Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(9):1113-1126. doi:10.7150/ijbs.12152 This issue Cite
Research Paper
Norisoboldine, an Anti-Arthritis Alkaloid Isolated from Radix Linderae, Attenuates Osteoclast Differentiation and Inflammatory Bone Erosion in an Aryl Hydrocarbon Receptor-Dependent Manner
Zhi-feng Wei1, Qi Lv1, Ying Xia1, Meng-fan Yue1, Can Shi1, Yu-feng Xia1, Gui-xin Chou2, Zheng-tao Wang2, Yue Dai1, ![]()
1. State Key Laboratory of Natural Medicine, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, China
2. Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China
Received 2015-3-16; Accepted 2015-6-12; Published 2015-7-17
Abstract

Norisoboldine (NOR), the primary isoquinoline alkaloid constituent of the root of Lindera aggregata, has previously been demonstrated to attenuate osteoclast (OC) differentiation. Accumulative evidence has shown that aryl hydrocarbon receptor (AhR) plays an important role in regulating the differentiation of various cells, and multiple isoquinoline alkaloids can modulate AhR. In the present study, we explored the role of NOR in the AhR signaling pathway. These data showed that the combination of AhR antagonist resveratrol (Res) or α-naphthoflavone (α-NF) nearly reversed the inhibition of OC differentiation through NOR. NOR could stably bind to AhR, up-regulate the nuclear translocation of AhR, and enhance the accumulation of the AhR-ARNT complex, AhR-mediated reporter gene activity and CYP1A1 expression in RAW 264.7 cells, suggesting that NOR might be an agonist of AhR. Moreover, NOR inhibited the nuclear translocation of NF-κB-p65, resulting in the evident accumulation of the AhR-NF-κB-p65 complex, which could be markedly inhibited through either Res or α-NF. Although NOR only slightly affected the expression of HIF-1α, NOR markedly reduced VEGF mRNA expression and ARNT-HIF-1α complex accumulation. In vivo studies indicated that NOR decreased the number of OCs and ameliorated the bone erosion in the joints of rats with collagen-induced arthritis, accompanied by the up-regulation of CYP1A1 and the down-regulation of VEGF mRNA expression in the synovium of rats. A combination of α-NF nearly completely reversed the effects of NOR. In conclusion, NOR attenuated OC differentiation and bone erosion through the activation of AhR and the subsequent inhibition of both NF-κB and HIF pathways.
Keywords: Norisoboldine, Aryl hydrocarbon receptor, Osteoclast differentiation, Rheumatoid arthritis, Inflammatory bone erosion
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by synovial inflammation and bone and cartilage erosion [1, 2]. Joint bone destruction ultimately results in movement disabilities, and agents for bone protection have received much research attention. In a phase II clinical trial, the anti-bone destruction agent Denosumab, a fully human anti-RANKL monoclonal antibody, halted the progression of subchondral bone erosion and systemic bone loss in patients with active RA. Accumulative evidence indicated that osteoclasts (OCs) are closely associated with local bone erosion. In RA, abundant OCs have been identified within the synovial tissues at sites adjacent to the bone, creating resorption pits and local bone erosion, followed by bone matrix degradation and calcium solubilization. In contrast, obvious bone erosion did not appear in OC-deficient mice in a serum transfer model of arthritis [3-7]. Therefore, OCs can be considered as a type of pivotal target cells for anti-bone destruction remedies.
Radix Linderae, the dry root of Lindera aggregata (Sims) Kosterm. (L. strychnifolia Vill), is frequently used in traditional Chinese medicine. This herb contains a series of components, including alkaloids, volatile oils and sesquiterpene esters [8]. Norisoboldine (NOR), the main isoquinoline alkaloid constituent of Radix Linderae, has been previously shown to ameliorate bone erosion in rats with adjuvant-induced arthritis at lower doses than required for the repression of systemic inflammation and exhibit a direct anti-joint bone destruction effect [9-11]. Moreover, Radix Linderae markedly inhibits the differentiation of OCs in vitro, an effect likely associated with the inhibition of the NF-κB pathway, however the precise mechanism remains unclear [11].
The aryl hydrocarbon receptor (AhR) is a transcription factor activated through ligands. After ligand binding, this protein enters the cell nucleus from the cytoplasm, forms a heterodimer with the AhR nuclear transporter (ARNT), and induces the transcription of target genes [12, 13]. AhR is considered an important mediator of toxic environmental substances, such as doxin, 2, 3, 7, 8-tetrachlorodibenzo-para-dioxin (TCDD), and benzopyrene (Bap). Recent evidence suggests that AhR participates in the differentiation and activation of various cells (including OCs) through the regulation of NF-κB, STATs, MAPKs, and tyrosine kinase signaling pathways. TCDD and Bap inhibit OC differentiation through AhR activation and subsequent inhibition of NF-κB activation [14-16]. Various natural products have been reported to react with AhR. Notably, many isoquinoline alkaloids, such as berberine, protopine, and allocryptopine, regulate AhR [17-19]. Based on these findings, we examined the mechanisms through which NOR inhibits the differentiation and activation of OCs and reduces bone erosion via the AhR pathway.
Materials and methods
Chemicals and reagents
NOR (purity>98%) was isolated and purified from Radix Linderae, and the structure was identified through the comparison of the spectral data (UV, IR, MS, 1H-, and 13C-NMR) with the literature data [20]; Resveratrol (Res, purity>95%) was purchased from Ze Lang Pharmaceutical Technology Co., Ltd. (Nanjing, China); α-naphthoflavone (α-NF, purity>95%), chicken type II collagen, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), and a leukocyte acid phosphatase kit were purchased from Sigma Chemical Co. (St. Louis, MO, USA); Mouse RANKL was purchased from PeproTech Int. (Connecticut, USA); Dulbecco's modified Eagle's medium (DMEM), and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, USA); Mycobacterium butyricum was purchased from Becton DriveCo. Ltd. (New Jersey, USA); AhR and ARNT monoclonal antibodies were purchased from Santa Cruz Biotechnology (CA, USA); HIF-1α, NF-κB-p65 and peroxidase-conjugated secondary antibodies were purchased from BioWorld (Georgia, USA); GAPDH monoclonal antibody was purchased from KangChen Bio-tech (Shanghai, China); nuclear and cytoplasmic protein extraction kits were purchased from Sangon Biotech (Shanghai, China); TRIzol reagent was purchased from Invitrogen (Carlsbad, CA); the pGL3-Promoter Vector was purchased from Promega (Madison WI, USA); the IScriptTM cDNA Synthesis Kit was purchased from Bio-Rad (CA, USA); enhanced chemiluminescent (ECL) plus reagent kits were purchased from MultiSciences (Hangzhou, China). The other chemicals and reagents used were of analytical grade.
OC differentiation
RAW264.7 cells (ATCC, Manassas, VA, USA) were seeded at a density of 2×105 cells/mL into 48-well plates and treated with Res (1 µM), α-NF (1 µM), NOR (30 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) in the presence or absence of RANKL (100 ng/mL) for five days. The OCs were identified through TRAP staining according to the manufacturer's instructions. TRAP-positive multinucleated cells with more than three nuclei were counted as OCs.
