Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Taxane Chemotherapy and...
The Role of AR Axis in CRPC
ABCG2-Mediated Chemoresistance
PCSC Plays a Pivotal Role in...
Key Signaling Involved in PCSC...
New Therapeutic Strategies: PCSC...
Conclusion Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(10):1160-1170. doi:10.7150/ijbs.11439 This issue Cite
Review
Insights into Chemoresistance of Prostate Cancer
Wei Zhang1#, Yan Meng2#, Na Liu3, Xiao-Fei Wen4, Tao Yang2 ![]()
1. Department of Pharmacology, School of Basic Medicine, Hebei University of Chinese Medicine, Shijiazhuang 050200, China
2. Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China
3. Department of Nephrology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China
4. Department of Urology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China
# These two authors contributed equally to this work.
Received 2014-12-26; Accepted 2015-6-29; Published 2015-8-1
Abstract

Prostate cancer (PCa) remains the most prevalent malignancy among males in the western world. Though hormonal therapies through chemical or surgical castration have been proposed many years ago, heretofore, such mainstay for the treatment on advanced PCa has not fundamentally changed. These therapeutic responses are temporary and most cases will eventually undergo PCa recurrence and metastasis, or even progress to castration-resistant prostate cancer (CRPC) due to persistent development of drug resistance. Prostate cancer stem cells (PCSCs) are a small population of cells, which possess unlimited self-renewal capacities, and can regenerate tumorigenic progenies, and play an essential role in PCa therapy resistance, metastasis and recurrence. Nowadays advanced progresses have been made in understanding of PCSC properties, roles of androgen receptor signaling and ATP-binding cassette sub-family G member 2 (ABCG2), as well as roles of genomic non-coding microRNAs and key signaling pathways, which have led to the development of novel therapies which are active against chemoresistant PCa and CRPC. Based on these progresses, this review is dedicated to address mechanisms underlying PCa chemoresistance, unveil crosstalks among pivotal signaling pathways, explore novel biotherapeutic agents, and elaborate functional properties and specific roles of chemoresistant PCSCs, which may act as a promising target for novel therapies against chemoresistant PCa.
Keywords: prostate cancer, chemoresistance, cancer stem cell, androgen receptor, cancer recurrence
Introduction
Prostate cancer (PCa) is the most prevalent solid tumor and a leading cause of cancer-related deaths among males in the United States, with an estimated 238,590 and 233,000 new cases diagnosed in 2013 and 2014 respectively, and 29,720 deaths in 2013 (1, 2). Also in China, the morbidity and mortality of PCa have increased dramatically (3). Due to technical improvement in screening and diagnostics, early surgical resection and introduction of novel anti-neoplasm agents, the incidence of PCa has been declining approximately 2.4% annually from 2002 to 2011. The mortality of PCa has dropped 3.3% each year over the last decade, with elevated overall survival rate from 66% in the 1970s to 99.6% nowadays (2, 4). Unfortunately, after routine treatment for 2-3 years, most patients will suffer castration-resistant prostate cancer (CRPC), which, as the lethal form of PCa, is often incurable nowadays, and remain the tough challenge in public health field (5, 6).
PCa cells depend on hormonal stimulation for proliferation. Androgen receptor (AR) is a nuclear hormone receptor, the discovery of which has allowed the identification of a subset of patients who are extremely sensitive to androgen deprivation therapy (ADT) (6, 7). However, after treatment for 2-3 years, most cases will eventually fail ADT, although some patients will respond initially to secondary hormonal manipulations before the unavoidable formation of CRPC (5-7). Instead of ADT, systemic chemotherapy with taxanes was applied to CRPC patients with metastasis as the treatment of choice. Mitoxantrone was the first chemotherapeutic agent approved by the US Food and Drug Administration (FDA) for CRPC treatment (8). In the late 1990s, docetaxel was prospectively evaluated in two phase 3 clinical trials. On this basis, docetaxel administered every 3 weeks with prednisone was approved as standard front-line medication for CRPC by FDA in 2004 and the European Medicines Agency (EMA) in 2005 (9-11). Although docetaxel prolonged survival of CRPC patients, the overall benefit was modest, since most patients would experience disease progression in around 7 months, followed by adverse effects including diarrhea, neutropenia and docetaxel resistance (9). Accordingly, on the premise of understanding mechanisms underlying CRPC, there are mounting demands for exploring alternative chemotherapeutic agents, or developing novel targeted drugs, or combinatorial regimens since then.
Tumor cell heterogeneity has been appreciated for decades. Like many other cancer types, PCa is heterogeneous (Fig.1). For example, most early-stage PCa tissues are mainly composed of cells positive for prostate-specific antigen (PSA) and AR; whereas advanced PCa mainly consists of poorly differentiated or undifferentiated cells which are largely negative for PSA and AR expression (12-14). To fully dissect heterogeneity of tumor cells, the xenograft models with human cancer cell grafts in mice were established, which suggest that cells in many human cancers are organized hierarchically, and only a small subpopulation is endowed with tumor-initiating and long term tumor-propagating activities. Such cells possess many phenotypic and functional properties associated with normal stem cells and are often termed cancer stem cells (CSCs), which possess unlimited self-renewal ability and can regenerate tumorigenic progenies, and have been demonstrated to play a pivotal role in tumor initiation, progression, metastasis and therapy resistance (15-17). Although significant progresses have been made in the field of PCa treatment, mechanisms underlying PCa chemoresistance and CRPC formation are incompletely understood (15).
Illustration of hypothetical heterogeneity of prostate cancer cells. After androgen deprivation therapy (ADT), the volume of prostate cancer shrinks, with highly differentiated PSA+ cells dying, during which process, more quiescent PSA-/lo cells (including miscellaneous subpopulations as indicated) survive ADT and become cells-of-origin for castration-resistant prostate cancer (CRPC).
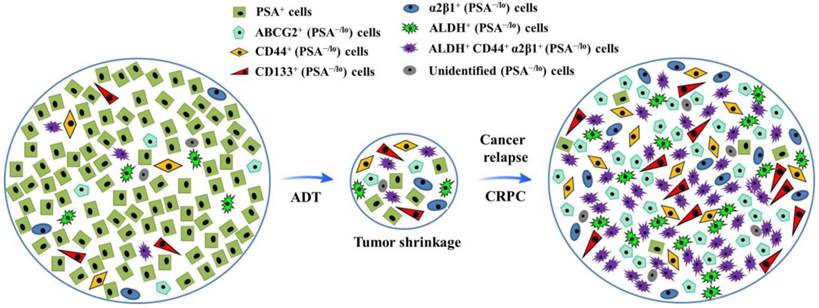
Consequently, elucidating the phenotypic and genetic properties as well as molecular regulators of prostate CSCs (PCSCs) may bring out novel insights into tracking of cells-or-origin for CRPC and shed light on CSC-based mechanisms underlying PCa chemoresistance, thus laying a foundation for development of novel therapeutics targeting PCSCs (17,18 ). Together, a complex series of molecular events, including the activation of aberrant AR, and/or ATP-binding cassette (ABC) family of membrane transporters, inactivation of tumor suppressor genes, evasion of apoptosis, cross-talk among key signaling pathways and miscellaneous microRNAs, have led to the development of chemoresistance (19, 20). This review will summarize roles of AR, ABC sub-family G member 2 (ABCG2), key signaling pathways, microRNAs and PCSCs in PCa chemoresistance development and discuss novel treatment strategies with an emphasis on PCSC-targeted therapies.
