Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(11):1257-1268. doi:10.7150/ijbs.12611 This issue Cite
Research Paper
NF-κB-DICER-miRs Axis Regulates TNF-α Expression in Responses to Endotoxin Stress
Yi Guan1,#, Hailan Yao2,#, Junfeng Wang2, Kailai Sun3, Liu Cao1 ![]() , Yizheng Wang2
, Yizheng Wang2 ![]()
1. Key Laboratory of Medical Cell Biology, China Medical University, Shenyang, 110122, China
2. Laboratory of Neural Signal Transduction, Institute of Neuroscience, State Key Laboratory of Neuroscience, SIBS, Chinese Academy of Sciences, Shanghai, 200031, China
3. Department of Medical Genetics, China Medical University, Shenyang, 110122, China
#These authors contributed equally to this work.
Received 2015-5-5; Accepted 2015-8-7; Published 2015-9-9
Abstract

Unbalanced tumor necrosis factor (TNF)-α production is associated with pathogenesis of a variety of human diseases. However, the molecular pathways maintaining TNF-α homeostasis remain elusive. Here, we report that NF-κB/p65-DICER-miRs axis negatively regulates TNF-α production. We demonstrated that NF-κB bound to DICER promoter and transcriptionally regulated DICER expression. In addition, the NF-κB/DICER signaling suppresses TNF-α expression by generating mature forms of miR-125b and miR-130a which negatively regulate TNF-α mRNA. Furthermore, we showed that the hepatocyte-specific depletion of Dicer in mice resulted in TNF-α overproduction and sensitized the mice to endotoxin, which could be corrected by administration of miR-125b mimics. These data suggest that NF-κB/p65-DICER-miRs axis involved in maintaining of TNF-α homeostasis, and injection of miR-125b as a potential therapeutic method for septic shock.
Keywords: TNF-α/DICER/NF-κB/microRNAs/septic shock
Introduction
Tumor necrosis factor (TNF)-α is a multifunctional cytokine involved in important biological processes, including cell survival, proliferation, differentiation and death [1]. It also plays a crucial role in the pathogenesis of a variety of human diseases. For example, TNF-α overproduction has been implicated in septic shock [2], autoimmune diseases [3] and neurodegenerative diseases [4]. Depending on its expression levels, TNF-α may exhibit both beneficial and detrimental effects. However, the mechanism by which a proper level of TNF-α is maintained remains largely unclear.
Production of TNF-α is regulated at both transcriptional and post-transcriptional levels in most systems [5, 6]. NF-κB, a transcriptional factor known important for cell survival, immunity and inflammation [7], plays a predominant role in transcriptional activation of TNF-α in response to several factors, including lipopolysaccharide (LPS) [5]. Excessive stimulation of innate immunity, including liver cells by LPS, can lead to TNF-α overproduction and endotoxin shock, a systemic disorder with a high mortality rate in humans and experimental mouse models [8].
MicroRNAs (miRNAs) are short RNAs (18-24 nt in length), function as important post-transcriptional regulators of gene expression through modulating the stability and/or translation of the target mRNAs [9-11]. They are involved in numerous physiological and pathological processes, including development, differentiation, metabolism, inflammation, viral infection, and tumorigenesis [12-15]. The miRNAs are involved in the post-transcriptional regulation of TNF-α production. For example, miR-125b targets the 3'-untranslated region (3'-UTR) of TNF-α transcripts to suppress TNF-α production [16].
Most miRNAs are initially transcribed by RNA polymerase II (poly II) as primary transcripts (pri-miRNAs), which are subsequently cleaved into a hairpin-structured precursor miRNA (pre-miRNA) by Drosha, a nuclear RNase III. Pre-miRNAs are then exported into the cytoplasm and further digested by DICER, another RNase III, to generate mature miRNAs [17, 18]. In human biliary epithelial cells, NF-κB/p65 activation induced by LPS regulates the expression of a subset of miRNA genes, including pri-miR-125b, pri-miR-21 and pri-miR-130a [19]. Moreover, LPS affects the expression of miR-125b, miR-146 and miR-155 in human monocytes [16, 20]. Therefore, NF-κB may regulate both pri-miRNA expression and miRNA formation.