Cytotoxicity assay
RAW264.7 cells were seeded at the density of 2×105 cells/mL onto 96-well plates and treated with Res (1 µM), α-NF (1 µM), NOR (30 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) in the presence or absence of RANKL (100 ng/mL) for three or five days. Subsequently, 20 μL of MTT solution (5 mg/mL) was added, and the cells were continuously incubated for an additional 4 h. Subsequently, the supernatants were removed and the formazone crystals were dissolved using 150 µL of DMSO. The optical absorbance at 570 nm was read using a Model 1500 Multiskan Spectrum Microplate Reader (Thermo, Waltham, MA, USA).
Molecular docking
The three-dimensional structure of AhR was downloaded from the protein database (protein data bank, PDB), serial number: IP97. According to the method of Bisson WH et al. [21], AhR ligand binding domain homology model was constructed. Subsequently, NOR was inserted into the active site of the model, and the binding ability was detected.
Immunoprecipitation (IP) assay
The accumulation of AhR-ARNT, NF-κB-p65-AhR, and ARNT-HIF-1α complexes was detected through IP assay using the following protocol. Briefly, RAW264.7 cells (2×105 cells/mL) were plated in cell culture flasks and pre-treated with NOR (3, 10 and 30 µM) for 24 h and subsequently stimulated with RANKL (100 ng/mL) for appropriate periods. The cells were washed once with ice-cold PBS and lysed in buffer. The cell extracts were centrifuged at 12,000 rpm for 5 min, followed by immunoprecipitation with the indicated antibodies. The co-precipitated proteins were detected through immunoblotting with the corresponding antibodies. The bands were visualized through film exposure with ECL reagent.
Report gene assay
RAW264.7 cells were plated at a density of 1.5×104 cells/mL onto 96-well plates and transfected with the pGL3-xenobiotic response element (XRE) promoter and 0.6 µL FuGene 6 according to the manufacturer's instructions. After transfection for approximately 24 h, the media was removed and the cells were treated with NOR (3, 10 and 30 μM) for an additional 24 h. Subsequently, the cells were lysed using 100 µL Glo lysis buffer and transferred into 96-well plates. Approximately 100 µL of luciferase substrate was added to each well, and the absorbance was read at 405 nm using a Spectromax 96-well plate reader.
Quantitative-polymerase chain reaction (Q-PCR) assay
In RAW264.7 cells and the synovial membrane tissues of CIA rats treated with NOR, the mRNA expression of CYP1A1 and VEGF (the target genes of AhR and HIF-1α, respectively) was assessed using Q-PCR assay. Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions, and the concentration was determined through spectrophotometric optical density measurement at 260 and 280 nm. RNA (2 µg) was reverse transcribed into cDNA using the iScriptTMcDNA Synthesis Kit (Bio-Rad, USA) according to the manufacturer's instructions. The cDNA template (2 μL) was added to the 20-μL PCR reaction containing sequence-specific primers and the SsoFastTMEvaGreenRSupermix Kit (Bio-Rad, USA). The cycling conditions included an initial step at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 55-60°C for 10 s. The threshold was set above the non-template control background within the linear phase of the target gene amplification to calculate the cycle number at which the transcript was detected. The following gene-specific primers were used: CYP1A1 forward: 5'-GGTTAACCATGACCGGGAACT-3', reverse: 5'-TGCCCAAACCAAAGAGAGTGA-3'; VEGF forward: 5'-CACTCCCTCAAATCACTTCg-3', reverse 5'-CCTgTCCCTCTCTCTgTTCg-3'; GAPDH forward: 5'- AAATGGTGAAGGTCGGTGTG-3', reverse 5'- TGAAGGGGTCGTTGATGG-3'.
Western blotting assay
RAW264.7 cells (2×105 cells/mL) were pre-treated with NOR (3, 10 and 30 µM), Res (1 µM), α-NF (1 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM), respectively. After 24 h, the cells were stimulated with RANKL (100 ng/mL) for the appropriate period. Subsequently, the cells were washed twice with PBS buffer (pH = 7.2). The proteins were extracted using a protein extraction kit according to the manufacturer's instructions, and the concentration was determined using the Bradford assay. The samples were fractionated through 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked for 1 h at room temperature with nonfat-milk and subsequently incubated with different antibodies. After washing, the blots were incubated with secondary antibodies for 1 h. The bands were visualized through film exposure using ECL reagent.
Animal
Male Wistar rats, weighing 130-150 g, were purchased from the Comparative Medicine Center of Yangzhou University (Yangzhou, China). The mice were fed standard laboratory chow and provided tap water ad libitum, and the husbandry room was maintained at 22 ± 2 °C with a 12 h light: 12 h dark cycle. The animal experiments were conducted under the guidelines of current ethical regulations for institutional animal care and use at China Pharmaceutical University.
Collagen-induced arthritis
Chicken type II collagen was emulsified in incomplete Freund's adjuvant. Arthritis was induced through two intradermal injections of collagen at the base of the tail of rats (on day 0, 200 μg; on day 7, 100 μg) [22, 23]. On day 14 after immunization, the rats were randomly divided into the following six groups: normal group, control group, NOR (15 mg/kg) group, α-NF (50 mg/kg) group, NOR (15 mg/kg)+α-NF (50 mg/kg) group and leflunomide (Lef, 2 mg/kg) group. NOR and Lef were orally administered, and α-NF was intraperitoneally injected for fourteen consecutive days from day 14 to day 28. The rats in the normal and control groups were administered an equal volume of vehicle in the same schedule.
Radiological scanning
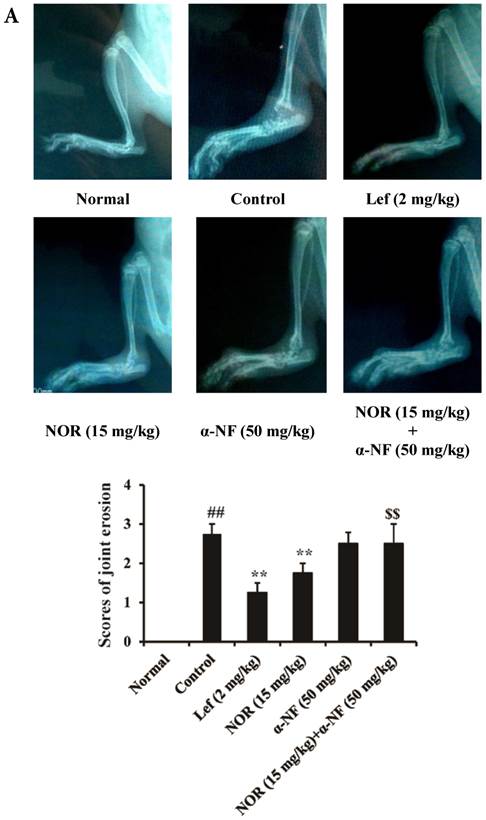
On day 28 after immunization, the rats were administered a general anesthetic of ether, and the joints of the left hind paws were X-rayed. The destruction of the ankle joints was scored on a scale of 0-3, where 0: no joint erosion; 1: mild joint erosion; 2: moderate joint erosion; and 3: severe joint erosion, and the mean scores of each rat was calculated.