Taxane Chemotherapy and Resistance
Upon the inevitable progression to CRPC with metastasis, chemotherapy with taxanes has been the main cure. Taxanes are microtubule-targeted chemotherapeutic agents which can stimulate microtubule depolymerization (21, 22). Based on two landmark randomized control trials, TAX327 and SWOG 99-16, docetaxel with prednisone is currently recommended treatment for CRPC, as optimized by the SWOG 99-16 trial in which docetaxel administered every 3 weeks in combination with estramustine (an AR antagonist) was compared with mitoxantrone plus prednisone, and the TAX327 trial which compared docetaxel plus prednisone to mitoxantrone with prednisone (9-11). The median overall survival time in above trials was 17.5 months vs. 15.6 months (p = 0.02) and 19.2 months vs 16.3 months (p = 0.009) respectively, demonstrating the survival advantage of docetaxel (9, 10). However, the overall benefit was modest. Docetaxel resistance develops afterwards and disease progresses in approximately 7.5 months. Many reports have suggested that multiple factors contribute to the development of PCa chemoresistance, such as ABCG2 activation, overexpression of P-glycoprotein and multidrug resistance gene 1, mutational alterations in the tubulin gene, and inhibition of apoptosis (23, 24). Since PCa resistance and multidrug resistance genes have attracted so much attention, this review will be mainly focused on deciphering of mechanisms underlying CRPC development.
The Role of AR Axis in CRPC
AR is a nuclear hormone receptor consisting of eight exons which encode four functional domains: the NH2-terminal domain (NTD), DNA-binding domain (DBD), the hinge region and ligand-binding domain (LBD) (7). The NTD accounts for majority of AR transcriptional activities, and the LBD binds androgens and transfers AR to the nucleus. The DBD, composed of two zinc fingers, is critical to DNA recognition and binding; whereas the hinge domain regulates the translocation of AR into the nucleus (25-27). PCa cells depend on androgens for growth and survival via AR axis, and the roles of AR in maintenance of prostate tissue lineage, as well as in PCa initiation and development, are the basis for the effectiveness of ADT (20, 25). Since Huggins and Hodges first demonstrated that PCa was hormone responsive and castration could be used as an effective therapy for patients in 1941, efforts focusing on ablating AR signaling have never been halted (28). Unfortunately, although surgical or chemical castration is highly effective in shrinking tumor burden (Fig. 1), decreasing serum PSA levels, and improving survival rate during initial treatment, PCa recurs after a median duration of response for 12 - 24 months and gradually develops into CRPC (29, 30). Previously, PCa with resistance to hormonal manipulation has been variously termed hormone refractory/resistant PCa (HRPC) or endocrine resistant PCa (ERPC), but now CRPC is used most widely, whose standard definition includes the following criteria: (i) Serum levels of testosterone after castration is less than 1.7 nM (with normal level at 10 - 35 nM); (ii) Three consecutive rises of PSA, 2 weeks apart, resulting in two 50% increases over the nadir; (iii) Anti-androgen withdrawal for at least 4 weeks; (iv) PSA progression, despite secondary hormonal manipulations; (v) Metastasis (31, 32). One of the key characteristics of CRPC is the ability to survive low level of androgen. It was noted that CRPC cells still rely on AR signaling, although the circulating level of testosterone after castration is less than 1.7 nM (31). Many studies have suggested that CRPC cells express mutated AR, which exhibited enhancement in both gene expression level and functional sensitivity. In clinical settings, AR amplification, promiscuity, and splice variant isoforms were more frequently observed in PCa previously treated with ADT, as compared to primary PCa without any treatment (7, 33, 34). Therefore, it is generally believed that most CRPC cases are not truly hormone refractory, in which AR transcription is aberrantly re-activated despite low serum level of androgen after castration (7, 20, 35). On the other hand, mutations involving the AR gene were found in up to around 44% of CRPC cases (36). Heretofore, hundreds of types of mutations in the AR have been identified, though 90% are non-sense and mis-sense mutations (37). AR mutations mainly occur in the LBD and NTD, with only 7% mutations in the DBD and 2% in the hinge region. Mutations in the LBD, which were demonstrated to increase the sensitivity and decrease the specificity of the ligand binding, are clinically significant (25, 38, 39). For example, T877A mutation, the most common point mutation as identified, allows activation of the receptor by the progestin, estrogen and hydroxyflutamide, which are anti-androgens (39). A withdrawal response can be also observed in some patients with T877A mutation, since discontinuous administration of anti-androgen bicalutamide could lead to a PSA decline (40, 41), further suggesting AR mutations may be the reason underlying acquired androgen-like agonistic properties exerted by continuous administration of anti-androgens such as bicalutamide.
However, there was also evidence suggesting CRPC may develop into a genuine androgen-independent tumor which does not express AR, PSA, or other androgen-regulated genes, but exhibits a phenotype of neuroendocrine features (42, 43). These AR-independent tumors are aggressive and most patients survive less than 1 year (29, 42). Recent studies have indicated that AR variants can be induced rapidly in response to ADT, and AR splice variants (AR-Vs) without LBD have been discovered (7, 44, 45). These AR-Vs are independent of ligand stimulation. AR antagonists, which target the LBD, have no effect on them. For instance, AR-V7, like other AR-Vs lacking an LBD, was shown to bind DNA through its nuclear localization and regulate a set of genes that target mitosis and therefore promote PCa progression even in the presence of AR antagonists (38, 45, 46). Given all that, ligand-independent activation of the AR signaling is a critical mechanism underlying PCa progression and CRPC development. Accordingly, further investigations are required to differentiate these distinctive types of CRPC and clarify corresponding underlying mechanisms, which may involve differential levels of AR amplification, AR mutation, overexpression of ligand-less AR splice isoforms and increased AR-independent and survival signaling (40, 41, 44-46).
ABCG2-Mediated Chemoresistance
ABCG2 is a member of the ABC transporter family and classified as the second member of the G subfamily, which was first recognized in a breast cancer cell line resistant to doxorubicin (47-49). The protein, known to be overexpressed in many cancer types, consists of 655 amino acids with six trans-membrane helices and one nucleotide binding domain (NBD). ABCG2 is a half-transporter, and thus requires at least two NBDs to function as an efflux pump (48, 49). Preferentially localized on the internal surface of organs involved in drug transport and metabolism, such as the apical surface of enterocytes (50), the blood-brain barrier (51), the luminal surface of liver canaliculi and kidney tubules (52), ABCG2 functions as an efflux pump for a wide variety of xenobiotics, including many approved therapeutic agents (53).
