-diallyl-2,2'-dihydroxybiphenyl) on diabetic nephropathy in type 2 diabetic Goto-Kakizaki rats. Life Sci. 2007;80:468-475
14. Choi SS, Cha BY, Lee YS, Yonezawa T, Teruya T, Nagai K. et al. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sci. 2009;84:908-914
15. Sun W, Zhang Z, Chen Q, Yin X, Fu Y, Zheng Y. et al. Magnolia extract (BL153) protection of heart from lipid accumulation caused cardiac oxidative damage, inflammation, and cell death in high-fat diet fed mice. Oxid Med Cell Longev. 2014;2014:205849
16. Zhang H, Hu Z, Yang G, Shi Z, Sun H. Separation and determination of magnolol and honokiol in crude and burn-fried Magnolia officinalis. Anal Lett. 1997;30:2327-2339
17. Lee YJ, Lee YM, Lee CK, Jung JK, Han SB, Hong JT. Therapeutic applications of compounds in the Magnolia family. Pharmacol Ther. 2011;130:157-176
18. Zhang Z, Chen J, Jiang X, Wang J, Yan X, Zheng Y. et al. The Magnolia Bioactive Constituent 4-O-Methylhonokiol Protects against High-Fat Diet-Induced Obesity and Systemic Insulin Resistance in Mice. Oxid Med Cell Longev. 2014;2014:965954
19. Feng M, Whitesall S, Zhang Y, Beibel M, D'Alecy L, DiPetrillo K. Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens. 2008;21:1288-1291
20. Bai Y, Cui W, Xin Y, Miao X, Barati MT, Zhang C. et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. J Mol Cell Cardiol. 2013;57:82-95
21. Wang Y, Feng W, Xue W, Tan Y, Hein DW, Li XK. et al. Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes. 2009;58:1391-1402
22. Jiang X, Zhang C, Xin Y, Huang Z, Tan Y, Huang Y. et al. Protective effect of FGF21 on type 1 diabetes-induced testicular apoptotic cell death probably via both mitochondrial- and endoplasmic reticulum stress-dependent pathways in the mouse model. Toxicol Lett. 2013;219:65-76
23. Cai L, Chen S, Evans T, Deng DX, Mukherjee K, Chakrabarti S. Apoptotic germ-cell death and testicular damage in experimental diabetes: prevention by endothelin antagonism. Urol Res. 2000;28:342-347
24. Zhao Y, Tan Y, Dai J, Li B, Guo L, Cui J. et al. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol Lett. 2011;200:100-106
25. Yoshida K, Kim-Mitsuyama S, Wake R, Izumiya Y, Izumi Y, Yukimura T. et al. Excess aldosterone under normal salt diet induces cardiac hypertrophy and infiltration via oxidative stress. Hypertens Res. 2005;28:447-455
26. Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK. et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530-3540
27. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774-785
28. Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33-39
29. Pucci G, Battista F, de Vuono S, Boni M, Scavizzi M, Ricci MA. et al. Pericardial fat, insulin resistance, and left ventricular structure and function in morbid obesity. Nutr Metab Cardiovasc Dis. 2014;24:440-446
30. Dirkx E, van Eys GJ, Schwenk RW, Steinbusch LK, Hoebers N, Coumans WA. et al. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J Mol Cell Cardiol. 2014;76:208-17
31. van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. 2008;94:231-241
32. Chou IP, Chiu YP, Ding ST, Liu BH, Lin YY, Chen CY. Adiponectin receptor 1 overexpression reduces lipid accumulation and hypertrophy in the heart of diet-induced obese mice - possible involvement of oxidative stress and autophagy. Endocr Res. 2014;39:173-9
33. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013;10:109-121
34. Pendergrass M, Koval J, Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R. et al. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes. 1998;47:387-394
35. Choi SS, Cha BY, Lee YS, Yonezawa T, Teruya T, Nagai K. et al. Honokiol and magnolol stimulate glucose uptake by activating PI3K-dependent Akt in L6 myotubes. Biofactors. 2012;38:372-377
36. Cohen MM Jr. The AKT genes and their roles in various disorders. Am J Med Genet A. 2013;161a:2931-2937
37. Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd. et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728-1731
38. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349-38352
39. Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A. et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A. 2007;104:16480-16485
40. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S. et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466-8471
41. Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113-120
42. Brandauer J, Vienberg SG, Andersen MA, Ringholm S, Risis S, Larsen PS. et al. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J Physiol. 2013;591:5207-5220
43. Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855-2862
44. Lee NJ, Oh JH, Ban JO, Shim JH, Lee HP, Jung JK. et al. 4-O-methylhonokiol, a PPARgamma agonist, inhibits prostate tumour growth: p21-mediated suppression of NF-kappaB activity. Br J Pharmacol. 2013;168:1133-1145
45. Padmanabhan M, Arumugam G. Effect of Persea americana (avocado) fruit extract on the level of expression of adiponectin and PPAR-gamma in rats subjected to experimental hyperlipidemia and obesity. J Complement Integr Med. 2014;11:107-119
46. Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ. et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518-524
47. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X. et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625-633
48. Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H. et al. Ca mishandling and cardiac dysfunction in obesity and insulin resistance: Role of oxidative stress. Cell Calcium. 2014;56:408-15
49. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX. et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457-2466
50. Hickson-Bick DL, Buja LM, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol. 2000;32:511-519
51. Ishii T, Itoh K, Yamamoto M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002;348:182-190
52. Apopa PL, He X, Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol. 2008;22:63-76
53. Zhuang CL, Mao XY, Liu S, Chen WZ, Huang DD, Zhang CJ. et al. Ginsenoside Rb1 improves postoperative fatigue syndrome by reducing skeletal muscle oxidative stress through activation of the PI3K/Akt/Nrf2 pathway in aged rats. Eur J Pharmacol. 2014;740:480-487
Author contact
![]() Corresponding authors: Dr. Yang Zheng, Department of Cardiology, the First Hospital of Jilin University, Changchun, China. E-mail: zhengyangedu.cn; Tel:+86-0431-88782217. Dr. Young Heui Kim at the Bioland R&D Center, 59 Songjeongni 2-gil, Byeongcheon, Dongnam, Cheonan, Chungnam 330-863, Republic of Korea, Telephone: 82-41-550-7791; Fax: 82-41-550-7707, Email: yhkim1stcom
Corresponding authors: Dr. Yang Zheng, Department of Cardiology, the First Hospital of Jilin University, Changchun, China. E-mail: zhengyangedu.cn; Tel:+86-0431-88782217. Dr. Young Heui Kim at the Bioland R&D Center, 59 Songjeongni 2-gil, Byeongcheon, Dongnam, Cheonan, Chungnam 330-863, Republic of Korea, Telephone: 82-41-550-7791; Fax: 82-41-550-7707, Email: yhkim1stcom