Hematoxylin and Eosin (H&E) and TRAP staining
After radiological scanning, the rats were sacrificed through ether anesthesia, and the paws of the right sides were harvested for further analysis. The formaldehyde-fixed hind paws were decalcified in a solution containing 5% EDTA, and the ankle tissues were sectioned, embedded in paraffin, and sliced (5 μm) for H&E and TRAP staining.
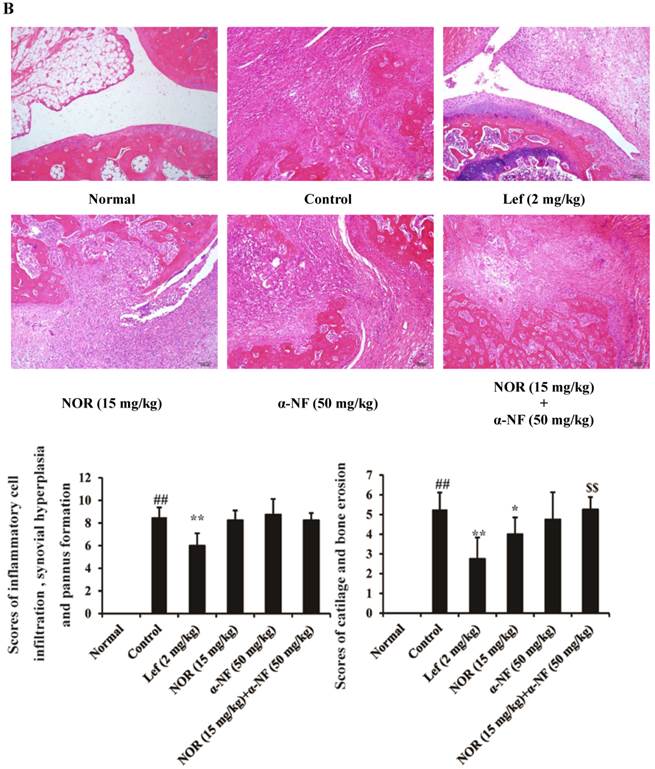
H&E staining: The histological scoring of the rat ankle joints was performed. A pathologist blinded to the experimental groups scored the tissue sections for inflammatory cell infiltration, synovial hyperplasia, pannus formation, cartilage and bone erosion, respectively, on a scale of 0-3 (0 = none, 1 = mild, 2 = moderate, and 3 = severe). Then, the mean of all scores was calculated.
TRAP staining: Staining was performed according to the manufacturer's instructions (Sigma, St. Louis, MO, USA).
Statistical analysis
The data were presented as the means ± S.E.M. Significant differences were assessed through one-way analysis of variance (ANOVA) followed by a post hoc Tukey's test. P values less than 0.05 (P < 0.05) were accepted as significant.
Results
NOR inhibited RANKL-induced osteoclastogenesis in an AhR-dependent manner
In previous studies, NOR (10 and 30 μM) showed the marked inhibition of RANKL-induced osteoclastogenesis from either RAW264.7 or bone marrow-derived macrophages (BMMs). The emerging evidence that AhR participates in the activation and function of OCs and many isoquinoline alkaloids regulate AhR, suggested the potential involvement of AhR in the NOR-mediated attenuation of osteoclastogenesis. To examine this idea, two different AhR antagonists Res and α-NF were employed in the present study. The data showed that NOR (30 µM) attenuated RANKL-induced OC differentiation from RAW264.7 cells. Neither Res (1 µM) nor α-NF (1 µM) affected OC differentiation, but these proteins dramatically suppressed the inhibitory effect of NOR (Fig. 1A). It has been suggested that NOR might exert anti-osteoclastogenesis activity in an AhR-dependent manner.
Influences of resveratrol (Res) and α-naphthoflavone (α-NF) on the inhibition of norisoboldine (NOR) against osteoclastogenesis. (A) Osteoclastogenesis assay. RAW264.7 cells were treated with Res (1 µM), α-NF (1 µM), NOR (30 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) in the presence or absence of RANKL (100 ng/mL) for 5 days. OCs were identified using TRAP staining according to the manufacturer's instructions. TRAP-positive multinucleated cells with more than three nuclei were counted as OCs. (B) The effects of NOR alone or combined with Res and α-NF on the viability of RAW264.7 cells. The cells were treated with Res (1 µM), α-NF (1 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) in the presence or absence of RANKL (100 ng/mL) for 3 or 5 days, respectively. Cell viability was determined through MTT assay. The data were expressed as the means ± S.E.M. of three independent experiments. ##p < 0.01 vs. normal; **p< 0.01 vs. control; $$p < 0.01 vs. NOR (30 µM).
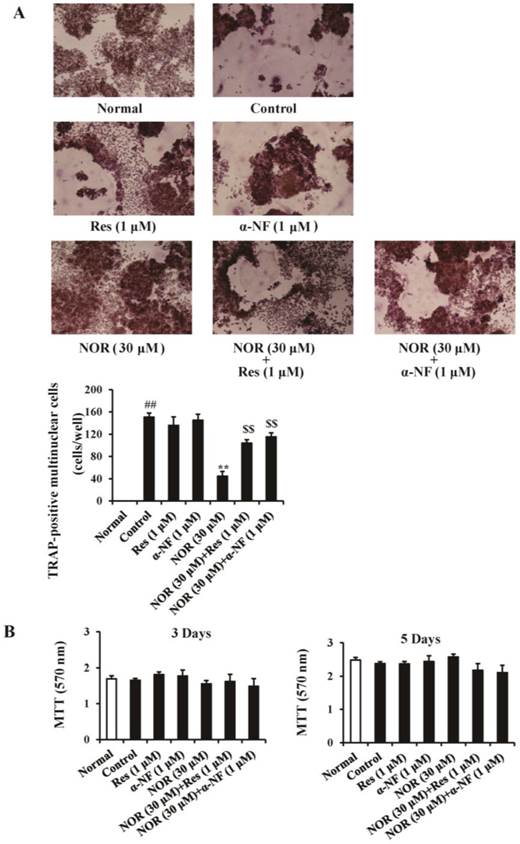
To exclude the possibility that the effect of NOR reflected cytotoxicity, the viability of RAW264.7 cells was examined using the MTT assay. The results showed that NOR alone or in combination with Res and α-NF for 3 and 5 days did not exhibit significant cytotoxicity against RAW264.7 cells (Fig. 1B).
NOR could activate AhR
Simulation of the docking between small molecular compounds and proteins using a computer program is an intuitive and fast method for the prediction of targets. To determine whether NOR directly interacts with AhR, we built the ligand-binding domain of AhR through homologous modeling and detected the docking between NOR and AhR. As shown in Fig. 2A, NOR docked onto the mouse and human AhR-LBD homology models, and the binding locus was between the cavities formed by two α-helixes. The hydroxy of NOR could come into being hydrogen bonding with Thr 54. In addition, the two aromatic rings of NOR solidly associated with Tyr 42 through pi-pi action.