Structural and functional studies have provided valuable insights into the molecular mechanisms of ABCG2-mediated chemoresistance. Through comparison of the cloning of ABCG2 cDNAs between drug-selected cells and normal cells (MCF7/AdVp3000 cell lines), functional variations based on the amino acid substitutions have been identified. ABCG2 protein in drug-selected cells belongs to a mutant form, and distinctive mutation has been found at amino acid position 482 (54). MCF7/AdVp3000 cells, highly resistant to both mitoxantrone and doxorubicin, have been demonstrated to express R482T and R482G variants respectively. Anthracycline resistance and a rhodamine efflux capacity are also characteristic phenotypes of these two ABCG2-overexpressing cell lines (54, 55). Subsequent structural studies using three-dimensional homology modeling of ABCG2 suggested that the transmembrane domain of ABCG2 functions as drug-recognition interface (56, 57) and R482 is located in the central cavity of the protein (54, 58). When it comes to PCa, the drug efflux activity of ABCG2 can be influenced by various mutations, which will necessarily affect the clinical efficacy of ABCG2-transportable anti-androgens such as bicalutamide (59). Numerous germ-line mutations in the ABCG2 gene have been found, such as C421A (60-62). It was hypothesized that PCa patients carrying the C421A single nucleotide polymorphism may have decreased survival as compared with those with the wild-type ABCG2 genotype. The C421A polymorphism was also demonstrated to play a crucial role in the determination of the pharmacokinetics and plasma concentrations of bicalutamide (63). However, in contrast to C421A, the C34G polymorphism does not influence the pharmacokinetics of bicalutamide (63, 64). Altogether, primary structural variations of ABCG2 were thought to associate with its drug-resistance function, which may be another critical mechanism underlying PCa resistance to chemotherapeutics and ADT.
PCSC Plays a Pivotal Role in CRPC Development
The existence of a CSC subpopulation was first proved in leukemia by Bonnet and Dick in 1997 (65). Since then, CSCs have been identified in a number of solid tumors, including breast cancer (66), glioblastoma multiforme (67) and also PCa (12, 68, 69). Multiple CSC-based mechanisms may be involved in PCa chemoresistance, such as drug-efflux pumps, enhanced DNA repair efficiency, detoxification enzyme expression and quiescence. Heretofore, several populations of PCSCs have been reported (12, 70). Tang's group, in terms of PSA expression level, has sorted bulk PCa cells, which were derived from either in vitro cell line cultures or human PCa cell line xenografts or primary PCa samples, into PSA-negative/low expressing (PSA-/lo) and PSA+ subpopulations (12). PSA-/lo cells were shown to fulfill all criteria defining PCSCs (70, 71). As compared to PSA+ cells which are highly proliferative contributing to tumor mass, PSA-/lo cells are relatively quiescent, remarkably tumorigenic and metastatic. This internally slow cycling rate of PSA-/lo cells was thought to provide inherent defense against chemotherapeutic agents, which are anti-proliferative and only can effectively target fast dividing PSA+ cells (12, 72). Furthermore, whole-genome transcriptome profiling has revealed distinct gene expression patterns in PSA-/lo cells, which could overexpress many genes involved in anti-stress responses, including detoxification-related genes (metallothioneins, GSTT2), hypoxia-responsive genes (THBS1, PLAU, APLN), p53 signaling components (ZBTB7A, PSME3), and DNA damage sensing/repair genes (MSH6, XPA, REV1), suggesting that the PSA-/lo cells are resistant to not only ADT and chemotherapeutic agents but also other stresses including radiotherapy (12, 73). Specifically, a subset of ALDH+CD44+α2β1+ cells from the PSA-/lo cells were shown to be even more tumorigenic and could regenerate serially transplantable tumors in fully castrated hosts (12). Additionally, cytokeratin (CK) 18 and CK19-negative (CK18-/CK19-) cells in both docetaxel-resistant PCa cell lines and primary as well as metastatic PCa tissue samples were demonstrated to survive docetaxel exposure and exhibit active chemoresistant ability (69). Another study indicates that the expression level of CD166, a cell surface marker, is much higher in human CRPC samples (74). Together, these data suggest that PCSCs, or PCSC pool which may contain miscellaneous subsets of chemoresistant cells, may function as cells-of-origin for CRPC, and mediate, at least partially, the chemoresistance of PCa.
Recently, PCSCs derived from human primary PCa explants were functionally characterized and analyzed for ABCG2-based drug resistance, which indicated that PCSC-derived tumorsphere cultures positive for ABCG2 transporter, CD133, CD44, CK5 and negative for AR and PSA showed a higher clonogenic capacity in soft agar, and a higher survival rate in the presence of ABCG2 substrate drugs, as compared to control PCa cells (75). Moreover, this high drug resistance declined dramatically once PCSC-derived tumorspheres were exposed to a selective inhibitor of ABCG2, suggesting drug resistance of PCSCs was attributable, or at least partially, to robust expression of ABCG2 (75, 76). On the other hand, certain co-factors are required for ABCG2 to fulfill its functions. A 44-kDa serine/threonine kinase termed Pim-1L was found to protect PCa cells from apoptosis and mediate resistance to chemotherapeutic drugs in PCa (77). Subsequent studies indicated that Pim-1L was co-localized with ABCG2 on the plasma membrane, and could induce phosphorylation of ABCG2 at threonine 362. Pim-1L knock-down in chemoresistant PCa cells was shown to resensitize resistant cells to chemotherapeutic drugs. Moreover, the plasma membrane localization and drug-resistant activity of ABCG2 were compromised by T362A mutation, suggesting ABCG2 phosphorylation induced by Pim-1L is essential for ABCG2 functionality (78).
Currently, ABCG2 is also thought to be a CSC marker. Side population (SP), characterized as a subpopulation of cells with self-renewal activity, tumorigenicity and invasiveness, as well as the capability to express CSC markers and stemness genes, is thought to function as CSCs in miscellaneous types of cancer. SP cells can exclude Hoechst 33342 and express the ABCG2 gene, which was considered responsible for the Hoechst 33342 dye-efflux activity of SP cells (79). Kruger et al demonstrated that SP cells from two murine carcinoma cell lines exhibited upregulation of ABCG2 and stem cell markers Wnt-1 and Sca-1, increased efflux of chemotherapeutic agents and augmented chemoresistance, and enhanced ability to regenerate tumors in vivo, as compared to parental cell lines (80). Also, the presence of ABCG2 expression in hepatocellular carcinoma cells appeared to be a critical indicator of self-renewal and differentiation capacity, since ABCG2+ cells could undergo asymmetric cell division and regenerate both ABCG2+ and ABCG2- cells; whereas ABCG2- cells could only derive cells of the same phenotype, further suggesting ABCG2 can be not only the molecular determinant for the SP cell phenotype but also a universal stem cell marker (81). ABCG2 expression in the prostate was found to locate in both the epithelium (82) and endothelium (50, 83). The SP of the prostate has been previously isolated and characterized as integrin α2+ population which contains a subpopulation of quiescent (~12%) cells (84, 85). Immunohistochemical analysis of both normal and cancerous ABCG2+ cells shows that this subset lacks the protein expression of AR, which was thought to protect ABCG2+ cells in PCa from ADT (86). Meanwhile, ABCG2+ PCSCs were found to conduct constitutive ABCG2-mediated efflux of androgen, thus protecting PCa cells from androgen deprivation to some extent and providing the nidus for recurrent PCa (86).
