Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Natural Sources
3. Targeting Apoptosis pathways...
4. Targeting Cancer Cells by...
5. Icariside II As an Adjuvant...
6. Conclusion and Future...
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(9):1100-1112. doi:10.7150/ijbs.11595 This issue Cite
Review
Targeting Apoptosis and Multiple Signaling Pathways with Icariside II in Cancer Cells
Muhammad Khan1, Amara Maryam1, Javed Iqbal Qazi2, Tonghui Ma1 ![]()
1. College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China.
2. Department of Zoology, University of the Punjab, Quaid-e-Azam Campus, Lahore 54590, Pakistan.
Received 2015-1-15; Accepted 2015-5-20; Published 2015-7-16
Abstract

Cancer is the second leading cause of deaths worldwide. Despite concerted efforts to improve the current therapies, the prognosis of cancer remains dismal. Highly selective or specific blocking of only one of the signaling pathways has been associated with limited or sporadic responses. Using targeted agents to inhibit multiple signaling pathways has emerged as a new paradigm for anticancer treatment. Icariside II, a flavonol glycoside, is one of the major components of Traditional Chinese Medicine Herba epimedii and possesses multiple biological and pharmacological properties including anti-inflammatory, anti-osteoporosis, anti-oxidant, anti-aging, and anticancer activities. Recently, the anticancer activity of Icariside II has been extensively investigated. Here, in this review, our aim is to give our perspective on the current status of Icariside II, and discuss its natural sources, anticancer activity, molecular targets and the mechanisms of action with specific emphasis on apoptosis pathways which may help the further design and conduct of preclinical and clinical trials.
Icariside II has been found to induce apoptosis in various human cancer cell lines of different origin by targeting multiple signaling pathways including STAT3, PI3K/AKT, MAPK/ERK, COX-2/PGE2 and β-Catenin which are frequently deregulated in cancers, suggesting that this collective activity rather than just a single effect may play an important role in developing Icariside II into a potential lead compound for anticancer therapy. This review suggests that Icariside II provides a novel opportunity for treatment of cancers, but additional investigations and clinical trials are still required to fully understand the mechanism of therapeutic effects to further validate it in anti-tumor therapy.
Keywords: Icariside II, Herba epimedii, Cancer, Multiple signaling pathways, Apoptosis
1. Introduction
Cancer is one of the most crucial public health problems in both developed and developing countries and it represents the second leading cause of deaths worldwide [1, 2], with approximately 14 million new cases and 8.2 million cancer related deaths in 2012 [3]. Regardless of whether a particular cancer results from genetic mutation or viral and bacterial infection, accumulating research evidence suggests that most cancers are caused by dysfunction of many genes coding for proteins such as anti-apoptotic proteins, inhibitors of apoptosis, transcriptional factors, growth factors, growth factor receptors and tumor suppressors; which constituted the targets for cancer treatment [4].
Current treatment options based on synthetic drugs/chemotherapy have limited therapeutic success in cancer; because they are highly toxic, expensive, and alter the functioning of cell signaling pathways [5]. In the past decade, several drugs have been developed that specifically block a single target in cancer cells. One such example of these drugs includes monoclonal antibodies that kill the cancer cells by specifically binding to the extracellular domains of receptor tyrosine kinase (RTKs), thereby preventing the endogenous ligand from binding and activating the receptor [6-10]. Recent evidence showed that emergence of secondary drug resistance has become one of the major limitations of these drugs [11-14]. As cancer development is a multi-step process which begins with initiation, followed by promotion and progression and characterized by multiple abnormalities rather than a single mutation, it is unlikely that a single target agent will effectively inhibit cancer growth [6, 15]. Cancer chemoprevention by natural compounds, especially phytochemicals, minerals and vitamins has emerged as a promising and pragmatic approach to reduce the burden of cancer and is gaining increasing attention because it is safe and cost effective alternative to cancer treatment [4, 16, 17]. Unlike pharmaceutical drugs that act as monotarget molecules, medicinal plants have multitarget molecules that can regulate the cancer growth and its progression [5].
Plants have a long history of use in the treatment of various ailments and provide an extensive reservoir of natural products, demonstrating outstanding structural diversity and offer a wide variety of novel and exciting chemical entities [4, 18, 19, 20]. The importance of natural products in health care can be estimated by a report of World Health Organization that approximately 80% of global population still relies on plant derived medicines for their primary health care. It is reported that 50% of all drugs in clinical use and 74% of the most important drugs are derived from natural sources [4, 21]. At present, more than 60% commercially available anticancer drugs are derived from natural sources including plants, marine organisms and micro-organisms [18, 22]. Recent research has shown that more consumption of vegetables and fruits can prevent 20% incident of cancer and about 200,000 cancer related deaths annually [24]. Extracts of plants have long been used in the treatment of cancer and there are more than 3000 plant species that have reportedly been used in the treatment of cancer [16, 18]. There is a continued interest in the investigation of plant extracts for anticancer drug development because of four main reasons. First, plants often produce bioactive compounds that exceed the current capacity of synthetic chemistry [25, 26]. Second, natural anticancer compounds fit into mechanism-based approach as perfectly as a hand fits into a glove. There is solid evidence that phytochemicals inhibit cancer by interfering with multiple mechanisms which are central to cancer progression [4, 27]. Third, there exists about 2.5 to 5.0 million plants species on earth and only about 10% have been thoroughly studied for their potential values as source of drugs [28, 29]. Thus identification of novel structures and understanding their molecular mechanisms will contribute to the faster and more specific strategies for the development of successful therapies. Fourth, natural compounds are already being used in cancer treatment and their popularity in cancer treatment appears to be growing rather than declining [30].
Although there are some new approaches to drug discovery, such as combinatorial chemistry and computer-based molecular modeling design, none of them can replace the importance of natural products in drug discovery and development. Indeed bioactive compounds isolated from natural products have played, and continue to play, a dominant role in the discovery of highly effective conventional drugs for the treatment of various cancers [4, 31, 32]. The aim of this review is to summarize the current knowledge and discuss the natural sources, anticancer activity, molecular targets, mechanisms of action and usefulness of a natural bioactive compound, “Icariside II ” in anticancer drug development. Icariside II is a flavonol glycoside which has been isolated from various plant species and exhibits a broad-spectrum of anticancer activity against various human cancer cell lines of different origin both in vitro and in vivo through interfering with multiple signaling pathways which are considered to be crucial and central to cancer development and metastasis. The anticancer activity and molecular targets of Icariside II in various cancer cell lines are summarized in Table 1.
Molecular targets of Icariside II in different cancer types
| Cancer type | Cell lines | Conc. range | Molecular targets | References |
|---|---|---|---|---|
| Lung cancer | A549 | 6.25-25 µM | Bax/bcl-2↑, ΔΨm↓, Cl- Caspase 3/9↑, Cl-PARP↑, ROS↑, p-p38↑, p-JNK↑ | [33] |
| Osteosarcoma | HOS,MG-63, Saos-2 | 0.1-30 µM | HIF-1α↓, VEGF↓, uPAR↓, ADM↓, MMP2↓, MCT4↓, Aldolase A↓, Enolase-1↓, p-EGFR↓, p-PI3K↓, p-Raf↓, p-mTOR↓, p-PDK-1↓, p-AKT↓, p-PRAS40↓, p-GSK3β↓, p-MEK↓, p-ERK↓ | [34, 35] |
| Prostate cancer | PC-3 | 10-40 µM | COX-2↓ PGE2↓, VEGF↓, iNOS↓ MMP↓, Cl-Caspase-3↑, Cl-PARP↑ | [36] |
| Multiple meyloma | U266 | 25-100 µM | p-STAT3↓, p-JAK2↓, p-Src↓, PTEN↑, SHP-1↑, Bcl-2↓, Bcl-xL↓, Cyclin D1↓, COX-2↓, VEGF↓, Cl-Caspase-3↑, Cl-PARP↑, | [37] |
| Melanoma | A375, B16, SK-MEL | 5-100 µM | p-STAT3↓, p-ERK↓, Survivin↓, CDK2↓, Cyclin B1↓, Cyclin E↓, p-CDK1↓, ROS↑, p-p38↑, p-p53↑, p21↑, Cl-Caspase-3↑, Cl-PARP↑, MyD88↓ | [38,39, 40] |
| Breast cancer | MCF-7 | 25-75 µM | Cl-Caspase-9,8,7,3↑,Cl-PARP↑,Bax↑, Bcl-xL↑, BimL↑, Fas↑, FasL↓, FADD↑, MMP↓ Cyto- C↑, AIF↑ | [41] |
| Epidermoid carcinoma | A431 | 10-100 µM | p-STAT3↓, p-ERK↓, p-AKT↑, Cl-Caspase-9↑, Cl-PARP↑,p-EGFR↓ | [42] |
| Esophageal carcinoma | Eca109 | 12-50 µg/ml | Survivin↓, Cyclin D1↓, β-catanin↓ | [43] |
| Acute Myeloid Leukemia | U937, HL-60 | 25-100 µM | p-STAT3↓, p-JAK2↓, p-Src↓, Bcl-2↓, bcl-xL↓, Survivin↓, COX-2↓, Cl-Caspase-3↑, Cl-PARP↑ | [44,45] |
↑: Up-regulation, ↓: Down-regulation, Cl: Cleaved
2. Natural Sources
Icariside II, a flavonol glycoside, also knowns as Baohuoside I (Figure 1) is one of the major components of Herba epimedii [40] and Cortex periplocae [43] which are Traditional Chinese Medicines. Herba epimedii which is called as Yin Yanghuo in Chinese and Horny Goat Weed in English, is the common name of dried aerial parts of Epimedium species such as E.brevicornum, E. sagittatum Maxim, E. koreanum Nakai, E. pubescens Maxim, and E.devidii [40, 46]. Herba epimedii exhibits a broad-spectrum of biological and pharmacological properties including anticancer activity and has traditionally been used for cardiovascular diseases, osteoporosis, and for improving sexual and neurological functions for many years in China [46].