Activation of aryl hydrocarbon receptor (AhR) through norisoboldine (NOR). (A) Molecular docking of NOR in the homology model of the AhR ligand binding domain. (B) Effect of NOR on the nuclear translocation of AhR. RAW264.7 cells were treated with NOR (3, 10 and 30 μM) for 24 h and subsequently lysed. The expression of AhR in the cytosol and nucleus was analyzed using western blotting assay. (C) Effect of NOR on the accumulation of the AhR-ARNT complex in RAW264.7 cells. RAW264.7 cells were treated with NOR (3, 10 and 30 μM) for 24 h and subsequently lysed. The cell extracts were immunoprecipitated with anti-AhR antibody. Co-precipitated ARNT was detected through immunoblotting. (D) Effect of NOR on AhR-mediated reporter gene activity. RAW264.7 cells were transiently transfected with a luciferase reporter gene expression vector under control of a promoter containing xenobiotic response elements (XRE-Luc) and treated with NOR (3, 10 and 30 μM) for 24 h. The absorbance at 405 nm was read using a Spectromax 96-well plate reader. (E) Effect of NOR on the mRNA expression of CYP1A1 in RAW264.7 cells. The cells were treated with NOR (3, 10 and 30 μM), and the cell lysates were collected at the indicated time points. The mRNA expression of CYP1A1 was detected using Q-PCR. The data were expressed as the means ± S.E.M. of three independent experiments. *p< 0.05, **p< 0.01 vs. normal.
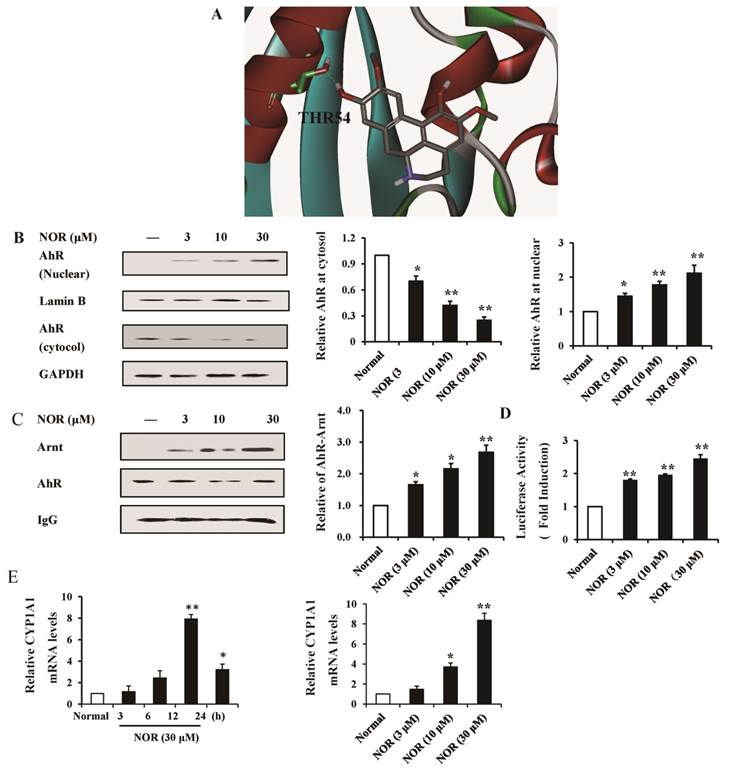
In the quiescent state, AhR primarily resides in the cytoplasm, and this receptor subsequently enters the nucleus after activation. In the present study, the nuclear translocation of AhR was assessed. The results showed that NOR (10 and 30 μM) treatment for 24 h markedly up-regulated AhR nuclear translocation in RAW264.7 cells, evidenced as the reduced amount of AhR in the cytoplasm and increased amount of AhR in the nucleus (Fig. 2B).
After entering the cellular nucleus, AhR functions through association with the molecular chaperone ARNT, forming a complex that specifically identifies and integrates with the XRE of target genes, such as CYP1A1, and regulates gene expression [24]. Therefore, we examined the effect of NOR on the accumulation of the AhR-ARNT complex. RAW264.7 cells were transiently transfected with a luciferase reporter gene expression vector under the control of a promoter containing XRE-Luc, and the binding activity was assessed. As shown in Fig. 2C, NOR (10 and 30 μM) treatments resulted in the significant accumulation of the AhR-ARNT complex and obviously increased the intensity of the XRE reporter in a concentration-dependent manner (Fig. 2D).
In RAW264.7 cells, NOR (30 μM) treatment resulted in the evident mRNA expression of CYP1A1 with a peak at 12 h. In addition, NOR (10 and 30 μM) increased the levels of CYP1A1 mRNA in RAW264.7 cells in a concentration-dependent manner (Fig. 2E).
NOR inhibited the RANKL-induced activation of NF-κB in RAW264.7 cells through AhR
These data strongly suggested that NOR might be an agonist of AhR. To understand the detailed mechanism through which NOR inhibits OC differentiation, we investigated the signaling transduction following AhR activation.
NF-κB, a transcriptional factor, is involved in osteoclastogenesis. The activation of NF-κB is associated with a sequential cascade that includes IKK-dependent IκBα phosphorylation, ubiquitination and proteolytic degradation and the translocation of cytosolic NF-κB-p65 to the nucleus. Notably, activated-AhR binds and inhibits the nuclear translocation of NF-κB-p65. In previous studies, we showed that NOR pre-treatment did not affect the RANKL-induced phosphorylation and degradation of IκBα, but rather markedly inhibited NF-κB-p65 nuclear translocation in RAW264.7 cells. Therefore, we examined the relationship between NF-κB-p65 inhibition and AhR activation through NOR during RANKL-induced osteoclastogenesis.
As shown in Fig. 3A, NOR (30 μM) alone nearly completely inhibited the RANKL-induced translocation of cytosolic NF-κB-p65 to nucleus in RAW264.7 cells. Neither α-NF (1 μM) nor Res (1 μM) itself exhibited a significant effect on nuclear translocation, but these two AhR antagonists reversed the effect of NOR (30 μM). This result suggested that NOR exerted inhibitory effects on NF-κB-p65 translocation in RANKL-stimulated RAW264.7 cells through AhR.
Effects of norisoboldine (NOR) on the crosstalk between aryl hydrocarbon receptor (AhR) and NF-κB signaling pathway in osteoclastogenesis. (A) Effects of resveratrol (Res) and α-naphthoflavone (α-NF) on the inhibition of nuclear translocation of NF-κB-p65 through NOR in RANKL-induced RAW264.7 cells. The cells were treated with Res (1 µM), α-NF (1 µM), NOR (30 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) for 24 h in the presence or absence of RANKL (100 ng/mL), respectively. The cells were subsequently stimulated with RANKL (100 ng/mL) for 15 min. The expression of NF-κB-p65 in the cytosol and nucleus were analyzed using western blotting. (B) Effect of NOR on the accumulation of the AhR-NF-κB-p65 complex in RANKL-induced RAW264.7 cells. The time course of the accumulation of AhR-NF-κB-p65 complex was investigated. RAW264.7 cells were treated with RANKL (100 ng/mL), and the cell lysates were collected at the indicated time points. The cell extracts were immunoprecipitated with an anti-AhR antibody. Co-precipitated NF-κB-p65 was detected through immunoblotting with the relevant antibody. Subsequently, RAW264.7 cells were treated with NOR (10 and 30 μM) for 24 h, and subsequently stimulated with RANKL (100 ng/mL) for 15 min. The cells were lysed, and the extracts were immunoprecipitated with an anti-AhR antibody. Co-precipitated NF-κB-p65 was detected through immunoblotting with the relevant antibody. The data were expressed as the means ± S.E.M. of three independent experiments. ##p< 0.01 vs. normal; *p< 0.05, **p< 0.01 vs. control; $$p < 0.01 vs. NOR (30 µM).