Key Signaling Involved in PCSC Development and CRPC Formation
Notch signaling is required for embryonic and postnatal prostatic growth and regeneration after castration and hormonal replacement as well (87, 88). In highly metastatic PCa and CRPC, down-regulation of Notch1 and its ligand Jagged1 was demonstrated to inhibit PCa development and metastasis and to induce cell apoptosis in vitro, which process was achieved through inactivation of AKT, mTOR and NF-κB signaling pathways (89, 90). However, the extent to which Notch, PI3K/AKT and RAS/MAPK signaling contribute towards a dynamic equilibrium remains to be elucidated. Development to CRPC or chemoresistant PCa is shown to be associated with elevated expression of PI3K/AKT, RAS/MAPK and STAT3 signaling, which are supposed to promote PCSC self-renewal and tumorigenesis (91). Recently, a reciprocal feedback loop is found to function between PI3K/AKT and AR signaling (92, 93). Oncogenic transformation of the prostate by autonomous PI3K/AKT signal activation arises in the absence of epithelial AR expression or when androgen has been withdrawn (93), confirming that ADT may not be able to eradicate PCSCs which were noted to display activated PI3K/AKT signaling and survive in an androgen-independent status. On the other hand, ectopic expression of h-RAS (T35S) in androgen-dependent PCa cells (LNCaP) can maintain MAPK signal activation under serum-free conditions and increase their tumorigenicity in vivo. Over two thirds of these RAS-expressing PCa xenografts were resistant to castration and displayed robust activation of MAPK signaling after castration (94). Consistently, inducible expression of dominant-negative h-RAS (S17N) in the androgen-independent, highly tumorigenic LNCaP cell line (C4-2) inhibited MAPK signaling and restored androgen sensitivity to these cells, resulting in tumor regression in surgically castrated mice (95). Moreover, activation of ERK1/2 is required to mediate RAF-induced AR down-regulation in PCa cells (96), suggesting RAS/MAPK signaling in PCa advancement towards CRPC.
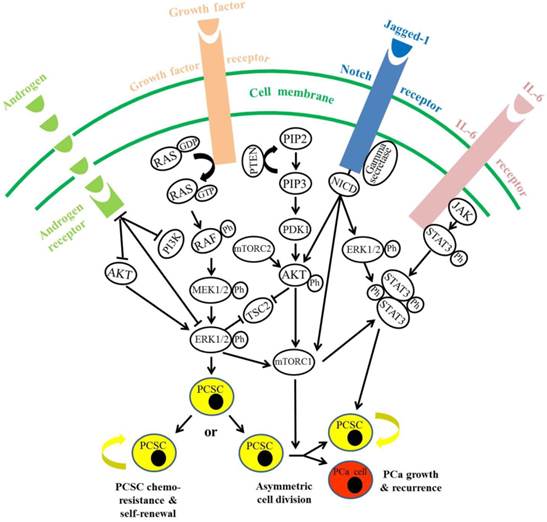
Activated STAT3 is found to be closely related with decreased survival in PCa patients (97), particularly in CRPC patients (98). Elevated serum levels of IL-6, an activator of STAT3, were also confirmed in CRPC and metastatic PCa patients, as compared to patients with benign counterparts (99-101). In human PCa cells, androgen-independent Du145 and PC3 cells exhibit higher STAT3 expression, as compared to androgen-dependent LNCaP cells (102), in which forced expression of STAT3 could promote androgen-independent PCa growth in castrated mice (103). CSCs have been demonstrated to secret IL-6, which could activate STAT3 signaling and induce the formation of a subpopulation of PCSCs from non-stem PCa cells within the tumor (104, 105). This process was regarded as a dynamic equilibrium between CSCs and non-CSCs used to maintain a constant population of tumor-initiating cells in PCa over successive generations or sequential transplantation assays (105, 106). Additionally, phosphorylation of STAT3 on Ser 727, which is thought to boost its transcriptional activity (107), arises in an ERK-dependent manner (108); whereas ERK1/2 was shown to activate mTOR complex 1 directly (109) or through inactivation of tuberous sclerosis factor 2 (110), to promote RAS-dependent mTOR signaling. Moreover, AKT could promote activation of B-RAF via phosphorylation on Ser 445, with ERK1/2 activation requiring androgen deprivation in androgen-dependent PCa cells (111). Down-regulation of AR signaling itself is demonstrated to promote a stemness phenotype and enhance the tumorigenicity of PCa cells via a STAT3 signal-dependent mechanism (112). Together, AR down-regulation is shown to promote activation and subsequent crosstalk among AKT/PI3K, RAS/MAPK and STAT3 signaling pathways to maintain PCSC subpopulation and tumorigenesis, with elevated expression of these signalings in CRPC (Fig. 2).
Androgen, RAS/MAPK, PI3K/AKT, Notch and STAT3 signaling pathways contribute to the regulatory network of prostate cancer stem cell chemoresistance and self-renewal. Through binding with corresponding ligands as indicated, activated RAS/MAPK, PI3K/AKT, Notch and STAT3 signaling pathways can promote prostate cancer stem cell (PCSC) chemoresistance and self-renewal by direct action, cascade activation or crosstalk activation. On the contrary, androgen receptor-mediated signaling plays an inhibitory role in regulating PCSC chemoresistance. ERK1/2, extracellular signal-regulated kinase 1/2; PI3K, phosphatidyl inositol 3-kinase; MEK1/2, dual specificity mitogen-activated protein kinase kinase 1/2; ph, phosphorylation; PIP2, phosphatidyl inositol biphosphate; PIP3, phosphatidyl inositol triphosphate; PDK1, 3-phosphoinositide-dependent protein kinase 1; mTORC1, mammalian target of rapamycin (mTOR) complex 1; mTORC2: mTOR complex 2; NICD: Notch intracellular domain; STAT3: signal transducer and activator of transcription 3; PCSC: prostate cancer stem cell; PCa: prostate cancer.