Chemical structure and natural sources of Icariside II. Icariside II is a major metabolite of Icariin. Substitution or removal of various groups at positions 1, 2, and 3 results in different flavonol compounds as described in Natural Sources section. Icariside II is major component of Herba epimedii and Cortex periplocae. Herba epimedii (A+B+C+D+E) is made up of dried aerial parts of Epimedium species such as E.brevicornum (A), E. pubescens Maxim (B), E. koreanum Nakai (C), E. sagittatum Maxim (D), and E.devidii (E). Cortex periplocae (F) is made up of dry roots of Chinese herb Periploca sepium Bunge (F).
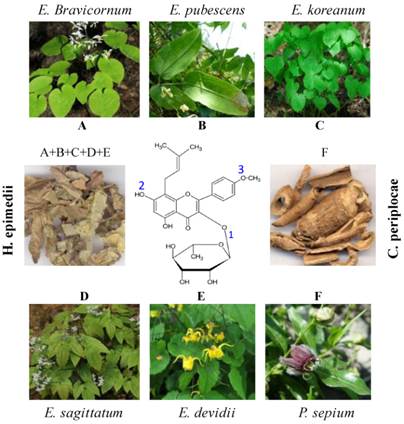
Cortex periplocae which is called as Xiang Jia Pi in Chinese is made up of dry roots of Chinese herb Periploca sepium Bunge. Cortex periplocae is a Traditional Chinese Medicine which is commonly used for a variety of clinical effects including inflammation, enhancing bone and muscles and stimulating nervous system [43, 47].
Several research reports indicate that Icariside II is a metabolite of Icariin [36, 38, 40 ] and can also be prepared from Icariin by enzymatic hydrolysis [40, 42, 48]. Other studies showed that Icariin is converted into several metabolites in vivo, including Icariside I, Icariside II, icaritin, and desmethylicaritin [49, 50, 51]. The chemical structure of Icariside II has been shown in Figure 1. The parent compound Icariin contains rhamnosyl, glucosyl, and methoxy group at positions 1, 2, and 3 respectively. Deglycosylation or demethylation of Icariin results in various metabolites. For example, removal of glucose residue from Icariin results in Icariside II while removal of rhamnose residue results in Icariside I. Deglycosylation of Icariin at position 1 & 2 results in icaritin while icaritin is demethylated to desmethylicaritin [52]. Icariin has also been reported to exhibit anticancer activity against various cancer cell lines. It mainly induced apoptosis via ROS generation and inhibiting NF-КB translocation into nucleus [53, 54]. Chemical profile of Icariin treated animals demonstrated that Icariside II is the major pharmacological active form of Icariin in vivo [50, 52, 55]. Therefore, Icariside II has been extensively studied for its antitumor properties.
3. Targeting Apoptosis pathways in Cancer with Icariside II
Apoptosis is extremely synchronized mode of cell death during which a series of intracellular events come into play to decommission the unwanted or dangerous cells [56-58]. Apoptosis is of widespread biological significance playing vital roles in development, homoeostasis, differentiation, regulation and functioning of the immune system, as well as the removal of defective, unwanted or harmful cells [58, 59]. Apoptosis is a necessary mechanism complementary to proliferation that has been recognized to play a key role in maintenance of tissue homeostasis by selective elimination of damaged or unwanted cells [60, 61]. The homeostasis in adult organs is maintained by a strict balance between apoptosis and cell proliferation. The disruption of apoptosis is implicated in tumors development, neurodegeneration and auto-immune diseases [57, 61].
It is well established that apoptosis can occur via two distinct pathways: the extrinsic or death receptor pathway and intrinsic or mitochondrial pathway, depending on stimulus. The extrinsic pathways is triggered by the activation and ligation of cell surface receptors (death receptors) of the tumor necrosis factor (TNF) receptor superfamily, such as fibroblast associated antigen (Fas/CD95) or TNF-related apoptosis-inducing ligand (TRAIL) receptors. The intrinsic pathway is triggered by a variety of extra- and intra-cellular stresses such as oxidative stress, irradiation, DNA damaging agents, chemotherapeutic drugs and growth factors depletion. Both pathways cause the activation of the initiator caspases, which then activate effector caspases. The effector caspases are the main executioner of apoptosis [58, 61, 62].
It is reported that many tumor promoters inhibit apoptosis [63]. Inhibition of apoptosis leads to tumor development and chemoresistance. Activation of apoptosis in cancer cells is thus considered as one of the most promising strategies in cancer therapy [63-65]. Since apoptotic programs can be manipulated to produce massive changes in cell death, the genes and proteins involved in apoptosis are potential drug targets. With the identification of an increasing number of molecular targets associated with programmed cell death, the identification of novel therapeutic agents capable of apoptosis regulation in cancer cells has been the focus of modern anticancer drug discovery [66]. It is currently accepted that natural compounds that have been approved for the clinical use in the treatment of cancer, inhibit the growth of cancer cells by inducing apoptosis through one or more than one mechanisms [22, 67, 68]. Recently, Icariside II has been extensively studied and found to exhibit a broad-spectrum of cytotoxicity against multiple cancer types both in vitro and in vivo via induction of apoptosis through various apoptotic pathways including extrinsic as well as intrinsic pathways [33-45]. The molecular targets of Icariside II have been represented in Figure 2. The review will further discuss the mechanisms of action by which Icariside II acts on apoptosis pathways in various types of cancer cells which have been characterized till now.
Icariside II inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple molecular targets.
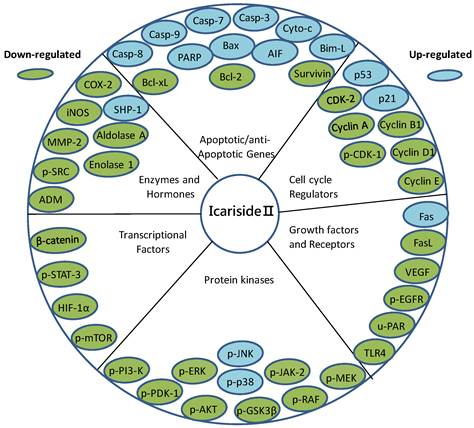
Icariside II is a flavonol glycoside. Flavonols are polyphenolic compounds widely distributed in plants and occur as aglycones or glycosides [69, 70]. They act as anti-oxidant as well as pro-oxidant [70, 71] and are known as receptor tyrosine kinase (RTK) inhibitors [72]. Quercetin and kaempferol are two major naturally occurring flavonol compounds. The anticancer activity of both these compounds is well documented. Both these compounds have been reported to inhibit growth and induce apoptosis in a variety of human cancer cells through various distinct mechanisms including ROS generation, tyrosine kinase inhibition and cell cycle arrest [71, 73]. Quercetin was the first tyrosine kinase inhibitor tested in human phase-1 trail [69, 74]. Bloking the EGFR signaling pathway by quercetin [75] and Icariside II [42] resulted in a significant growth inhibition in A431 cells via the induction of apoptosis. Lee et al., (2010) showed that occurrence, position, and type of sugar moieties play important role in anti-oxidant and anticancer activity of flavonoids. For example, the growth inhibitory effect of quercetin 3-glycoside was significantly higher in Caco-2 cells as compared to quercetin or quercetin 4-glycoside. Similarly, a greater growth inhibition by quercetin 3-glycoside than quercetin 3-rutinoside has been observed in breast cancer cells [70]. However, no data is available to highlight the importance of rhamnose residue in Icariside II. Like other well known anticancer flavonols (quercetin and kaempferol), Icariside II seems to hold its anticancer effects through induction of apoptosis via ROS generation, blocking receptor tyrosine kinase (EGFR)-induced signaling and cell cycle arrest.