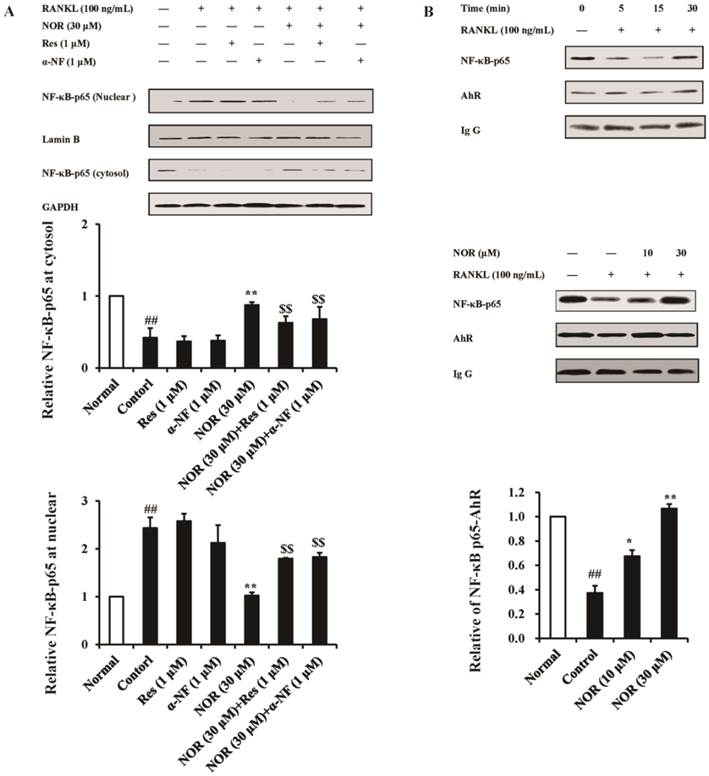
After being activation, AhR associates with NF-κB-p65 and interferes with the translocation of NF-κB-p65 from the cytoplasm to nucleus [25]. In RAW264.7 cells, RANKL (100 ng/mL) stimulation for 15 min markedly decreased the formation of the AhR-NF-κB-p65 complex, whereas NOR (30 μM) treatment markedly augmented the accumulation of this complex (Fig. 3B).
NOR inhibited the RANKL-induced activation of HIF-1α signaling pathway in RAW264.7 cells through AhR
Hypoxia and the associated transcription factor HIF (hypoxia inducer factor) regulate angiogenic-osteogenic coupling and osteoclast-mediated bone resorption. In RANKL-induced OC differentiation from RAW264.7 cells, HIF signaling pathway is significantly activated [26, 27]. Moreover, HIF-1α has the same partner molecule ARNT (HIF-1β) as AhR, suggesting that the activation of the HIF-1α pathway might interfere with AhR signal transduction through competition with AhR for ARNT [28].
VEGF is the target gene of HIF-1α, and the HIF-1α/VEGF signaling pathway has been implicated in the regulation of OC differentiation. In the present study, RANKL (100 ng/mL) stimulation markedly up-regulated the expression of HIF-1α in RAW264.7 cells, and NOR (3, 10 and 30 μM) showed little effect on the expression of HIF-1α. In contrast, NOR (10 and 30 μM) treatment significantly inhibited the mRNA expression of VEGF in RANKL-induced RAW264.7 cells (Fig. 4A). In-depth studies revealed that AhR antagonists Res (1 μM) and α-NF (1 μM) dramatically reversed the inhibition of VEGF expression through NOR (30 μM) (Fig. 4B). These data indicated that although NOR did not affect the expression of HIF-1α, and this factor inhibited the function of HIF-1α through AhR.
Effects of norisoboldine (NOR) on the crosstalk between aryl hydrocarbon receptor (AhR) and HIF-1α signaling pathway in osteoclastogenesis. (A) Effect of NOR on the expression of HIF-1α protein and VEGF mRNA in RANKL-induced RAW264.7 cells. The cells were treated with NOR (3, 10 and 30 µM) for 24 h in the presence or absence of RANKL (100 ng/mL). The cells were lysed, and the expression of HIF-1α in RAW264.7 cells was analyzed using western blotting. The mRNA expression of VEGF was detected using Q-PCR. (B) Effects of resveratrol (Res) and α-naphthoflavone (α-NF) on the inhibition of VEGF expression through NOR in RANKL-induced RAW264.7 cells. The cells were treated with Res (1 µM), α-NF (1 µM), NOR (30 µM), NOR (30 µM)+Res (1 µM), and NOR (30 µM)+α-NF (1 µM) for 24 h in the presence or absence of RANKL (100 ng/mL). The cells were lysed, and the expression of VEGF was analyzed using western blotting. (C) Effect of NOR on the accumulation of the ARNT-HIF-1α complex. RAW264.7 cells were pre-treated with NOR (10 and 30 μM) for 24 h and subsequently stimulated with RANKL (100 ng/mL) for 30 min. The cells were lysed, and the extracts were immunoprecipitated with an anti-Arnt antibody. Co-precipitated HIF-1α was detected through immunoblotting with the relevant antibody. The data were expressed as the means ± S.E.M. of three independent experiments. ##p< 0.01 vs. normal; *p< 0.05, **p< 0.01 vs. control; $$p < 0.01 vs. NOR (30 µM).
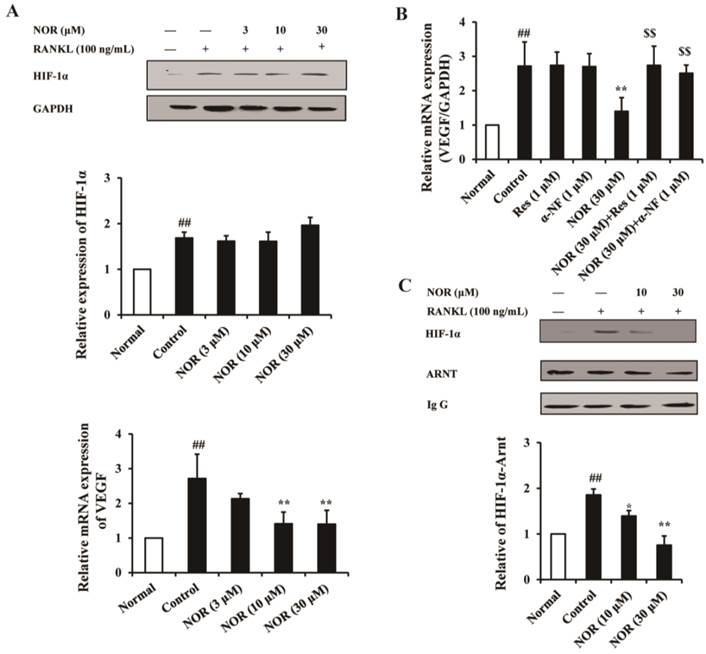
The dimerization of ARNT (HIF-1β) with HIF-1α is required for the expression of downstream genes, such as VEGF. The results of the present study showed that RANKL stimulation significantly facilitated the accumulation of the ARNT-HIF-1α complex, whereas NOR (10 and 30 μM) treatment obviously inhibited the accumulation of this complex (Fig. 4C). These results suggested that NOR likely inhibits VEGF mRNA expression in RANKL-induced RAW264.7 cells through the inhibition of ARNT-HIF-1α complex accumulation.