New Therapeutic Strategies: PCSC as a Promising Therapeutic Target
Comprehension of roles of androgen-mediated signaling in PCa development led to the eventual approval of abiraterone acetate and other potent androgen synthesis inhibitors into the clinic. Despite these advances, the median survival of CRPC patients is no more than 2 years (29). In this regard, it is necessary to find novel and more effective reagents to treat chemoresistant PCa. PCSCs, which mediate PCa chemoresistance and PCa relapse, are therefore taken into account in future clinical trial design. Recent studies have proposed some potential approaches in targeting PCSCs, including blockade of essential PCSC signaling pathways, disruption of PCSC self-renewal machinery, and utilization of newly identified tumor-suppressive microRNAs.
Targeting CSC signaling pathways
Notch and Hedgehog signalings are demonstrated to be a potential target, since these signaling pathways play a role in regulating PCSC self-renewal, proliferation and differentiation (69). Domingo-Domenech et al have demonstrated that the CK18-/CK19- PCa cells are highly resistant to docetaxel treatment as a result of activation of both Hedgehog and Notch signaling, and targeting these two signaling pathways abrogates docetaxel resistance in CK19- cells via inhibition of AKT and Bcl-2 (69). Another work from Dubrovska et al has provided evidence that PCSCs may also be regulated by the PTEN/PI3K/AKT pathways, which suggests that a CD133+/CD44+ population of cells enriched in prostate cancer progenitors possess tumor-initiating potential and undergo preferential activation of the PI3K/AKT signaling pathway (113). Consistently, treatment with PI3K inhibitor LY294002 could reduce sphere formation of CD133/AC141+CD44+ subpopulation derived from Du145 cells (113); whereas the dual PI3K/mTOR inhibitor NVP-BEZ235 decreases CD133/AC141+CD44+ cell population in vivo and partially abolishes tumor formation (113, 114). In human PCa xenograft models used to mimic clinical settings, NVP-BEZ235 administered in combination with chemotherapeutic drug Taxotere is shown to be more effective than chemotherapy alone in decreasing human PCSC subpopulation and inhibiting tumor formation (114), suggesting these studies may hold promise for treating chemoresistant PCa or CRPC with combinatorial strategies in regard to blockade of Notch, PI3K/AKT/mTOR, RAS/ MAPK and/or STAT3 signalings, which could exert multifaceted inhibition over PCSC maintenance and propagation, resulting in a decrease in PCSC burden within PCa parenchyma via disruption of dynamic conversion between non-PCSCs and PCSC populations, and subsequently consecutive self-renewal of PCSCs.
Targeting essential molecules in PCSCs
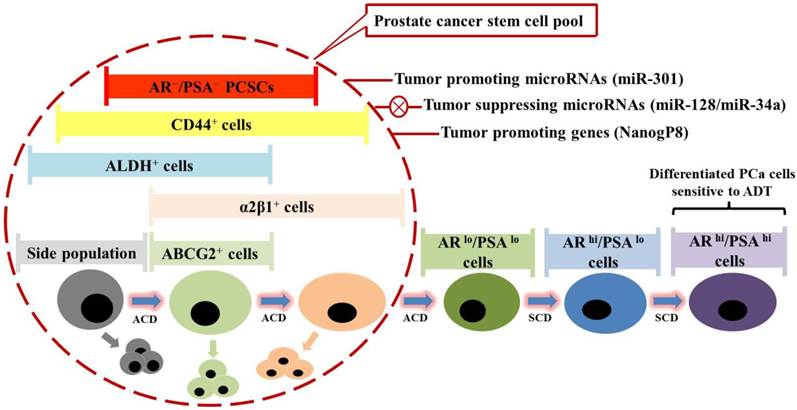
Evidence has implicated roles of several molecules in regulating PCSC activities. Nanog, a homeodomain transcription factor essential for embryonic stem cell self-renewal and pluripotency, was shown to play a critical role in positively regulating PCSC-mediated chemoresistance (115, 116). With Nanog involvement, molecular oncogenesis can be considered as a process of spontaneous cellular reprogramming. Unlike engineered reprogramming to generate induced pluripotent stem cells, aberrant expression of Nanog could elicit oncogenic reprogramming to facilitate acquisition of tumor cell-like adaptability to microenvironmental changes during tumor initiation and progression (115). Due to the dediffentiation and plasticity, Nanog-driven cells are supposed to be intrinsically resistant to chemotherapeutics and enriched upon clinical treatments. More specifically, CSCs are noted to express Nanog mRNA primarily from the NanogP8 locus on chromosome 15q14 (115). NanogP8 mRNA and protein are highly enriched in many CSC populations such as the CD44+ prostate (115) and breast (117) CSC subpopulations, as well as CD133+ brain (118, 119), prostate (115) and ovarian (120) CSC subpopulations, which is thought to be closely associated with worse clinical prognosis in these malignancies. Also, castration-tolerant PCa repopulating cells from early passage xenografts have been demonstrated to highly express NanogP8 (121), which appears to inversely correlate with AR expression (115), suggesting a possible mechanism by which NanogP8 may mediate castration resistance. Consistently, RNA interfere-mediated knockdown of endogenous NanogP8 inhibits PCa development and sensitizes PCa to ADT by restricting PCSC properties including sphere formation capability and clonogenic efficiency (115, 116). By contrast, inducible expression of NanogP8 is sufficient to confer PCSC properties and to promote CRPC development by increasing the frequency of CSCs within cancer parenchyma (116, 122, 123). From a drug development perspective, these studies suggested that NanogP8 may be a pivotal regulatory element in mediating castration resistance, and consequently represent a clinically relevant target for cure of CRPC and late-stage PCa (Fig. 3).
Prostate cancer stem cells (PCSCs) are hypothetically considered a novel therapeutic target in prostate cancer (PCa) treatment. Prostate cancer comprises miscellaneous subpopulations of PCSCs at different stages of differentiation, which do not express androgen receptor (AR) and prostate specific antigen (PSA), but bear specific markers as indicated. PCSCs can undergo asymmetric cell division (ACD) to duplicate themselves and simultaneously give rise to differentiated PCa cells, which are PSA+, and exhibit high proliferation rate via symmetric cell division (SCD) and sensitivity to androgen deprivation therapy (ADT). Recent studies have identified several tumor-promoting microRNAs (e.g. miR-301) and specific genes (e.g. NanogP8), as well as tumor-suppressing microRNAs (e.g. miR-128 and miR-34a), which might be taken into account for future clinical trial design.