Although both MITF [21] and Tap63 [22], two transcriptional factors, have been implicated in regulation of DICER expression, the transcriptional regulation of DICER remains largely unclear. Since NF-κB regulates both TNF-α transcription and miRNA formation, we asked whether transcription of DICER could be regulated by NF-κB to control TNF-α production in septic shock. Here, we reported that NF-κB/p65 controls DICER transcription and that a novel NF-κB/p65-DICER-miRs regulatory circuitry is essential to TNF-α homeostatic production.
Results
NF-κB/p65 regulates DICER expression
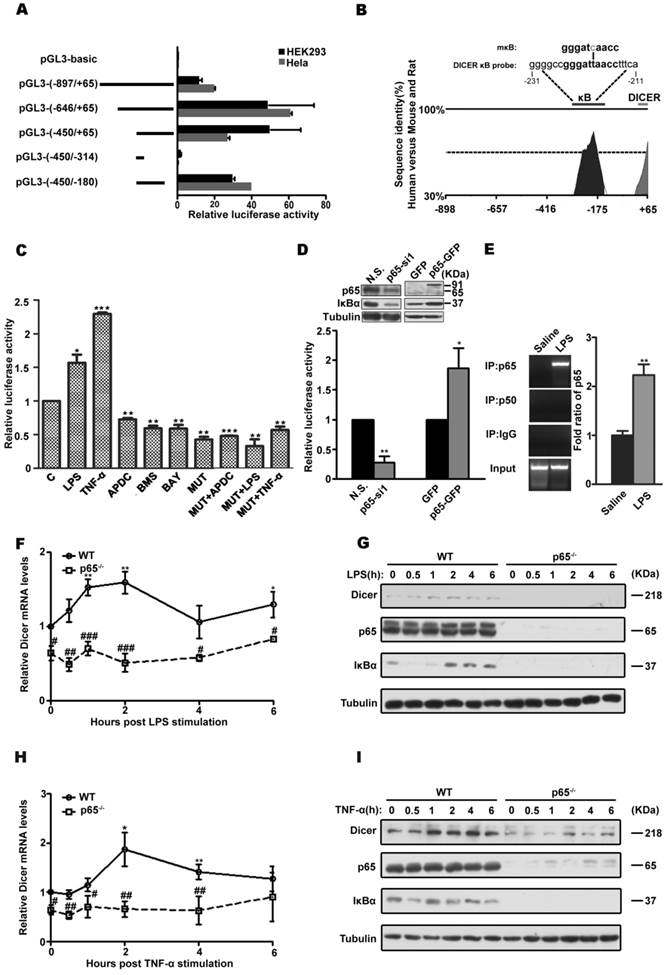
To analysis the transcriptional regulation of DICER, we first mapped the regions of DICER promoter and found that its core promoter is located at -450 to -180 bp relative to its transcription start site (Figure 1A, 1B and Supplementary Figure 1A), a region highly conserved among human, mouse and rat. Further the TRANSFAC analysis revealed the presence of an NF-κB binding site in this region (Figure 1B). Mutagenic analysis confirmed this putative NF-κB regulatory element in DICER promoter was functional in both Huh7 cells, a human hepatocarcinoma cell line (Figure 1C), and HEK293 cells (Supplementary Figure 1B). Further, siRNA knockdown and overexpression experiments showed that the p65 subunit rather than the p50 subunit was important for DICER promoter activity (Figure 1D, Supplementary Figure 1C and 1D, and Supplemental Figure 2). Additionally, electrophoretic mobility shift assays (EMSA) (Supplementary Figure 1E and 1F) and chromatin immunoprecipitation (ChIP) assays (Figure 1E and Supplementary Figure 1G) revealed that DICER promoter was physically associated with NF-κB/p65. Consistently, DICER expression induced by LPS (Figure 1F and 1G), a bacterial endotoxin responsible for septic shock [23], and by TNF-α (Figure 1H and 1I) in mouse embryonic fibroblasts (MEF) deficient for p65 was prevented. Taken together, these results demonstrate that NF-κB/p65 regulates DICER expression.