3.1. Targeting Cancer Cells by Mitochondrial Mediated Apoptosis
It is well established that mitochondria play an important role in the regulation of cell proliferation and apoptosis [76]. Mitochondria have become an important component of the apoptosis execution machinery [56]. Unlike the extrinsic pathway, the intrinsic or mitochondrial pathway is mediated by down-regulation of Bcl-2 or Bcl-XL and translocation and insertion of Bax/Bak into mitochondrial membrane. The modulation of Bcl-2 family proteins results in dissipation of mitochondrial membrane potential (MMP) and subsequent release of many pro-apoptotic proteins such as cytochrome c, apoptosis inducing factor (AIF), and second mitochondrial activator of caspases (Smac/DIABLO) from the mitochondrial inter-membrane space into the cytosol. Cytochrome c, once released into the cytosol, interacts with apoptotic protease activating factor-1 (Apaf-1), leading to the activation of caspase-9. Active caspase-9 then activates caspase-3, which in turn leads to the degradation of vital cellular proteins and thus apoptosis. Smac/DIABLO promotes caspases activation through neutralizing the inhibitory effects of inhibitor of apoptosis proteins such as xiap and survivin while AIF and Endo G induce caspase-independent apoptosis by directly inducing DNA damage and condensation [77, 78 ].
Several reports indicate that the effect of Icariside II has been examined in many cancers including lung cancer, breast cancer, prostate cancer, melanoma, osteosarcoma, multiple myeloma, epidermoid carcinoma, esophageal carcinoma and acute myeloid leukemia [33-45]. Icariside II has been shown to induce apoptosis in A549 lung adenocarcinoma [33], MCF breast carcinoma [41], A375 human melanoma [39] and PC-3 prostate cancer cells [36] through mitochondrial pathway. Icariside II has significantly reduced MMP and increased the expression of cleaved caspase-3 and PARP in A549, MCF and PC-3 cells [33, 36, 41]. Icariside II also modulated the expression of Bcl-2 family proteins in A549 and MCF-7 cells. Huang et al., (2012) found that Icariside II effectively induced caspase-independent apoptosis by releasing AIF from mitochondria into nucleus in MCF-7 cells [41].
Based on available data, it appears that in cancerous cells Icariside II interacts directly or indirectly with Bcl-2 family proteins. Modulation of Bcl-2 family proteins leads to the opening of mitochondrial permeability transition pores (PTP). The opening of PTP leads to the release of cytochrome c and other pro-apoptotic molecules such as AIF. Cytochrome c binds and activates caspase-9 which then activates the effector caspase-3 which ultimately executes the mitochondrial caspase-dependent apoptosis. While AIF is translocated into nucleus where it induces DNA damage which leads to caspase-independent apoptosis.
3.2. Targeting Cancer Cells by ROS Mediated Apoptosis
Reactive oxygen species (ROS) has been shown to play vital role in different cellular processes such as proliferation, gene expression, and differentiation. It has now been widely accepted that intracellular ROS generation play important role in cancer cells apoptosis. Reactive oxygen species are produced inside the cells during normal physiological processes of the cells which are being neutralized by the anti-oxidant system of the cells. A precise balance between ROS production and anti-oxidant system's ability to scavenge ROS is critical for normal cellular functions. Over production of ROS results in oxidative damage including; lipid peroxidation, protein oxidation and DNA damage. In addition, ROS are known to act as second messengers to activate diverse redox-sensitive signaling cascades including mitochondrial intrinsic apoptotic cascade through interaction with Bcl-2 family proteins, MAPK family member p38 and its downstream transcription factors. Other studies have shown that ROS eliminate cancer cells by arresting the cell cycle at various check points and thereby inducing apoptosis [79-82].
An increasing body of literature evidence indicated that phytochemical targeting ROS metabolism can selectively kill cancer cells [79, 83]. Icariside II has been shown to induce ROS-mediated apoptosis in A375 melanoma as well as A549 lung cancer cells. In A375 melanoma cells, Icariside II induced G0/G1 and G2/M phase cell cycle arrest and apoptotic cell death. This effect was mediated through Icariside II activated ROS-p38-p53 signaling pathway. These effects were confirmed by using specific inhibitors of ROS, p38, and p53 [39]. While in A549 lung cancer cells, the apoptotic effects of Icariside II has been associated with activation of ROS downstream effectors, p38 MPAK, and JNK. Pretreatment of cells with NAC (ROS inhibitors), SB203580 (p38 inhibitor) and SP600125 (JNK inhibitor) effectively abrogated the apoptotic effects of Icariside II, indicating the potential involvement of ROS/MAPK pathway in Icariside II -mediated apoptosis in A549 cells. NAC supplementation not only diminished apoptotic effect of Icariside II but also reduced the phosphorylation of p38 and JNK. The data is evident that Icariside II is a potential phytochemical that targets cancer cells through ROS generation [33].
3.3. Targeting cancer cells by Death Receptor Mediated apoptosis
The extrinsic pathways is triggered by the activation and ligation of cell surface receptors (death receptors) of the tumor necrosis factor (TNF) receptor superfamily, such as fibroblast associated antigen (Fas) also called CD95 or TNF-related apoptosis-inducing ligand (TRAIL) receptors [58]. Upon ligand binding, the receptors interact with Fas associated death domain (FADD) and procaspase-8 to form death inducing signaling complex (DISC). Within this complex, FADD recruits procaspase‑8, which becomes activated. Activated caspase-8 then directly activates the downstream effector caspase-3, resulting in cell death via the type I extrinsic apoptotic pathway, or cleaves Bid, a pro-apoptotic member of the Bcl-2 family, leading to activation of the type II extrinsic apoptotic pathway [84, 85].
Icariside II also has a stimulatory effect on the extrinsic apoptotic pathway. Icariside II has been shown to increase the level of Fas, and FADD without changing the expression of Daxx, another Fas binding protein that induces apoptosis via JNK pathway in MCF-7 breast cancer cells. Moreover, Icariside II treatment significantly induced cleavage of caspase-8, caspase-3 and PARP which are characteristic features of apoptotic cell death. Based on available data, it can be concluded that Icariside II induced apoptosis by stimulating Fas/FADD/caspase-8 extrinsic pathway. Moreover, this Icariside II -mediated apoptosis in MCF-7 breast cancer cells appears to occur via the type I extrinsic apoptotic pathway as evident by the absence of truncated Bid (tBid) [41].
3.4. Targeting Cancer Cells by Cell Cycle Mediated Apoptosis
Cell cycle control is the major regulatory process of cell growth. Cell cycle is regulated at various checkpoints by a coordinated interaction of a variety of cyclins with their respective cyclin-dependent kinases (CDKs) to form active complexes and these checkpoints ensure that processes at each stage of the cell cycle have been accurately completed before progression into next phase [86]. Moreover, the activity of CDKs is negatively regulated by a variety of cyclin-dependent kinases inhibitors (CDKIs), of which p21 has been shown to play a major role in regulating the cell cycle at various checkpoints [86, 87]. Checkpoint failure often causes mutations and genomic arrangements resulting in genetic instability which is the major cause of cancer development [4, 88]. With the identification of increasing number of CDKs associated with cell cycle checkpoints, the identification of novel natural compounds capable of selective inhibition of these kinases present a potential attractive strategy to tumor therapy. Accumulating evidence suggests that many anticancer agents arrest the cell cycle at a particular checkpoint and thereby induce apoptosis in cancer cells [89, 90].
Icariside II has been reported to arrest the cell cycle in different cells at G0/G1 and G2/M phases through interaction with various cyclins and CDKs [39, 91]. In human T cells, it has been shown to arrest the cell cycle at G1-S phase transition by down-regulating the protein expression of cyclin A, D and p33cdk2 proteins [91]. In A375 human melanoma cells, Icariside II was found to arrest the cell cycle at G0/G1 and G2/M phases. The induction of cell cycle arrest at G0/G1 phase was found to be associated with decreased expression of Cyclin E/CDK2 complex while at G2/M phase with decreased expression of Cyclin B1/CDC2 complex. These effects were mediated by p53 through its downstream activator p21 [39].