NOR ameliorated joint bone destruction in CIA rats through AhR
OCs are unique effector cells for bone erosion in RA, and NOR has been previously shown to potently inhibit the joint bone destruction in AIA rats at a minimum effective dose lower than that required for repressing systemic inflammation. Herein, we examined the involvement of AhR in the protection of NOR against joint bone destruction in CIA rats.
The results of radiological scanning showed that the rats in the CIA model group presented dramatic joint destruction, which could be ameliorated through both NOR (15 mg/kg) and Lef (an identified agonist of AhR, 2 mg/kg) treatment (Fig. 5A). Notably, the NOR-mediated amelioration could be remarkably reversed through α-NF (50 mg/kg). Furthermore, histological assessments of the tissue sections were performed to examine the changes in rat ankles on day 28 after the induction of CIA. The ankle joints of the rats in the model group presented pronounced infiltration of granulocytes and mononuclear cells, synovial hyperplasia, pannus formation, and bone and cartilage erosion compared with the normal group. NOR treatment only slightly affected the infiltration of inflammatory cells and markedly ameliorated the destruction of bone and cartilage in CIA rats, and this effect was nearly and completely reversed through the combination of α-NF (Fig. 5B).
Effects of α-naphthoflavone (α-NF) on the anti-bone destruction effect of norisoboldine (NOR) in collagen-induced arthritis (CIA) rats. The rats were intradermally injected with CII emulsion on day 0 and day 7, and administered with NOR (15 mg/kg, i.g.), α-NF (50 mg/kg, i.p.), NOR (15 mg/kg, i.g.)+α-NF (50 mg/kg, i.p.), and leflunomide (Lef, 2 mg/kg, i.g.) from day 14 to day 28. (A) On day 28 after the intradermal injection of CII emulsion, X-rays were taken after the rats received a general anesthetic of ether. The degree of joint erosion on the radiographs was scored on a scale of 0-3 as described in the Materials and Methods. (B) On day 28 after the intradermal injection of CII emulsion, the ankle joints of AIA rats were collected. Histological changes were detected using H&E staining. Pathohistological grades were evaluated according to the infiltration of inflammatory cells, synovial hyperplasia, pannus formation, and cartilage and bone erosion. The histological scores of all groups were summarized (original magnification × 200). (C) The OCs in joints of CIA rats were identified using TRAP staining according to the manufacturer's instructions. (D) The expression of CYP1A1 and VEGF in the synovium of CIA rats. The data were presented as the means ± S.E.M., n = 8. ##p< 0.01 vs. normal; *p< 0.05, **p< 0.01 vs. control; $$p < 0.01 vs. NOR (15 mg/kg).
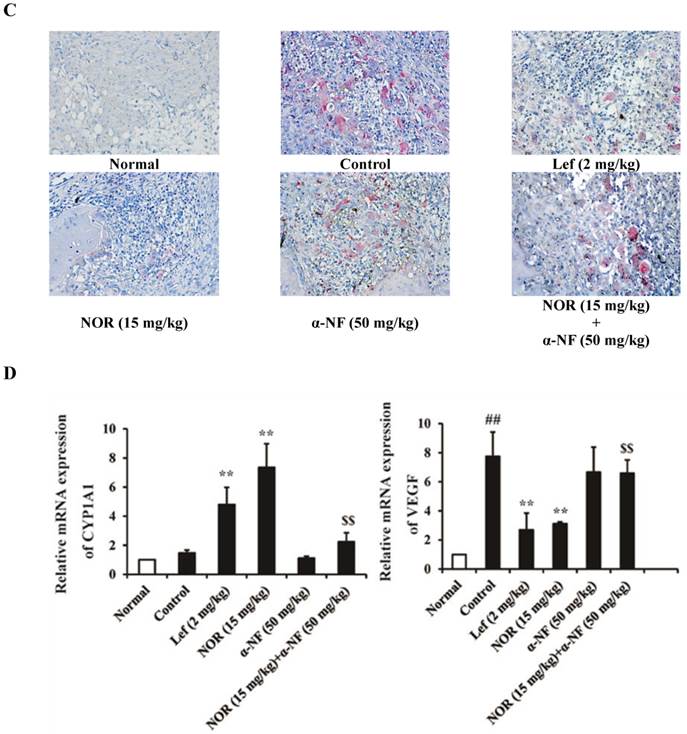
Subsequently, we detected the number of OCs in the joint sections of CIA rats through TRAP staining and observed the marked accumulation of OCs. NOR (15 mg/kg) treatment significantly reduced the number of OCs, and this effect could be significantly disrupted through α-NF (50 mg/kg) treatment (Fig. 5C).
As shown in Fig. 5D, the expression of CYP1A1 and VEGF mRNA in the synovium of CIA rats was detected through Q-PCR. NOR (15 mg/kg) treatment increased the expression of CYP1A1 mRNA and decreased the expression of VEGF mRNA. In combination with α-NF, the significant reduction of the effect of NOR was observed.
Taken together, these findings strongly suggested that NOR protects against bone and cartilage destruction resulting from OC differentiation and activation in CIA rats through the activation of AhR and subsequent inhibition of NF-κB and HIF-1α signaling pathways.
Discussion
The two primary hallmarks of RA are synovial inflammation and cartilage and bone impairment. The goal of anti-rheumatic treatment is not only to inhibit the clinical symptoms of joint inflammation but also to suppress the progression of joint destruction. Increasing evidence has shown that joint destruction in RA primarily reflects elevated pro-inflammatory cytokines, synovial neovascularization, proteinase-mediated the articular cartilage matrix dissolution, and OC-mediated bone resorption. There is also growing evidence indicating that OCs are key players in the erosive and inflammatory events leading to joint destruction in arthritis with special regard to RA. NOR, the major isoquinoline alkaloid constituent of Radix Linderae, indeed attenuated the destruction of cartilage and bone in rat AIA at a dose lower than that required for repressing systemic inflammation [29]. In vitro, NOR could markedly inhibit RANKL-induced OC differentiation from RAW264.7 cells. These findings indicate that NOR has a direct bone-protective effect, and the underlying mechanism likely involves the inhibition of differentiation and activation of OCs. In the present study, we identified the potential target proteins of NOR.