NF-κB /p65 regulation of DICER expression. (A) DICER promoter analysis. HEK293 and HeLa cells were transiently transfected with firefly luciferase reporter constructs (pGL3-basic) containing the indicated genomic fragments (Left, schematic diagram) upstream of DICER and analyzed for luciferase activities. All luciferase data were normalized to β-galactosidase. Fold changes were shown with respect to pGL3-basic, where the normalized activity was set to value of 1. (B) VISTA schematic diagram showed the phylogenetic conservation between human, mouse and rat in the genomic region upstream of DICER. The sequences of predicted κB site (DICER κB probe) and mutated κB (mκB) site. (C) Huh7 cells were transfected with the proximal promoter construct pGL3 (-450/-180) (C) or mκB-pGL3 (-450/-180) (MUT) and subsequently treated with LPS for 4h, TNF-α for 2h, APDC for 8h, BMS or BAY for 1h, and luciferase activity was determined. *P<0.05, **P<0.01, ***P<0.001 vs C. (D) Luciferase activities in HEK293 cells transfected with pGL3 (-450/-180) and nonsense siRNA (N.S.), p65-specific siRNA (p65-si1), p65-GFP or empty vector (GFP) each. The activities were relative to those in N.S. or GFP groups. Immunoblots (inset) showing the knockdown or overexpression of p65. *P<0.05, **P<0.01 vs N.S. or GFP. (E) NF-κB/p65 interaction with DICER promoter analyzed by ChIP assay. EB staining, qPCR products on a 2% agarose gel; bar graph, statistical analysis of PCR. Data were normalized with the input DNA. **P<0.01 vs Saline. Wild-type (WT) or p65 KO (p65-/-) MEFs were treated with LPS or TNF-α for the indicated times and harvested for qRT-PCR (F and H) or immunoblot (G and I). The mRNA levels of DICER were normalized to those of actin and relative to those in non-stimulated cells. *P<0.05, **P<0.01 vs 0h. #P<0.05, ##P<0.01, ###P<0.001 vs WT. All quantitative data, unless stated, were means ± SEM of three independent experiments in triplicates and tubulin served as a loading control in all immunoblots.
DICER suppress TNF-α production via NF-κB/p65 activation
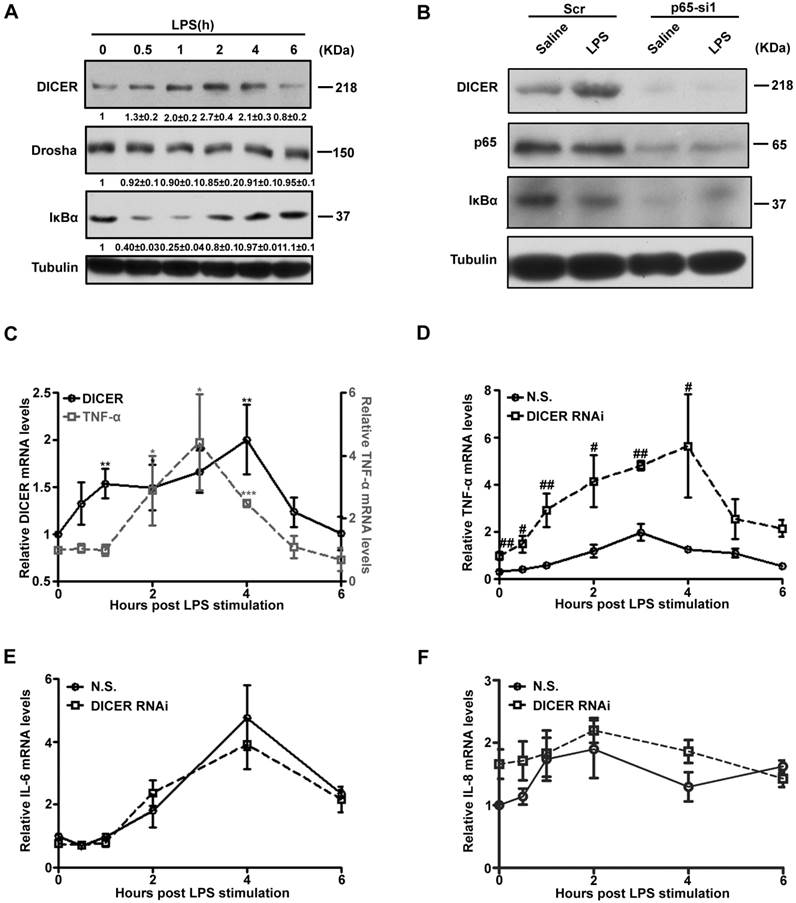