4. Targeting Cancer Cells by Regulating Multiple Signaling Pathways and Transcription Factors
Cancer development is a multi-step process which is characterized by multiple abnormalities rather than a single mutation; it is therefore, unlikely to achieve therapeutic window by using single target therapy [6, 15]. Targeting multiple signaling pathways in cancer may therefore be a successful treatment option. Targeting various signaling pathways in cancer may reduce the chances of drug resistance which has become the major drawback of most of the anticancer drugs that are designed to specifically block a particular signaling pathway.
Epidermal growth factor receptor (EGFR/HER1) is abnormally expressed in various cancers and its expression is correlated with poor clinical prognosis [92]. Upon ligand binding (such as EGF, TGF-β), EGFR becomes activated. Activated EGFR recruits a number of downstream signaling molecules, leading to activation of major pro-survival signaling pathways such as Ras-Raf-MEK, PI3k/Akt, and JAK2/STAT3 which play key role in cell proliferation, survival, angiogenesis, anti-apoptosis, migration, adhesion, and metastasis [93, 94].
Icariside II has been investigated for its inhibitory effects on EGFR signaling in A431 human epidermoid carcinoma cells and in MG-63 and Saos-2 human osteosarcoma cells. Icariside II has demonstrated an inhibitory effect on EGFR expression and its downstream signaling molecules in all three cell lines which are discussed in detail in PI3K/AKT /mTOR and MAPK/ERK pathways [35, 42].
4.1. STAT3 Pathway
Signal transducer and activator of transcription 3 (STAT3) is a cytoplasmic transcription factor which has been implicated in many cellular processes such as development, differentiation, immune function, proliferation, survival and epithelial to mesenchymal transition (EMT) [95]. STAT3 is activated by phosphorylation at tyrosine 705 (Y705) or serine 727 (S727) [96, 97]. STAT3 can be activated by growth factor receptors, cytokine receptors; Janus activated kinases (JAK), Abl family kinases, and Src family kinases as shown in Figure 3 [98, 99]. STAT3 is constitutively expressed in a wide range of tumors [100-102]. Accumulating evidence strongly implicates the critical role of constitutive activation of STAT3 in malignant transformation, tumorigenesis, and resistance to chemotherapy [103, 104]. These cumulative findings have validated STAT3 as a novel target for cancer therapy and hence provided the rational for exploring small molecule STAT3 inhibitors for anticancer drug development. As STAT3 is activated through different signaling pathways, identification of small molecules that can inhibit STAT3 activation by modulating more than one target might have a potential therapeutic scope.
Interaction of Icariside II with apoptosis signaling pathways: Binding of Ligand such as growth factor (EGF) to the growth factor receptor (EGFR) promotes the activation of downstream pro-survival signaling pathways. These include the SRC and Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3), PI3K/AKT, and RAS/RAF/MEK/ERK pathways. Activation of these pathways promotes survival, proliferation, invasion and metastasis. Icariside II inhibits all these three pathways at multiple levels. Activated AKT activates NF-КB and mTOR while inactivate GSK-3β via phosphorylation. Suppression of GSK-3β leads to stabilization of β-catenin which induces the expression of downstream target genes such as cyclin D1 and survivin and thus promotes survival and tumorigenesis. ( Activation;
Activation;  Inhibition;
Inhibition;  Activation by Icariside II;
Activation by Icariside II;  Inhibition by Icariside II )
Inhibition by Icariside II )
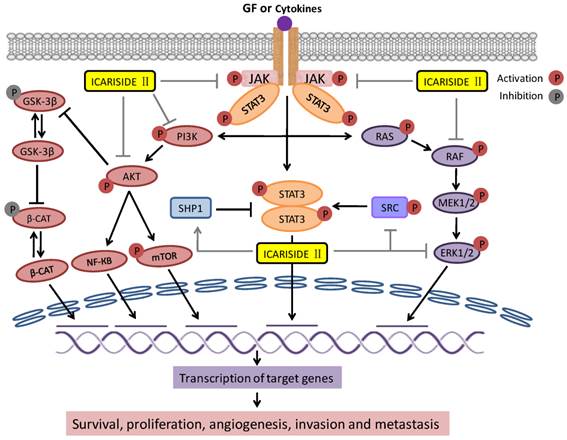
Recently Icariside II has been extensively investigated for its inhibitory effects on STAT3 expression in various cancer cell lines [37, 40, 42, 44, 45]. Icariside II has been shown to inhibit STAT3 activation through different mechanisms. In A375 human melanoma cells, Icariside II decreased the phosphorylation of STAT3 and reduced the expression of its downstream target survivin in a dose-dependent manner. Further in vivo study indicated that Icariside II effectively reduced the tumor volume at a dose of 50 mg/kg in two different mouse models bearing A375 and B16 cells [40]. Wu et al., [42] recently showed that Icariside II inhibited the constitutive and EGF-induced activation of STAT3 in A431 human epidermoid carcinoma cells. In U266 multiple myeloma cells and U937 acute myeloid leukemia cells, Icariside II has been shown to inactivate STAT3 signaling pathway through dephosphorylation of JAK2 and Src and induction of protein tyrosine phosphatase SHP-1 in a dose- and time-dependent manner. Sodium pervanadate, a broad-spectrum tyrosine phosphatases inhibitor attenuated the Icariside II -induced STAT3 inactivation and cleavage of caspase-3 and PARP in U266 and U937 cells. Since SHP-1 is a known upstream protein tyrosine phosphatase for JAK2 and JAK2 and Src are STAT3 upstream kinases; it can therefore be concluded that Icariside II -mediated STAT3 inhibition and induction of apoptosis is associated with induction of SHP-1 by Icariside II. Icariside II also suppressed the expression of various downstream effectors of STAT3 including cyclin D1, Bcl-2, Bcl-xL, survivin, VEGF and COX-2 in these cell lines [37, 44]. The effect of Icariside II on inhibition of STAT3 activation was further validated using MDA-MB-231 breast adenocarcinoma and DU145 prostate carcinoma cells in which STAT3 is constitutively expressed [37]. These effects of Icariside II on inhibition of STAT3 activation through various kinases and tyrosine phosphatases has been shown in Figure 3.
4.2. PI3K/AKT /mTOR Pathway
Phosphatidylinositol-3-Kinase/protein kinase B (PI3K/AKT) signaling promotes cell growth and survival by several ways [105]. AKT is activated via phosphorylation of two residues; threonine 308 (Thr 308) and serine 473 (Ser473) [106].After activation, AKT is translocated into nucleus [107, 108] where it affects the activity of several transcriptional regulators such as E2 transcription factor (E2F) and nuclear factor КB (NF-КB) leading to increased transcription of anti-apoptotic and pro-survival genes [90]. Aside from transcription factor, it has been reported to regulate a number of other molecules such as Bcl-2-associated death promoter (BAD) and Bax, and glycogen synthase kinase-β (GSK-3β) to affect the survival state of cell. GSK-3β regulates β-catenin protein stability [105, 107, 109]. Thus PI3k/AKT pathway is connected to wnt/β-catenin pathway as shown in Figure 3. Mammalian target of rapamycin (mTOR) is also phosphorylated by PI3k/AKT signaling, the overexpression of which has been associated with poor prognosis [107]. Figure 3 shows the activation and inhibition of major signaling molecules of this pathway via phosphorylation.
PI3k/AKT pathway is frequently overexpressed in a wide variety of human cancers via several different mechanisms [111-114]. The most common genetic alteration in PI3k/AKT signaling is inactivation of phosphatase and tensin homologue (PTEN) protein [105, 107]. Thus PTEN is an enciting therapeutic target for activation to regulate the PI3k/AKT signaling in cancer [115, 116].
Recently Icariside II has been found to have a dramatic effects on several components of PI3k/AKT signaling pathway. In U266 multiple myeloma cells, it has been reported to induce the expression of PTEN [37]. Other researchers found that in MG-63 and Saos-2 osteosarcoma cells, Icariside II inhibited the growth of cells by reducing the phosphorylation (Inhibition) of PI3K, PI3K-dependent kinase-1 (PDK-1), AKT, proline-rich Akt substrate of 40-kDa (PRAS-40), and mTOR. Icariside II also activated GSK-3β by dephosphorylation at Serine 9 (Ser9). [35]. As GSK-3β is inactivated by phosphorylation at Ser9 by p-AKT, activation of GSK-3β (dephosphorylation) in MG-63 and Saos-2 osteosarcoma cells appears to be mediated through inhibitory effect of Icariside II on AKT.
These results were further validated by animal mouse model study. Icariside II significantly reduced the tumor volume in mice bearing osteosarcoma sarcoma-180 cells at a dose of 20 and 30 mg/kg. The expression of phosphorylated PI3K, PDK-1, AKT, PRAS-40, and mTOR in tumor tissues was comparable to in vitro cells study results.