AhR is a ligand-dependent transcription factor activated through ligands. Ligand binding leads to the dimerization of AhR with ARNT, and the transcriptional activation of several xenobiotic-metabolizing enzymes, including CYP1A1. In addition, accumulating evidence has demonstrated that AhR plays an important role in the differentiation of various cells. For example, the silencing of AhR gene expression promotes the differentiation of stage 3 innate lymphoid cells to natural killer cells, and AhR shRNA promotes N-myc proto-oncogene protein expression and inhibits neuroblastoma cell differentiation [30, 31]. In addition, the AhR agonists TCDD and Bap could inhibit OC differentiation. TCDD decreased the number of TRACP+ multinucleated cells, accompanied by corresponding decreases in the number of F-actin rings and the area of resorption; Bap inhibited OC differentiation and bone resorption, and these mechanisms could be mediated through crosstalk between RANKL and the AhR signaling cascade, which compete for NF-κB, a common transcription factor for both pathways [32]. These data indicate that AhR is an important regulator for OC differentiation.
Notably, abundant isoquinoline alkaloids regulate the activation of AhR. For example, berberine activates AhR in human (HepG2) and rat hepatoma cells stably transfected with a dioxin responsive element fused to the luciferase gene (H4IIE.luc), improving the mRNA expression of CYP1A1 but eliminating its catalytic activity, displaying dual-directional regulation [17]. Protopine and allocryptopine obviously up-regulate the mRNA expression of CYP1A1, but only show a slight effect on the activation of AhR [18], and chelerythrine significantly inhibits the dioxin-induced activation of AhR in rat hepatoma cells. In addition, other isoquinoline alkaloids, such as arecoline, rutaecarpine, and harmine, also exhibit regulatory effects on AhR [19]. Based on these findings, in the present study, we investigated the relationship between the activation of AhR and inhibition of OC differentiation through NOR. RANKL-stimulated RAW264.7 cells were adopted to establish a model of OC differentiation. NOR treatment significantly reduced the number of TRAP-positive multinuclear OCs, and AhR antagonists Res and α-NF nearly completely reversed the inhibitory effect of NOR, suggesting that AhR might play an important role in the inhibition of osteoclastogenesis through NOR.
The action mediated through AhR involves a multi-step, ligand-induced signal transduction process. The binding of a ligand to AhR triggers the dissociation of AhR with associated proteins, including heat shock protein 90 and the immunophilin-type chaperone, AhR-interacting protein (AIP). The activated AhR translocates into the nucleus, dimerizes with ARNT, and activates the transcription of target genes through binding to specific enhancer sequences in the regulatory regions of the genes [7, 8]. In the present study, the data obtained from molecular docking showed that NOR could stably associate with AhR. Furthermore, the nuclear translocation of AhR, accumulation of the AhR-ARNT complex, activation of the AhR-mediated reporter gene and mRNA expression of CYP1A1 were all up-regulated after NOR treatment. These data indicated that NOR is an activator of AhR.
The NF-κB family regulates multiple physiological and pathological responses, such as cell differentiation, tissue development, response to environmental stress, inflammatory response, and cell apoptosis. This protein family comprises five proteins: p65/RelA, c-Rel, RelB, NF-κB1 (p105/p50), and NF-κB2 (p100/p52). Among them, NF-κB-p65 plays an important role in the biological behavior of cells, including the differentiation of OCs. Mice lacking NF-κB-p65 show global embryonic lethality, but radiation chimeras with p65-deficient bone marrow are viable. These p65-/- chimeras have fewer OCs and a significantly repressed osteoclastogenic response to RANKL [33]. Notably, NF-κB-p65 is a common transcription factor for both RANKL and AhR signaling pathways, which compete for p65. After activation through ligands, AhR is dissociated from HSP90 and exists in a free form. Subsequently, AhR regulates the activation of NF-κB, STATs and other signal transduction pathways, and controls the migration, survival, and differentiation of cells. AhR associates with NF-κB-p65, inhibits the nuclear translocation of NF-κB-p65, and mediates the inhibition of Bap in OC differentiation [33-35]. In the present study, both Res and α-NF were shown to markedly interfere with the inhibition of NF-κB-p65 nuclear translocation through NOR. Moreover, NOR obviously up-regulated the accumulation of the AhR-NF-κB-p65 complex, indicating that the inhibition of NF-κB-p65 nuclear translocation by NOR might be mediated through the accumulation of the AhR-NF-κB-p65 complex.
Hypoxia is an important regulator of bone biology and stimulates the differentiation of OCs from monocytic precursors. HIF is a key pro-tumorigenic transcription factor mediating pathways of hypoxia-inducible gene expression. HIF-1 transcription factor comprises an oxygen-regulated alpha subunit (HIF-1α) and a constitutively expressed beta subunit (HIF-1β, ARNT). Both AhR and HIF-1α belong to members of the class I bHLH/PAS (basic helix-loop-helix/PER-ARNT-SIM) protein family. In general, bHLH proteins are important regulators of gene expression. To perform their respective functions, bHLH proteins must form heterodimers with other bHLH proteins. AhR and HIF-1α form obligate heterodimers with a member of the class II bHLH/PAS protein family to form a functional DNA binding complex. The different domains in bHLH/PAS proteins regulate various functions. The PAS A and PAS B domains act as secondary dimerization domains and interact with ARNT. Therefore, ARNT acts as a pivotal mediator between AhR and HIF signaling pathways. In addition, the oral administration of the HIF-1α inhibitor could protect ovariectomized mice from OC activation and bone loss [35]. HIF-1α stimulates the transcription of multiple genes, including angiogenic VEGF, an important growth factor involved in tumor angiogenesis. The HIF-1α-VEGF pathway has been implicated in the development of multiple tumors and OC differentiation [36]. Hypoxia induces OC differentiation, and the HIF-1α-mediated induction of VEGF makes important contributions, including the recruitment of monocytes and OCs, the promotion of monocyte-OC differentiation and support of OCs survival and activity. In the present study, NOR did not affect the expression of HIF-1α, but dramatically inhibited the expression of VEGF. In addition, the accumulation of the ARNT-HIF-1α complex was significantly down-regulated through NOR treatment. These data indicated that NOR likely suppresses VEGF expression through the down-regulation of ARNT-HIF-1α complex accumulation, resulting in the inhibition of OC differentiation.
OC-mediated subchondral bone resorption has been implicated in joint bone destruction in RA. We next verified whether the suppression of NOR on joint bone destruction in RA was dependent on AhR. In CIA model rats, the obvious joint destruction could be observed. NOR and Lef (an identified agonist of AhR) could markedly attenuate bone destruction and reduce the number of OCs in the joints of rats, accompanied by the up-regulation of CYP1A1 expression and the down-regulation of VEGF expression in the synovium. Interestingly, the effect of NOR nearly disappeared when administered in combination with α-NF.
In conclusion, NOR suppresses the differentiation of OCs and consequent joint bone impairment in RA in an AhR-dependent manner and might be an agonist of AhR. NOR functions through the inhibition of NF-κB and HIF-1α signaling pathways.