To explore the functional regulation of NF-κB/p65-DICER axis, we treated Huh7 cells with LPS and followed DICER and TNF-α expression. DICER protein levels in Huh7 cells were increased between 1h and 4h after LPS treatment (Figure 2A). Interestingly, its levels in RAW 264.7 cells, a mouse macrophage cell line known abundant for TNF-α production [24], were not changed (Supplementary Figure 3A and 3B). LPS did not increase DICER expression when p65 was down-regulated (Figure 2B). In the time course analysis of expression of DICER and TNF-α, we found that DICER mRNA levels were enhanced 1h with the peak at 4h, while TNF-α was at 2h with the peak level at 3h after LPS treatment (Figure 2C). This LPS-increased TNF-α mRNA level was greatly enhanced with the peak level at 4h when DICER was down-regulated by DICER RNAi (Figure 2D), while NF-κB/p65 activation remained unaffected (Supplementary Figure 3C and 3D). In contrast, the mRNA levels of IL-6 (Figure 2E) and IL-8 (Figure 2F), two inflammatory cytokines also regulated by NF-κB [25, 26], remained unaffected by DICER knockdown in response to LPS. These results suggested that up-regulation of DICER following NF-κB/p65 activation may play an important role in preventing overproduction of TNF-α in Huh7 cells.
NF-κB /p65-induced DICER expression suppressed TNF-α production. (A) Huh7 cells treated with LPS for the indicated times were harvested for immunoblot. (B) Huh7 cells transfected with Scr or p65-si1 for 72h and treated with LPS for 4h were lysated for immunoblot. (C) The mRNA levels of DICER and TNF-α in Huh7 cells stimulated by LPS and normalized to those of actin. *P<0.05, **P<0.01, ***P<0.001 vs 0h. The mRNA levels of TNF-α (D), IL-6 (E) and IL-8 (F) in Huh7 cells transfected with N.S. or DICER RNAi plasmids (DICER RNAi) for 72h and treated with LPS and normalized to those of actin. #P<0.05, ##P<0.01 vs N.S..
NF-κB/DICER axis regulates MiR-125b and miR-130a expression
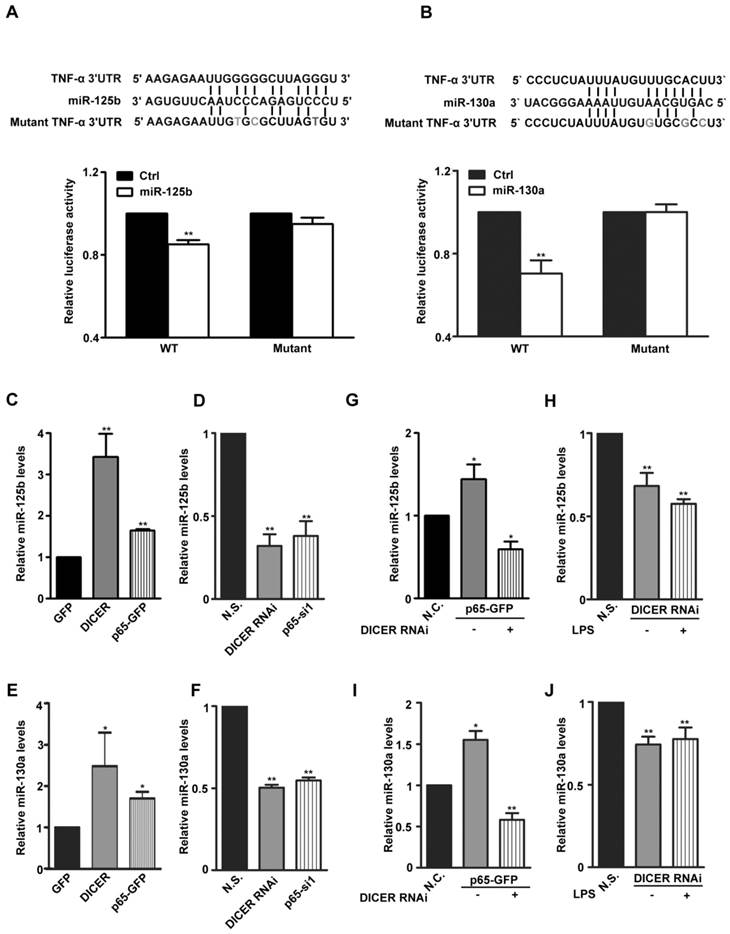
DICER is an RNase III responsible for production of mature miRNAs. To determine which miRNAs mediate the potential inhibitory effects of DICER on TNF-α production, we analyzed the 3'-UTR of TNF-α mRNA and found that, miR-125b, miR-19, miR-181, miR-130a and miR-16 target sequences which were reported previously affecting TNF-α expression [16, 27-30] (Supplementary Figure 4A). Through expressing these miRNAs in HEK293 