4.3. MAPK/ERK (Ras-Raf-MEK-ERK) Pathway
The mitogen-activated protein kinase (MAPK/ERK) pathway (also known as Ras-Raf-MEK-ERK pathway) comprises several key signaling cascades (Figure 3) of which Ras-Raf-Mek-extracellular signal-regulated kinase-1 and 2 (ERK1/2) is one of the most dysregulated in human cancers [117]. This pathway regulates multiple key cellular functions including cell growth, proliferation, differentiation, apoptosis, migration and senescence [90, 117, 118]. The signaling molecules of this pathway are activated via phosphorylation as shown in Figure 3. Once activated, ERK can translocate to nucleus where it phsophorylate additional transcription factors that bind promoters of several genes including growth factor and cytokine genes which play important role in promoting cell proliferation and inhibiting apoptosis in several cells [90,119]. Abnormal signaling of this pathway leads to tumorigenesis, senescence, and drug resistance [118, 120, 121]. Abnormal signaling of this pathway has frequently been detected in a wide variety of human tumors [122, 123]. Hence, targeting the MAPK/ERK pathway may provide a useful strategy in cancer therapy.
Icariside II has been reported to inhibit growth both in vitro and in vivo and suppressed the activation of ERK in A375 melanoma cells in a dose- and time-dependent manner [40]. A dose-dependent suppression of ERK activation by Icariside II has also been investigated in A431 human epidermoid human carcinoma cells. The suppression of ERK activation in A431 by Icariside II appears to result from inhibition of EGFR signaling by Icariside II. Treatment of cells with AG1478 (EGFR inhibitor) reduced the expression of phosphorylated ERK in a similar fashion. Therefore, growth inhibitory effect of Icariside II seems to be associated in part with regulation of apoptosis through inhibiting EGFR-induced activation of MAPK pathway [42]. In MG-63 and Saos-2 osteosarcoma cells, it has been reported to induce apoptosis and inhibit both the constitutive as well as EGF-induced activation of Raf, MEK and ERK in a dose-dependent manner. In animal mouse model study, it reduced the expression of Ki-67 (cells proliferation marker) and the tumor size and inhibited the expression of Raf, MEK and ERK in tumor tissues in a dose-dependent manner [35]. This set of data from various studies show that Icariside II is a potent inhibitor of EGFR-induced activation of signaling pathways.
4.4. Wnt/β-Catenin Pathway
Wnt/β-catenin signaling has been implicated in several important cellular mechanisms including cell proliferation, polarity and cell death during embryonic development and in tissue homeostasis in adult [124, 125]. Aberrant signaling of Wnt/β-catenin pathway can lead to a variety of human diseases ranging from birth defects to cancer [125, 126]. β-catenin is a key component of this pathway. Perturbations of β-catenin signaling are associated with many cancers, including hepatocellular carcinoma, colorectal carcinoma, lung cancer, malignant breast tumors, ovarian cancer, endometrial cancer and esophageal cancer [124, 127-129]. The cellular level of β-catenin is tightly regulated by a multiple destruction complex consisting of Adenomatous polyposis coli (APC), axin, and glycogen synthase kinase-3β (GSK-3β). In the absence of Wnt signaling, β-catenin is phosphorylated by GSK-3β; the phosphorylated β-catenin is recognized and destroyed by proteasomal degradation [130-132]. In the presence of Wnt signaling, β-catenin (non-phosphorylated) is translocated from cytoplasm into nucleus where it induces the expression of downstream target genes such as cyclin D1, c-Myc and survivin and thus promotes survival and tumorigenesis [132-134].
Several reports indicate that esophageal squamous cell carcinoma (ESCC) has abnormal nuclear accumulation of β-catenin accompanied with increased cyclin D1 expression [135]. β-catenin mRNA expression has appeared a new prognostic marker for ESCC [136]. Overexpression of β-catenin and its target proteins is commonly observed in human Eca109 tumor xenografts. Wang et al. [43] investigated the effect of Icariside II in esophageal squamous cell carcinoma using in vitro cell study and animal mouse model. Icariside II has been found to inhibit the growth of Eca109 cells in vitro as well as in vivo. These effects were associated with decreased expression of β-catenin and its downstream target genes cyclin D1 and survivin both at mRNA and protein levels. The suppressive activity of Icariside II on the expression of β-catenin and its downstream target protein was also verified in tumor xenografts. As shown in Figure 3, GSK-3β is the direct target of AKT. Activation of AKT leads to phosphorylation and thus inactivation of GSK-3β. Inhibition of AKT activation results in activation of GSK-3β, which then triggered phosphorylation-mediated degradation of β-catenin. Therefore, the inhibitory effect of Icariside II on AKT may at least partially responsible for observed decreased expression of β-catenin and its downstream target genes cyclin D1 and survivin in Eca109 cancer cells.
4.5. Hypoxia-inducible Factor-1α (HIF-1α)
HIF-1 is a heterodimer transcription factor that consists of a constitutively expressed HIF-1β subunit and a HIF-1α subunit, the expression of which is highly regulated [137, 138]. Hypoxia is a characteristic feature of malignant tumors which has been associated with treatment failure, invasion, metastasis and patient mortality. The principal mechanism by which cancer cells achieve hypoxic microenvironment is through the activity of HIF-1 [139-141]. The transcription factor HIF-1 regulates the expression of hundreds of genes in response to hypoxia which are involved in several aspects of cancer progression including proliferation, angiogenesis, invasion, metastasis, apoptosis, erythropoiesis, and glucose metabolism [142, 143]. Among these genes, the most important are VEGF, which encodes vascular endothelial growth factor, a key regulator of angiogenesis [90, 137]; MCT4 (Monocarboxylate transporter 4) [144], GLUT1 (glucose transporter 1), HK1 and HK2 (Hexokinase1 & 2) which play important role in glucose metabolism [137]; ADM (adrenomedullin) and MMP2 (matrix metalloproteinase 2) which are key regulators of invasion and metastasis [137].
As HIF-1 has been documented to regulate the expression of a wide range of essential genes associated with tumorigenesis and cancer progression, the inhibition of HIF-1 may be a useful strategy in the treatment of cancer. Icariside II has been studied to evaluate its effects on HIF-1 expression and its target genes by Choi et al [34]. The findings demonstrated that Icariside II decreased the protein expression of HIF-1α without affecting mRNA expression and decreased transcriptional activity of its target genes in human HOS osteosarcoma cells. The inhibitory effect of Icariside II on HIF-1α protein level appears to come from post-translational modifications. The post-translational modifications that regulate the activity and stability of HIF-1α include hydroxylation, ubiquitination, acetylation and phosphorylation [34]. Icariside II increased the activity of Prolyl 4-hydroxylase-2 (PHD2) and enhanced the binding of von-Hippel Lindau (VHL) to HIF-1α in HOS cells [34]. In the presence of molecular oxygen, PHDs induces proline hydroxylation of HIF-1α which facilitates its interaction with VHL resulting in ubiquitination and subsequent proteasomal degradation of HIF-1α [145]. On the basis of available data, it can be concluded that Icariside II decreased the protein level of HIF-1α through an anti-oxidant mechanism.
Among the target genes, it inhibited the expression of VEGF, and tube formation in HUVECs, confirming its inhibitory effect on angiogenesis. It also inhibited the mRNA expressions of several genes involved in cancer invasion such as Urokinase-type plasminogen activator receptor (uPAR), ADM and MMP2. Moreover, Icariside II has significantly inhibited the mRNA expression of MCT4 which mediates lactic acid efflux in glycolysis for energy production and the expression of other glycolytic enzymes such as aldolase A and enolase 1.
4.6. The COX-2/PGE2 pathway
Extensive research reports indicate that cyclooxygenase-2 (COX-2) over-expresses in a series of human cancers including colorectal, gastric, pancreatic, lung, breast, prostate and head and neck cancers [146-148]. Elevated expression of COX-2 is an early event during carcinogenesis, and is mostly associated with poor prognosis [146]. COX-2 is closely associated with inflammation and cancer progression. A large number of research reports indicated that anti-inflammatory drugs exert anticancer activity via COX-2 inhibition [149, 150].
It is widely accepted now that deregulation of COX-2 expression leads to an increased abundance of its enzymatic product prostaglandin E2 (PGE2), the most abundant prostaglandin in human body [151]. Tumor development by COX-2 is believed to be mediated by PGE2 which stimulates proliferation, invasion, angiogenesis and migration [152].