Targeting the microRNAs
Recently, microRNAs have also emerged as critical regulators of chemoresistant PCSCs. Multiple microRNAs were identified to be commonly and differentially expressed in miscellaneous PCa stem/progenitor subpopulations such as ABCG2+, CD44+, CD133+ and integrin α2β1+ subpopulations, among which four microRNAs (miR-34a, let-7b, miR-106a and miR-141) were under-expressed, and two microRNAs (miR-301 and miR-452) were over-expressed as compared to the corresponding marker-negative subpopulations (124). Moreover, the expression patterns of miR-34, let-7b, miR-141 and miR-301 were further confirmed in the CD44+ human primary PCa samples. Subsequent studies demonstrate that miR-34a, as a direct target of p53, functions as a potent negative regulator of chemoresistant PCSCs and PCa growth/metastasis by directly repressing CD44 (125). Similarly, let-7 shows similar suppressive effects on PCa development. However, differential mechanisms between let-7 and miR-34a on the cell cycle were found, with miR-34a mainly inducing G1 cell cycle arrest followed by cell senescence and let-7 inducing G2/M arrest (124, 125). Another tumor-suppressing microRNA identified recently, miR-128, was shown to target stem cell regulatory factors including BMI-1, NanogP8 and TGF-β receptor 1, expressions of which vary inversely with miR-128 expression in PCSC subpopulations, with BMI-1 defined as a direct and functionally relevant target of miR-128 (126). Besides, it was reported that miR-99b, miR-205, miR-221 were also significantly down-regulated in PCa (127). However, further evidence is still required to clarify the roles of these 3 microRNAs in regulating chemoresistance and PCSC properties. Taken together, these studies suggested that different microRNAs collaboratively regulate various functional aspects of chemoresistant PCSCs, laying a scientific foundation for developing microRNA-based anti-cancer therapeutic methods (Fig. 3). Meanwhile, microRNAs could also be involved in NanogP8 regulation. The miR-214 was demonstrated to regulate ovarian cancer stem cell (OCSC) properties by targeting p53/NanogP8 axis (120). OCSC population and self-renewal would be enhanced by enforcing expression of miR-214 and decreased by inhibition of miR-214. The miR-214 regulates NanogP8 through p53 which could be directly repressed by miR-214, and reinforced expression of p53 abrogates miR-214-induced OCSC properties, suggesting the critical role of miR-214 in conferring OCSC properties by regulating the p53-NanogP8 axis (120). However, whether miR-214 could regulate chemoresistant PCSCs via p53-NanogP8 axis, and whether miR-214 could be defined as a therapeutic target for CRPC, remain to be elucidated.
Challenges and prospects in PCSC-targeted therapy
Although significant progresses have been made in PCa treatment, mechanisms underlying PCa chemoresistance and CRPC cell origin are incompletely understood, which partially explains why the CSC concept has attracted much attention. The experimental strategies for sorting chemoresistant PCSC subpopulations and subsequent transplantation assays indicate that PCSCs can be a potential therapeutic target. However, the applicability of this approach is limited, considering some important issues. First, cell surface markers on PCSCs may be co-expressed by non-cancer stem cells, and patients with the same malignancy (say, CRPC) may express different markers. Second, since most studies about PCSCs are based on long-term cultured cell lines, xenograft models or mouse PCa models, the facts concerning whether human primary PCa harbors chemoresistant PCSC-like cells are little known. Third, though many advances have been made, the phenotypic and functional properties of PCSCs are not known clearly, and a full understanding of the biology of these cells would be necessary to the development of the more standardized assays to target them. However, despite the existence of many limitations, there is no doubt that the application of PCSC-based studies will help change the understanding of the essence of cancer and supply a new way for treatment against chemoresistant PCa.
Conclusion Remarks
In clinical settings, current chemotherapeutics primarily target either differentiated or proliferating PCa cells, and may not be efficient in eradicating chemoresistant PCSCs which are relatively undifferentiated and mostly quiescent in cell cycle progression. Considering that CSCs and non-CSCs were demonstrated to reciprocally regulate and protect each other (128, 129), it would be beneficial to synchronously target undifferentiated and chemoresistant PCSCs as well as relatively differentiated non-PCSCs, so as to abrogate heterogeneity and plasticity of PCa cells, since many conventional anti-cancer therapies may actually enrich PCSCs possibly as a result of elimination of proliferative PCa cells as well as by inducing dedifferentiation or transdifferentiation of non-PCSCs (130, 131). Nowadays, although multiple new drugs have been officially approved, improvement in overall survival rate of chemoresistant PCa patients remains modest. In addition to androgen signaling components, multiple molecular and cellular targets promise to provide the next generation of advances. The main obstacle is to screen novel therapeutic agents that can specifically target chemoresistant PCSCs, which are supposed to be identifiable by exploring phenotypic traits or genetic signatures. Ultimately, it might be envisioned that PCSC-targeting therapies can be applied in conjunction with the current therapeutics to synergistically prevent PCa chemoresistance and recurrence.
Acknowledgements
The work was financially supported by National Natural Science Foundation of China (grant No. 81372750), Shanghai Natural Science Foundation of China (grant No. 12ZR1425200), Science & Technology Development Fund of Shanghai Pudong (grant No. PKJ2014-Y10), and Scientific Research Foundation for the Returned Overseas Chinese Scholars from State Education Ministry of China (grant No. 2013-1792) to TY.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30
2. Siegel R, Ma J, Zou Z. et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
3. Zhang L, Wu S, Guo LR. et al. Diagnostic strategies and the incidence of prostate cancer: reasons for the low reported incidence of prostate cancer in China. Asian J Androl. 2009;11:9-13
4. DeSantis CE, Lin CC, Mariotto AB. et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252-271
5. Schweizer MT, Zhou XC, Wang H. et al. The influence of prior abiraterone treatment on the clinical activity of docetaxel in men with metastatic castration-resistant prostate cancer. Eur Urol. 2014;66:646-652
6. Malik R, Khan AP, Asangani IA. et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344-352
7. Kahn B, Collazo J, Kyprianou N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int J Biol Sci. 2014;10:588-595
8. Kantoff PW, Halabi S, Conaway M. et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. J Clin Oncol. 1999;17:2506-2513
9. Tannock IF, de Wit R, Berry WR. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502-1512
10. Petrylak DP, Tangen CM, Hussain MH. et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513-1520
11. de Bono JS, Oudard S, Ozguroglu M. et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147-1154
12. Qin J, Liu X, Laffin B. et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556-569
13. Patrawala L, Calhoun-Davis T, Schneider-Broussard R. et al. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67:6796-6805