Acknowledgements
This work was financially supported through grants from the Fundamental Research Funds for the Central Universities (NO. PY2014ZY0007), the Natural Science Foundation of Jiangsu Province of China (NO. BK20140662), the University Innovation Research and Training Program of China Pharmaceutical University (G14067), and the Priority Academic Program Development of Jiangsu Higher Education Institutions, and it was partially supported through the Program of the National Natural Science Foundation of China (NO. 81373426).
Conflict of interest
There are no conflicts of interest associated with this work for any of the authors.
References
1. Jung SM, Kim KW, Yang CW. et al. Cytokine-Mediated Bone Destruction in Rheumatoid Arthritis. J Immunol Res. 2014;2014:263625
2. Ngian GS. Rheumatoid arthritis. Aus Fam Physician. 2010;39:626-8
3. Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin North Am. 2010;36:385-404
4. Pettit AR, Walsh NC, Manning C. et al. RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology (Oxford). 2006;45:1068-76
5. Schett G. Cells of the synovium in rheumatoid arthritis. OCs. Arthritis Res Ther. 2007;9:203
6. Lacey DL Timms E, Tan HL. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165-76
7. Kong YY, Yoshida H, Sarosi I. et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315-23
8. Wang C, Dai Y, Yang J. et al. Treatment with total alkaloids from Radix Linderae reduces inflammation and joint destruction in type II collagen-induced model for rheumatoid arthritis. J Ethnopharmacol. 2007;111:322-8
9. Luo Y, Liu M, Xia Y. et al. Therapeutic effect of norisoboldine, an alkaloid isolated from Radix Linderae, on collagen-induced arthritis in mice. Phytomedicine. 2010;17:726-31
10. Wei ZF, Wang FY, Song J. et al. Norisoboldine inhibits the production of interleukin-6 in fibroblast-like synoviocytes from adjuvant arthritis rats through PKC/MAPK/NF-κB-p65/CREB pathways. J Cell Biochem. 2012;113:2785-95
11. Wei ZF, Tong B, Xia YF. et al. Norisoboldine suppresses osteoclast differentiation through preventing the accumulation of TRAF6-TAK1 complex and activation of MAPKs/NF-κB/c-Fos/NFATc1 Pathways. PLoS One. 2013;8:e59171
12. Julliard W, Fechner JH, Mezrich JD. The aryl hydrocarbon receptor meets immunology: friend or foe? A little of both. Front Immunol. 2014;5:458
13. Gasiewicz TA, Singh KP, Bennett JA. The Ah receptor in stem cell cycling, regulation, and quiescence. Ann N Y Acad Sci. 2014;1310:44-50
14. Voronov I, Li K, Tenenbaum HC, Manolson MF. Benzo[a]pyrene inhibits osteoclastogenesis by affecting RANKL-induced activation of NF-kappaB. Biochem Pharmacol. 2008;75:2034-2044
15. Voronov I, Heersche JN, Casper RF. et al. Inhibition of osteoclast differentiation by polycyclic aryl hydrocarbons is dependent on cell density and RANKL concentration. Biochem Pharmacol. 2005;70:300-7
16. Zhou Y, Tung HY, Tsai YM. et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood. 2013;121:3195-204
17. Vrzal R, Zdarilová A, Ulrichová J. et al. Activation of the aryl hydrocarbon receptor by berberine in HepG2 and H4IIE cells: Biphasic effect on CYP1A1. Biochem Pharmacol. 2005;70:25-36
18. Vrba J, Vrublova E, Modriansky M. et al. Protopine and allocryptopine increase mRNA levels of cytochromes P450 1A in human hepatocytes and HepG2 cells independently of AhR. Toxicol Lett. 2011;203:135-41
19. Dvorák Z, Sovadinová I, Bláha L. et al. Quaternary benzo[c]phenathridine alkaloids sanguinarine and chelerythrine do not affect transcriptional activity of aryl hydrocarbon receptor: analyses in rat hepatoma cell line H4IIE.luc. Food Chem Toxicol. 2006;44:1466-73
20. Chou GX, Norio N, Ma CM. et al. Isoquinoline alkaloids from Lindera aggregata. Chin J Nat Med. 2005;5:272-5
21. Bisson WH, Koch DC, O'Donnell EF. et al. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem. 2009;52:5635-41
22. Wang T, Leng DD, Gao FF. et al. A LC-ESI-MS method for the simultaneous determination of madecassoside and its metabolite madecassic acid in rat plasma: comparison pharmacokinetics in normal and collagen-induced arthritic rats. Chin J Nat Med. 2014;12:943-51
23. Liu D, Li P, Song S. et al. Pro-apoptotic effect of epigallo-catechin-3-gallate on B lymphocytes through BAFF/PI3K/Akt/mTOR signaling in rats with collagen-induced arthritis. Eur J Pharmacol. 2012;690:214-25
24. Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275:8432-8
25. Øvrevik J, Låg M, Lecureur V. et al. AhR and Arnt differentially regulate NF-κB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun Signal. 2014;12:48
26. Bhaskara VK, Mohanam I, Gujrati M. et al. Intermittent hypoxia effect on osteoclastogenesis stimulated by neuroblastoma cells. PLoS One. 2014;9:e105555
27. Knowles HJ, Cleton-Jansen AM, Korsching E. et al. Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: role of angiopoietin-like 4. FASEB J. 2010;24:4648-59
28. Vorrink SU, Domann FE. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1α signaling node. Chem Biol Interact. 2014;218:82-8
29. Wei ZF, Jiao XL, Wang T. et al. Norisoboldine alleviates joint destruction in rats with adjuvant-induced arthritis by reducing RANKL, IL-6, PGE2, and MMP-13 expression. Acta Pharmacol Sin. 2013;34:403-13
30. Hughes T, Briercheck EL, Freud AG. et al. The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep. 2014;8:150-62
31. Wu PY, Liao YF, Juan HF. et al. Aryl hydrocarbon receptor downregulates MYCN expression and promotes cell differentiation of neuroblastoma. PLoS One. 2014;9:e88795
32. Ohtake F, Fujii-Kuriyama Y, Kawajiri K. et al. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J Steroid Biochem Mol Biol. 2011;127:102-7
33. Novack DV. Role of NF-κB in the skeleton. Cell Res 2011; 21: 1669-82. 34. Ohtake F, Fujii-Kuriyama Y, Kato S. AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem Pharmacol. 2009;77:474-84
34. Korkalainen M, Kallio E, Olkku A. et al. Dioxins interfere with differentiation of osteoblasts and osteoclasts. Bone. 2009;44:1134-42
35. Miyauchi Y, Sato Y, Kobayashi T. et al. HIF1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci U S A. 2013;110:16568-73
36. Knowles HJ, Athanasou NA. Hypoxia-inducible factor is expressed in giant cell tumour of bone and mediates paracrine effects of hypoxia on monocyte-osteoclast differentiation via induction of VEGF. J Pathol. 2008;215:56-66
Author contact
![]() Corresponding author: Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, China. Tel.: +86 25 83271400; Fax: +86 25 85301528. E-mail address: yuedaicpucom
Corresponding author: Department of Pharmacology of Chinese Materia Medica, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, China. Tel.: +86 25 83271400; Fax: +86 25 85301528. E-mail address: yuedaicpucom