Icariside II has been shown to exhibit anti-inflammatory and anti-tumor activities. COX-2 is constitutively expressed in PC-3 prostate cancer cells [36]. Icariside II was investigated for its effects on various cancer cell lines expressing COX-2 including PC-3 and LNCaP prostate cancer, H596 adenocarcinoma, U937 leukemia, and U266 and MM1.S myeloma cells. PC-3 cells appeared to be more sensitive towards Icariside II -induced cytotoxicity as PC-3 cells express higher level of constitutive COX-2 compared to other cell lines. The findings demonstrate that cytotoxic effect of Icariside II is more specific. Further investigation showed that Icariside II induced apoptosis in PC-3 cells by inhibiting the protein expression of COX-2, its enzymatic product PGE2, and other inflammation related proteins such as inducible nitric oxide synthase (iNOS), and VEGF which led to mitochondrial dysfunction, cytochrome c release, activation of caspase-8, -9, and -3 and cleavage of PARP in PC-3 cells. Addition of exogenous PGE2 attenuated Icariside II -mediated PARP cleavage while COX-2 siRNA transfection augmented Icariside II -mediated PARP cleavage. Taken together, Icariside II induced mitochondrial apoptosis in PC-3 cells by inhibiting COX-2/PGE2 pathway [36].
5. Icariside II As an Adjuvant Therapy
While extensive studies of Icariside II as single agent in treatment of a wide variety of human cancers has demonstrated promising results; there is a growing interest in potentially using this compound as an adjuvant agent in combination with standard chemotherapeutic drugs. Paclitaxel is a widely used first line cancer chemotherapeutic drug which exhibits a considerable clinical activity in a variety of human malignancies [153]. A number of research reports from different laboratories indicate that paclitaxel induces the activation of EGFR and toll like receptors-4 (TLR4) signaling cascades which are functionally associated with cell survival, tumor growth and drug resistance [154,155].
Icariside II has demonstrated an increased growth inhibitory effect in A375 human melanoma cells when used in combination with paclitaxel compared to treatment with either agent alone [38]. This suppressive effect of Icariside II has been shown to derive from inhibition of TLR4 and its adaptor protein Myeloid differentiation primary response gene 88 (MyD88), and ERK activity. Of note, paclitaxel alone significantly increased the expression of TLR4, MyD88 and phosphorylate ERK in A375 human melanoma cells [38]. Different mechanisms of paclitaxel and Icariside II provide a rational for the clinical use of subtherapeutic doses of paclitaxel in combination with Icariside II to accomplish effective suppression of tumor growth while minimizing paclitaxel resistance.
Bortezomib and thalidomide are the standard chemotherapeutic drugs which have been used to treat Multiple Meyloma patients [37]. Icariside II has been found to augment the efficacy of thalidomide from 20% to 50% and that of bortezomib from 25% to 60% in U266 cells. Moreover, caspase-3 activation and PARP cleavage were further increased by co-treatment of Icariside II with thalidomide and bortezomib than monotherapy in U266 cells [37]. Thus, Icariside II may be developed into a potential lead compound for treating multiple meyloma in combination with standard chemotherapeutic drugs.
6. Conclusion and Future Perspectives
In this review, we have summarized the recent progress of Icariside II in various in vitro and in vivo cancer models. Collective data from different studies indicate that Icariside II is a potential anticancer compound. Following observations make Icariside II a unique therapeutic agent for treatment of various cancers: (I) It exhibits a broad-spectrum of toxicity toward a wide range of human cancer cells of different origins; (II) It induces apoptosis by interfering with multiple mechanisms which are considered central to cancer development and progression; (III) It can inhibit multiple signaling pathways such as STAT3, PI3k/AKT, β-catenin, COX-2/PGE2 and MAPK pathways which are frequently deregulated in human cancers and associated with drug resistance; (IV) It can be used as adjuvant agent in combination therapy with standard drugs to overcome drug resistance and improve the treatment outcome in patients with apoptosis resistant tumors; (V) It is one of the major components of Herba epimedii and Cortex periplocae which in Traditional Chinese Medicines are clinically being used for many years, therefore Icariside II may be a safe chemotherapeutic candidate for the treatment of cancer; (VI) Poor bioavailability of natural compounds is one of the major challenges in development of anticancer drugs. Icariside II is readily available from various plant species which can be easily extracted.
In light of aforementioned findings, it can be speculated that Icariside II may become a potential lead compound for future development of anticancer therapy. However, preclinical and clinical trials are yet required to elucidate the full spectrum of anticancer effects of Icariside II either alone or in synergistic combination with other standard drugs to validate the further usefulness as potent anticancer agent.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Siegel R, Ward E. et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96
2. Siegel R, Naishadham D, Jemal A. Cancer statistics for Hispanics/Latinos. CA Cancer J Clin. 2012;62:283-98
3. Ferlay J, Soerjomataram I, Dikshit R. et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-386
4. Millimouno FM, Dong J, Yang L. et al. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev Res (Phila). 2014;7:1081-1107
5. Rahmani AH, Alzohairy MA, Khan MA. et al. Therapeutic Implications of Black Seed and Its Constituent Thymoquinone in the Prevention of Cancer through Inactivation and Activation of Molecular Pathways. Evid Based Complement Alternat Med. 2014;2014:724658
6. Faivre S, Djelloul S & Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33:407-420
7. Reynolds NA, Wagstaff AJ. Cetuximab: In the treatment of metastatic colorectal cancer. Drugs. 2004;64:109-118
8. Cunningham D, Humblet Y, Siena S. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337-345
9. Cohen MH, Williams GA, Sridhara R. et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839;Iressa) tablets. Clin Cancer Res. 2004;10:1212-1218
10. Johnson JR, Cohen M, Sridhara R. et al. Approval summary for erlotinib for treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen. Clin Cancer Res. 2005;11:6414-6421
11. Coco S, De Mariano M, Valdora F. et al. Identification of ALK germline mutation (3605delG) in pediatric anaplastic medulloblastoma. J Hum Genet. 2012;57:682-684
12. Shin S, Kim J, Yoon SO. et al. ALK-positive anaplastic large cell lymphoma with TPM3-ALK translocation. Leuk Res. 2012;36:e143-145
13. Lievre A, Bachet JB, Le Corre D. et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992-3995
14. Holohan C, Van Schaeybroeck S, Longley DB. et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-726
15. Shu L, Cheung KL, Khor TO. et al. Phytochemicals: cancer chemoprevention and suppression of tumor onset and metastasis. Cancer Metastasis Rev. 2010;29:483-502
16. Amin AR, Kucuk O, Khuri FR. et al. Perspectives for cancer prevention with natural compounds. J Clin Oncol. 2009;27:2712-2725
17. Weng CJ & Yen GC. Chemopreventive effects of dietary phytochemicals against cancer invasion and metastasis: phenolic acids, monophenol, polyphenol, and their derivatives. Cancer Treat Rev. 2012;38:76-87
18. Cragg GM & Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. 2005;100:72-79
19. Balunas MJ & Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005;78:431-441
20. Chin YW, Balunas MJ, Chai HB. et al. Drug discovery from natural sources. AAPS J. 2006;8:E239-253
21. Rasul A, Millimouno FM, Ali Eltayb W. et al. Pinocembrin: a novel natural compound with versatile pharmacological and biological activities. Biomed Res Int. 2013;2013:379850
22. Dall'Acqua S. Natural products as antimitotic agents. Curr Top Med Chem. 14. 2014:2272-2285
23. Chinembiri TN, du Plessis LH, Gerber M. et al. Review of natural compounds for potential skin cancer treatment. Molecules. 2014;19:11679-11721
24. Pratheeshkumar P, Sreekala C, Zhang Z. et al. Cancer prevention with promising natural products: mechanisms of action and molecular targets. Anticancer Agents Med Chem. 2012;12:1159-1184
25. Koch MA, Schuffenhauer A, Scheck M. et al. Charting biologically relevant chemical space: a structural classification of natural products (SCONP). Proc Natl Acad Sci U S A. 2005;102:17272-17277
26. Monks NR, Li B, Gunjan S. et al. Natural Products Genomics: A novel approach for the discovery of anti-cancer therapeutics. J Pharmacol Toxicol Methods. 2011;64:217-225