14. Li H, Chen X, Calhoun-Davis T. et al. PC3 human prostate carcinoma cell holoclones contain self-renewing tumor-initiating cells. Cancer Res. 2008;68:1820-1825
15. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967-2000
16. Shackleton M, Quintana E, Fearon ER. et al. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-829
17. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717-728
18. Nakouzi NA, Cotteret S, Commo F. et al. Targeting CDC25C, PLK1 and CHEK1 to overcome Docetaxel resistance induced by loss of LZTS1 in prostate cancer. Oncotarget. 2014;5:667-678
19. Mousses S, Wagner U, Chen Y. et al. Failure of hormone therapy in prostate cancer involves systematic restoration of androgen responsive genes and activation of rapamycin sensitive signaling. Oncogene. 2001;20:6718-6723
20. Yuan X, Cai C, Chen S. et al. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33:2815-2825
21. Madan RA, Pal SK, Sartor O. et al. Overcoming chemotherapy resistance in prostate cancer. Clin Cancer Res. 2011;17:3892-3902
22. Vrignaud P, Sémiond D, Lejeune P. et al. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19:2973-2983
23. Fitzpatrick JM, de Wit R. Taxane mechanisms of action: potential implications for treatment sequencing in metastatic castration-resistant prostate cancer. Eur Urol. 2014;65:1198-1204
24. Pal SK, Sartor O. Prostate cancer: the best fit for enzalutamide in metastatic prostate cancer. Nat Rev Clin Oncol. 2014;11:504-506
25. Egan A, Dong Y, Zhang H. et al. Castration-resistant prostate cancer: adaptive responses in the androgen axis. Cancer Treat Rev. 2014;40:426-433
26. Tanner TM, Denayer S, Geverts B. et al. A 629RKLKK633 motif in the hinge region controls the androgen receptor at multiple levels. Cell Mol Life Sci. 2010;67:1919-1927
27. Clinckemalie L, Vanderschueren D, Boonen S. et al. The hinge region in androgen receptor control. Mol Cell Endocrinol. 2012;358:1-8
28. Huggins C, Hodges CV. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;168:9-12
29. Beltran H, Beer TM, Carducci MA. et al. New therapies for castration-resistant prostate cancer: efficacy and safety. Eur Urol. 2011;60:279-290
30. Schrader AJ, Boegemann M, Ohlmann CH. et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014;65:30-36
31. Heidenreich A, Aus G, Bolla M. et al. EAU guidelines on prostate cancer. Eur Urol. 2008;53:68-80
32. Pienta KJ, Walia G, Simons JW. et al. Beyond the androgen receptor: new approaches to treating metastatic prostate cancer. Report of the 2013 Prouts Neck Prostate Cancer Meeting. Prostate. 2014;74:314-320
33. Sun F, Chen HG, Li W. et al. Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. J Biol Chem. 2014;289:1529-1539
34. Guo Z, Yang X, Sun F. et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305-2313
35. Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res. 2009;15:3251-3255
36. Beltran H, Yelensky R, Frampton GM. et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920-926
37. Mostaghel EA, Plymate S. New hormonal therapies for castration-resistant prostate cancer. Endocrinol Metab Clin North Am. 2011;40:625-642
38. Shafi AA, Yen AE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol Ther. 2013;140:223-238
39. Schrecengost R, Knudsen KE. Molecular pathogenesis and progression of prostate cancer. Semin Oncol. 2013;40:244-258
40. Yoshida T, Kinoshita H, Segawa T. et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65:9611-9616
41. Sun C, Shi Y, Xu LL. et al. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006;25:3905-3913
42. Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32:65-71
43. Nordin A, Wang W, Welén K. et al. Midkine is associated with neuroendocrine differentiation in castration-resistant prostate cancer. Prostate. 2013;73:657-667
44. Sun S, Sprenger CC, Vessella RL. et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715-2730
45. Hu R, Lu C, Mostaghel EA. et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457-3462
46. Antonarakis ES, Lu C, Wang H. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028-1038
47. Noguchi K, Katayama K, Mitsuhashi J. et al. Functions of the breast cancer resistance protein (BCRP/ABCG2) in chemotherapy. Adv Drug Deliv Rev. 2009;61:26-33
48. Robey RW, Medina-Pérez WY, Nishiyama K. et al. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001;7:145-152
49. Imai Y, Tsukahara S, Asada S. et al. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2-mediated multidrug resistance. Cancer Res. 2004;64:4346-4352
50. Maliepaard M, Scheffer GL, Faneyte IF. et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001;61:3458-3464
51. Aronica E, Gorter JA, Redeker S. et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia. 2005;46:849-857
52. Asashima T, Hori S, Ohtsuki S. et al. ATP-binding cassette transporter G2 mediates the efflux of phototoxins on the luminal membrane of retinal capillary endothelial cells. Pharm Res. 2006;23:1235-1242
53. Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. 2003;22:7340-7358
54. Honjo Y, Hrycyna CA, Yan QW. et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635-6639
55. Volk EL, Farley KM, Wu Y. et al. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002;62:5035-5040
56. Hazai E, Bikádi Z. Homology modeling of breast cancer resistance protein (ABCG2). J Struct Biol. 2008;162:63-74
57. Jones PM, George AM. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc Natl Acad Sci U S A. 2002;99:12639-12644
58. Cai X, Bikadi Z, Ni Z. et al. Role of basic residues within or near the predicted transmembrane helix 2 of the human breast cancer resistance protein in drug transport. J Pharmacol Exp Ther. 2010;333:670-681
59. Colabufo NA, Pagliarulo V, Berardi F. et al. Bicalutamide failure in prostate cancer treatment: involvement of Multi Drug Resistance proteins. Eur J Pharmacol. 2008;601:38-42
60. Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109:238-246
61. Sugimoto Y, Tsukahara S, Ishikawa E. et al. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci. 2005;96:457-465
62. Cascorbi I. Role of pharmacogenetics of ATP-binding cassette transporters in the pharmacokinetics of drugs. Pharmacol Ther. 2006;112:457-473
63. Kim KA, Cha YJ, Lee HM. et al. Effects of the ABCG2 and ABCB1 drug transporter polymorphisms on the pharmacokinetics of bicalutamide in humans. Clin Chim Acta. 2015;438:7-11
64. Sparreboom A, Gelderblom H, Marsh S. et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther. 2004;76:38-44
65. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737
66. Al-Hajj M, Wicha MS, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-3988
67. Lottaz C, Beier D, Meyer K. et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70:2030-2040
68. Patrawala L, Calhoun T, Schneider-Broussard R. et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696-1708
69. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V. et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373-388
70. Yang T, Rycaj K, Liu ZM. et al. Cancer stem cells: constantly evolving and functionally heterogeneous therapeutic targets. Cancer Res. 2014;74:2922-2927
71. Laffin B, Tang DG. An old player on a new playground: bmi-1 as a regulator of prostate stem cells. Cell Stem Cell. 2010;7:639-640
72. Yan J, Tang D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp Cell Res. 2014;328:132-142
73. Chang L, Graham PH, Hao J. et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875
74. Jiao J, Hindoyan A, Wang S. et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PLoS One. 2012;7:e42564
75. Castillo V, Valenzuela R, Huidobro C. et al. Functional characteristics of cancer stem cells and their role in drug resistance of prostate cancer. Int J Oncol. 2014;45:985-994