27. Ghantous A, Sinjab A, Herceg Z. et al. Parthenolide: from plant shoots to cancer roots. Drug Discov Today. 2013;18:894-905
28. Borris RP. Natural products research: perspectives from a major pharmaceutical company. J Ethnopharmacol. 1996;51:29-38
29. Juarez P. Plant-derived anticancer agents: a promising treatment for bone metastasis. BoneKEy Rep. 2014;3:doi 10.1038/bonekey.2014.94
30. Veeresham C. Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res. 2012;3:200-201
31. Koehn FE & Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206-220
32. Balunas MJ & Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005;78:431-441
33. Song J, Shu L, Zhang Z. et al. Reactive oxygen species-mediated mitochondrial pathway is involved in Baohuoside I-induced apoptosis in human non-small cell lung cancer. Chem Biol Interact. 2012;199:9-17
34. Choi HJ, Eun JS, Kim DK. et al. Icariside II from Epimedium koreanum inhibits hypoxia-inducible factor-1alpha in human osteosarcoma cells. Eur J Pharmacol. 2008;579:58-65
35. Geng YD, Yang L, Zhang C. et al. Blockade of epidermal growth factor receptor/mammalian target of rapamycin pathway by Icariside II results in reduced cell proliferation of osteosarcoma cells. Food Chem Toxicol. 2014;73:7-16
36. Lee KS, Lee HJ, Ahn KS. et al. Cyclooxygenase-2/prostaglandin E2 pathway mediates Icariside II induced apoptosis in human PC-3 prostate cancer cells. Cancer Lett. 2009;280:93-100
37. Kim SH, Ahn KS, Jeong SJ. et al. Janus activated kinase 2/signal transducer and activator of transcription 3 pathway mediates Icariside II -induced apoptosis in U266 multiple myeloma cells. Eur J Pharmacol. 2011;654:10-16
38. Wu J, Guan M, Wong PF. et al. Icariside II potentiates paclitaxel-induced apoptosis in human melanoma A375 cells by inhibiting TLR4 signaling pathway. Food Chem Toxicol. 2012;50:3019-3024
39. Wu J, Song T, Liu S. et al. Icariside II inhibits cell proliferation and induces cell cycle arrest through the ROS-p38-p53 signaling pathway in A375 human melanoma cells. Mol Med Rep. 2015;11:410-416
40. Wu J, Xu J, Eksioglu EA. et al. Icariside II induces apoptosis of melanoma cells through the downregulation of survival pathways. Nutr Cancer. 2013;65:110-117
41. Huang C, Chen X, Guo B. et al. Induction of apoptosis by Icariside II through extrinsic and intrinsic signaling pathways in human breast cancer MCF7 cells. Biosci Biotechnol Biochem. 2012;76:1322-1328
42. Wu J, Zuo F, Du J. et al. Icariside II induces apoptosis via inhibition of the EGFR pathways in A431 human epidermoid carcinoma cells. Mol Med Rep. 2013;8:597-602
43. Wang L, Lu A, Liu X. et al. The flavonoid Baohuoside-I inhibits cell growth and downregulates survivin and cyclin D1 expression in esophageal carcinoma via beta-catenin-dependent signaling. Oncol Rep. 2011;26:1149-1156
44. Kang SH, Jeong SJ, Kim SH. et al. Icariside II induces apoptosis in U937 acute myeloid leukemia cells: role of inactivation of STAT3-related signaling. PLoS One. 2012;7:e28706
45. Kim SH, Ahn KS, Jeong SJ. et al. Janus activated kinase 2/signal transducer and activator of transcription 3 pathway mediates Icariside II -induced apoptosis in U266 multiple myeloma cells. Eur J Pharmacol. 2011;654:10-16
46. Sze SC, Tong Y, Ng TB. et al. Herba epimedii: anti-oxidative properties and its medical implications. Molecules. 2010;15:7861-7870
47. Stoner GD & Gupta A. Etiology and chemoprevention of esophageal squamous cell carcinoma. Carcinogenesis. 2001;22:1737-1746
48. Xia Q, Xu D, Huang Z. et al. Preparation of Icariside II from Icariin by enzymatic hydrolysis method. Fitoterapia. 2010;81:437-442
49. Liu J, Ye H, Lou Y. Determination of rat urinary metabolites of Icariin in vivo and estrogenic activities of its metabolites on MCF-7 cells. Pharmazie. 2005;60:120-5
50. Ming LG, Chen KM, Xian CJ. Functions and action mechanisms of flavonoids genistein and Icariin in regulating bone remodeling. J Cell Physiol. 2013;228:513-21
51. Xu W, Zhang Y, Yang M. et al. LC-MS/MS method for the simultaneous determination of Icariin and its major metabolites in rat plasma. J Pharm Biomed Anal. 2007;45:667-72
52. Shen P, Wong SP, Li J. et al. Simple and sensitive liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of five Epimedium prenylflavonoids in rat sera. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:71-8
53. Li S, Dong P, Wang J. et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via a ROS/JNK-dependent mitochondrial pathway. Cancer Lett. 2010;298:222-30
54. Shi DB, Li XX, Zheng HT. et al. Icariin-mediated inhibition of NF-kappaB activity enhances the in vitro and in vivo antitumour effect of 5-fluorouracil in colorectal cancer. Cell Biochem Biophys. 2014;69:523-30
55. Cai WJ, Huang JH, Zhang SQ. et al. Icariin and its derivative Icariside II extend healthspan via insulin/IGF-1 pathway in C. elegans. PLoS One. 2011;6:e28835
56. Rasul A, Khan M, Ali M. et al. Targeting apoptosis pathways in cancer with alantolactone and isoalantolactone. Scientific World Journal. 2013;2013:248532
57. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742-58
58. Wu H, Che X, Zheng Q. et al. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci. 2014;10:1072-83
59. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nature Reviews Cancer. 2009;9(7):501-7
60. Larsson DE, Wickstrom M, Hassan S. et al. The cytotoxic agents NSC-95397, brefeldin A, bortezomib and sanguinarine induce apoptosis in neuroendocrine tumors in vitro. Anticancer Res. 2010;30:149-56
61. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
62. Duprez L, Wirawan E, Vanden Berghe T. et al. Major cell death pathways at a glance. Microbes Infect. 2009;11:1050-62
63. Kuno T, Tsukamoto T, Hara A. et al. Cancer chemoprevention through the induction of apoptosis by natural compounds. Journal of Biophysical Chemistry. 2012;3:156-173
64. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178-94
65. Roth W, Reed JC. Apoptosis and cancer: when BAX is TRAILing away. Nat Med. 2002;8:216-8
66. Hassan M, Watari H, AbuAlmaaty A. et al. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845
67. Liu H, Chen X, Sun J. et al. The Efficacy and Toxicity of Paclitaxel Plus S-1 Compared with Paclitaxel Plus 5-Fu for Advanced Gastric Cancer: A PRISMA Systematic Review and Meta-analysis of Randomized Controlled Trials. Medicine (Baltimore). 2014;93:e164
68. Huang Y, Fang Y, Wu J. et al. Regulation of Vinca alkaloid-induced apoptosis by NF-kappaB/IkappaB pathway in human tumor cells. Mol Cancer Ther. 2004;3:271-7
69. Kumar S, Pandey AK. Chemistry and biological activities of flavonoids: an overview. Scientific World Journal. 2013;2013:162750
70. Lea MA, Ibeh C, Deutsch JK. et al. Inhibition of growth and induction of alkaline phosphatase in colon cancer cells by flavonols and flavonol glycosides. Anticancer Res. 2010;30:3629-35
71. Dajas F. Life or death: neuroprotective and anticancer effects of quercetin. J Ethnopharmacol. 2012;143:383-96
72. Teillet F, Boumendjel A, Boutonnat J. et al. Flavonoids as RTK inhibitors and potential anticancer agents. Med Res Rev. 2008;28:715-45
73. Chen AY, Chen YC. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013;138:2099-107
74. Ferry DR, Smith A, Malkhandi J. et al. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996;2:659-68
75. Kandaswami C, Lee LT, Lee PP. et al. The antitumor activities of flavonoids. In Vivo. 2005;19:895-909
76. Jiang Y, Yang J, Yang C. et al. Vitamin K4 induces tumor cytotoxicity in human prostate carcinoma PC-3 cells via the mitochondria-related apoptotic pathway. Pharmazie. 2013;68:442-8
77. Ghavami S, Hashemi M, Ande SR. et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497-510
78. LaCasse EC, Mahoney DJ, Cheung HH. et al. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252-75
79. Khan M, Ding C, Rasul A. et al. Isoalantolactone induces reactive oxygen species mediated apoptosis in pancreatic carcinoma PANC-1 cells. Int J Biol Sci. 2012;8:533-47
80. Khan M, Yi F, Rasul A. et al. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life. 2012;64:783-94
81. Khan M, Li T, Ahmad Khan MK. et al. Alantolactone induces apoptosis in HepG2 cells through GSH depletion, inhibition of STAT3 activation, and mitochondrial dysfunction. Biomed Res Int. 2013;2013:719858
82. Di W, Khan M, Rasul A. et al. Isoalantolactone inhibits constitutive NF-kappaB activation and induces reactive oxygen species-mediated apoptosis in osteosarcoma U2OS cells through mitochondrial dysfunction. Oncol Rep. 2014;32:1585-93
83. Juan ME, Wenzel U, Daniel H. et al. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem. 2008;56:4813-4818
84. Siegelin MD, Reuss DE, Habel A. et al. Quercetin promotes degradation of survivin and thereby enhances death-receptor-mediated apoptosis in glioma cells. Neuro Oncol. 2009;11:122-131