76. Zhang L, Jiao M, Li L. et al. Tumorspheres derived from prostate cancer cells possess chemoresistant and cancer stem cell properties. J Cancer Res Clin Oncol. 2012;138:675-686
77. Xie Y, Xu K, Dai B. et al. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene. 2006;25:70-78
78. Xie Y, Xu K, Linn DE. et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349-3356
79. Smith PJ, Furon E, Wiltshire M. et al. ABCG2-associated resistance to Hoechst 33342 and topotecan in a murine cell model with constitutive expression of side population characteristics. Cytometry A. 2009;75:924-933
80. Kruger JA, Kaplan CD, Luo Y. et al. Characterization of stem cell-like cancer cells in immune-competent mice. Blood. 2006;108:3906-3912
81. Zen Y, Fujii T, Yoshikawa S. et al. Histological and culture studies with respect to ABCG2 expression support the existence of a cancer cell hierarchy in human hepatocellular carcinoma. Am J Pathol. 2007;170:1750-1762
82. Fetsch PA, Abati A, Litman T. et al. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett. 2006;235:84-92
83. Diestra JE, Scheffer GL, Català I. et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol. 2002;198:213-219
84. Bhatt RI, Brown MD, Hart CA. et al. Novel method for the isolation and characterisation of the putative prostatic stem cell. Cytometry A. 2003;54:89-99
85. Brown MD, Gilmore PE, Hart CA. et al. Characterization of benign and malignant prostate epithelial Hoechst 33342 side populations. Prostate. 2007;67:1384-1396
86. Huss WJ, Gray DR, Greenberg NM. et al. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005;65:6640-6650
87. Previs RA, Coleman RL, Harris AL. et al. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. 2015;21:955-961
88. Kashat M, Azzouz L, Sarkar SH. et al. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res. 2012;4:432-442
89. Ma J, Meng Y, Kwiatkowski DJ. et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J Clin Invest. 2010;120:103-114
90. Zhu H, Zhou X, Redfield S. et al. Elevated Jagged-1 and Notch-1 expression in high grade and metastatic prostate cancers. Am J Transl Res. 2013;5:368-378
91. Taylor BS, Schultz N, Hieronymus H. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11-22
92. Carver BS, Chapinski C, Wongvipat J. et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575-586
93. Mulholland DJ, Tran LM, Li Y. et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792-804
94. Bakin RE, Gioeli D, Sikes RA. et al. Constitutive activation of the Ras/mitogen-activated protein kinase signaling pathway promotes androgen hypersensitivity in LNCaP prostate cancer cells. Cancer Res. 2003;63:1981-1989
95. Bakin RE, Gioeli D, Bissonette EA. et al. Attenuation of Ras signaling restores androgen sensitivity to hormone-refractory C4-2 prostate cancer cells. Cancer Res. 2003;63:1975-1980
96. Hong SK, Kim JH, Lin MF. et al. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res. 2011;317:2671-2682
97. Liu X, He Z, Li CH. et al. Correlation analysis of JAK-STAT pathway components on prognosis of patients with prostate cancer. Pathol Oncol Res. 2012;18:17-23
98. Tam L, McGlynn LM, Traynor P. et al. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br J Cancer. 2007;97:378-383
99. Akimoto S, Okumura A, Fuse H. Relationship between serum levels of interleukin-6, tumor necrosis factor-alpha and bone turnover markers in prostate cancer patients. Endocr J. 1998;45:183-189
100. Adler HL, McCurdy MA, Kattan MW. et al. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161:182-187
101. Nakashima J, Tachibana M, Horiguchi Y. et al. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6:2702-2706
102. Mora LB, Buettner R, Seigne J. et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659-6666
103. DeMiguel F, Lee SO, Lou W. et al. Stat3 enhances the growth of LNCaP human prostate cancer cells in intact and castrated male nude mice. Prostate. 2002;52:123-129
104. Han Z, Wang X, Ma L. et al. Inhibition of STAT3 signaling targets both tumor-initiating and differentiated cell populations in prostate cancer. Oncotarget. 2014;5:8416-8428
105. Iliopoulos D, Hirsch HA, Wang G. et al. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108:1397-1402
106. Chaffer CL, Brueckmann I, Scheel C. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950-7955
107. Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628-2637
108. Chung J, Uchida E, Grammer TC. et al. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508-6516
109. Carriere A, Romeo Y, Acosta-Jaquez HA. et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem. 2011;286:567-577
110. Ma L, Chen Z, Erdjument-Bromage H. et al. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179-193
111. Hong SK, Jeong JH, Chan AM. et al. AKT upregulates B-Raf Ser445 phosphorylation and ERK1/2 activation in prostate cancer cells in response to androgen depletion. Exp Cell Res. 2013;319:1732-1743
112. Schroeder A, Herrmann A, Cherryholmes G. et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014;74:1227-1237
113. Dubrovska A, Kim S, Salamone RJ. et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A. 2009;106:268-273
114. Dubrovska A, Elliott J, Salamone RJ. et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res. 2010;16:5692-5702
115. Jeter CR, Badeaux M, Choy G. et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993-1005
116. Jeter CR, Liu B, Liu X. et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833-3845
117. Bourguignon LY, Peyrollier K, Xia W. et al. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008;283:17635-17651
118. Zbinden M, Duquet A, Lorente-Trigos A. et al. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659-2674
119. Niu CS, Li DX, Liu YH. et al. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol Rep. 2011;26:593-601
120. Xu CX, Xu M, Tan L. et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J Biol Chem. 2012;287:34970-34978
121. Germann M, Wetterwald A, Guzmán-Ramirez N. et al. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem Cells. 2012;30:1076-1086
122. Costa Y, Ding J, Theunissen TW. et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370-374
123. Iv Santaliz-Ruiz LE, Xie X, Old M. et al. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014;135:2741-2748
124. Liu C, Kelnar K, Vlassov AV. et al. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res. 2012;72:3393-3404
125. Liu C, Kelnar K, Liu B. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211-215
126. Jin M, Zhang T, Liu C. et al. miRNA-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiating cells. Cancer Res. 2014;74:4183-4195
127. Srivastava A, Goldberger H, Dimtchev A. et al. MicroRNA profiling in prostate cancer--the diagnostic potential of urinary miR-205 and miR-214. PLoS One. 2013;8:e76994
128. Inda MM, Bonavia R, Mukasa A. et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731-1745
129. Emmink BL, Van Houdt WJ, Vries RG. et al. Differentiated human colorectal cancer cells protect tumor-initiating cells from irinotecan. Gastroenterology. 2011;141:269-278
130. Yan H, Chen X, Zhang Q. et al. Drug-tolerant cancer cells show reduced tumor-initiating capacity: depletion of CD44 cells and evidence for epigenetic mechanisms. PLoS One. 2011;6:e24397
131. Yu F, Yao H, Zhu P. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-1123
Author contact
![]() Corresponding author: Dr. Tao Yang, Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China. Phone: 86-21-61569714; Fax: 86-21-33923060; E-mail: taoyangedu.cn
Corresponding author: Dr. Tao Yang, Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China. Phone: 86-21-61569714; Fax: 86-21-33923060; E-mail: taoyangedu.cn