85. Khan M, Bi Y, Qazi JI. et al. Evodiamine sensitizes U87 glioblastoma cells to TRAIL via the death receptor pathway. Mol Med Rep. 2015;11:257-62
86. Khan M, Rasul A, Yi F. et al. Jaceosidin induces p53-dependent G2/M phase arrest in U87 glioblastoma cells. Asian Pac J Cancer Prev. 2011;12:3235-8
87. Lu MC. Yang SH, Hwang SL, et al. Induction of G2/M phase arrest by squamocin in chronic myeloid leukemia (K562) cells. Life sci. 2006;78:2378-83
88. Yang G, Chang B, Yang F. et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010;16:3171-81
89. Khan M, Zheng B, Yi F. et al. Pseudolaric Acid B induces caspase-dependent and caspase-independent apoptosis in U87 glioblastoma cells. Evid Based Complement Alternat Med. 2012;2012:957568
90. Khan M, Yu B, Rasul A. et al. Jaceosidin Induces Apoptosis in U87 Glioblastoma Cells through G2/M Phase Arrest. Evid Based Complement Alternat Med. 2012;2012:703034
91. Ma A, Qi S, Xu D. et al. Baohuoside-1 inhibits activated T cell proliferation at G(1)-S phase transition. Transpl Immunol. 2005;15:55-62
92. Little SE, Bax DA, Rodriguez-Pinilla M. et al. Multifaceted dysregulation of the epidermal growth factor receptor pathway in clear cell sarcoma of the kidney. Clin Cancer Res. 2007;13:4360-4
93. Han W, Lo HW. Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012;318:124-34
94. Seshacharyulu P, Ponnusamy MP, Haridas D. et al. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:15-31
95. Siveen KS, Sikka S, Surana R. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845:136-54
96. Qin HR, Kim HJ, Kim JY. et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008;68:7736-41
97. Huang G, Yan H, Ye S. et al. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells. 2014;32:1149-60
98. Harada D, Takigawa N, Kiura K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers (Basel). 2014;6:708-22
99. Kim JE, Patel M, Ruzevick J. et al. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers (Basel). 2014;6:376-95
100. Demaria M, Giorgi C, Lebiedzinska M. et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY). 2010;2:823-42
101. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41-51
102. Zhou J, Ong CN, Hur GM. et al. Inhibition of the JAK-STAT3 pathway by andrographolide enhances chemosensitivity of cancer cells to doxorubicin. Biochem Pharmacol. 2010;79:1242-50
103. Zhao M, Gao FH, Wang JY. et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73:366-74
104. O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368:161-70
105. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075-83
106. Vincent EE, Elder DJ, Thomas EC. et al. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J Cancer. 2011;104:1755-61
107. Steelman LS, Chappell WH, Abrams SL. et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 2011;3:192-222
108. Martelli AM, Evangelisti C, Chiarini F. et al. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89-103
109. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-1657
110. Herberger B, Puhalla H, Lehnert M. et al. Activated mammalian target of rapamycin is an adverse prognostic factor in patients with biliary tract adenocarcinoma. Clin Cancer Res. 2007;13:4795-9
111. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87-90
112. Samuels Y, Wang Z, Bardelli A. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554
113. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221-1224
114. Kang S, Denley A, Vanhaesebroeck B. et al. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289-1294
115. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Ann Rev Pathol Mech Dis. 2009;4:127-150
116. McCubrey JA, Steelman LS, Kempf CR. et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011;226(11):2762-81
117. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK- RAS- RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103-19
118. Chang F, Steelman LS, Lee JT. et al. Signal transduction mediated by the Ras/ Raf/ MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263-93
119. McCubrey JA, Steelman LS, Abrams SL. et al. Targeting Survival Cascades Induced by Activation of Raf/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708-722
120. Martelli AM, Evangelisti C, Chiarini F. et al. The emerging role of the phosphatiylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogensis. Biochim Biophys Act. 2010;1803:991-1002
121. Martelli AM, Evangelisti C, Chiarini F. et al. The phosphatidylinositol 3-kinase/Akt/ mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89-103
122. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928-42
123. Dhillon AS, Hagan S, Rath O. et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279-3290
124. Saito-Diaz K, Chen TW, Wang X. et al. The way Wnt works: components and mechanism. Growth Factors. 2013;31:1-31
125. Cadigan KM. Wnt-beta-catenin signaling. Curr Biol. 2008;18:R943-7
126. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17:9-26
127. Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606-1609
128. Suarez MI, Uribe D, Jaramillo CM. et al. Wnt/beta-catenin signaling pathway in hepatocellular carcinomas cases from Colombia. Ann Hepatol. 2015;14:64-74
129. Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1-24
130. Sadot E, Conacci-Sorrell M, Zhurinsky J. et al. Regulation of S33/S37 phosphorylated beta-catenin in normal and transformed cells. J Cell Sci. 2002;115:2771-80
131. Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175-1186
132. McCubrey JA, Steelman LS, Bertrand FE. et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881-911
133. Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways. Science. 2004;303:1483-1487
134. Castellone MD, Teramoto H, Williams BO. et al. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504-10
135. Kudo J, Nishiwaki T, Haruki N. et al. Aberrant nuclear localization of β-catenin without genetic alterations in β-catenin or Axin genes in esophageal cancer. World J Surg Oncol. 2007;5:21-29
136. Ji L, Cao XF, Wang HM. et al. Expression level of beta-catenin is associated with prognosis of esophageal carcinoma. World J Gastroenterol. 2007;13:2622-2625
137. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
138. Luo D, Wang Z, Wu J. et al. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed Res Int. 2014;2014:409272
139. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist 9 Suppl. 2004;5:10-7
140. Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist 9 Suppl. 2004;5:4-9
141. Powis G, Kirkpatrick L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther. 2004;3:647-54
142. Burroughs SK, Kaluz S, Wang D. et al. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med Chem. 2013;5:553-72
143. Hong SS, Lee H, Kim KW. HIF-1alpha: a valid therapeutic target for tumor therapy. Cancer Res Treat. 2004;36:343-53
144. Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030-9037
145. Siddiq A, Aminova LR, Ratan RR. Prolyl 4-hydroxylase activity-responsive transcription factors: from hydroxylation to gene expression and neuroprotection. Front Biosci. 2008;13:2875-87
146. Jendrossek V. Targeting apoptosis pathways by Celecoxib in cancer. Cancer Lett. 2013;332:313-24
147. Bu X, Zhao C, Dai X. Involvement of COX-2/PGE(2) Pathway in the Upregulation of MMP-9 Expression in Pancreatic Cancer. Gastroenterol Res Pract. 2011;2011:214269
148. Park ES, Do IG, Park CK. et al. Cyclooxygenase-2 is an independent prognostic factor in gastric carcinoma patients receiving adjuvant chemotherapy and is not associated with EBV infection. Clin Cancer Res. 2009;15:291-8
149. Xiao H, Yang CS. Combination regimen with statins and NSAIDs: a promising strategy for cancer chemoprevention. Int J Cancer. 2008;123:983-90
150. Jana NR. NSAIDs and apoptosis. Cell Mol Life Sci. 2008;65:1295-301
151. Greenhough A, Smartt HJ, Moore AE. et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377-86
152. Takahashi H, Li A, Dawson DW. et al. Cyclooxygenase-2 confers growth advantage to syngeneic pancreatic cancer cells. Pancreas. 2011;40:453-9
153. McDaid HM, Horwitz SB. Selective potentiation of paclitaxel (taxol)- induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol. 2001;60:290-301
154. Wang AC, Su QB, Wu FX. et al. Role of TLR4 for paclitaxel chemotherapy in human epithelial ovarian cancer cells. Eur J Clin Invest. 2009;39:157-64
155. Szajnik M, Szczepanski MJ, Czystowska M. et al. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene. 2009;28:4353-63
Author contact
![]() Corresponding author: Tonghui Ma, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel:+86-411-86110278, Fax:+86-411-86110378, E-mail: tonghuimaedu.cn
Corresponding author: Tonghui Ma, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning 116044, P.R. China. Tel:+86-411-86110278, Fax:+86-411-86110378, E-mail: tonghuimaedu.cn