cells, we found that miR-125b and miR-130a could decrease both TNF-α mRNA levels (Supplementary Figure 4B) and the luciferase activities of a luciferase construct with the TNF-α 3'-UTR (Figure 3A and 3B). Furthermore, through overexpression or RNAi knockdown of either p65 or DICER, we did find that the expression of both miR-125b and miR-130a was regulated by p65 as well as DICER (Figure 3C-3F). Knockdown experiments further verified the requirement of DICER for either LPS-induced and p65-dependent expression of both miR-125b and miR-130a (Figure 3G-3J). These results suggest that up-regulation of miR-125b and miR-130a by the NF-κB/p65/DICER pathway may be an important mechanism preventing overproduction of TNF-α.
MiR-125b and miR-130a expression was regulated by NF-κB/DICER pathway. (A) Top: Sequence alignment of miR-125b and its target sites in 3′-UTR of TNF-α in human. Gray Characters: the mutant seed sequence. Lower: A luciferase reporter construct containing a ~240bp fragment of the TNF-α 3′-UTR with the miR-125b binding site was made. The luciferase activity in HEK293 cells transfected with Wild type (WT) or mutant reporter plasmids (Mutant) and miR-125b (miR-125b) or empty plasmid (Ctrl) for 48h was determined. **P<0.01 vs Ctrl. (B) Top: Sequence alignment of miR-130a and its target sites in 3′-UTR of TNF-α in human. Gray Characters: the mutant seed sequence. Lower: A luciferase reporter construct containing a ~240bp fragment of the TNF-α 3′-UTR with the miR-130a binding site was made. The luciferase activity in HEK293 cells transfected with Wild type (WT) or mutant reporter plasmids (Mutant) and miR-130a (miR-130a) or empty plasmid (Ctrl) for 48h was determined. **P<0.01vs Ctrl. The expression of miR-125b or miR-130a in Huh7 cells transfected with GFP, DICER or p65-GFP (C and E) for 48h; N.S., DICER RNAi or p65-si1 (D and F) for 72h was determined by qRT-PCR. *P<0.05, **P<0.01 vs GFP or N.S. The levels of miR-125b (G) or miR-130a (I) in Huh7 cells cotransfected with N.S. and GFP (N.C.) or DICER RNAi and p65-GFP for 72h were determined. *P<0.05, **P<0.01 vs N.C. MiR-125b (H) or miR-130a (J) levels in Huh7 cells transfected with N.S. or DICER RNAi for 72h and treated with LPS for 4h were determined., **P<0.01 vs N.S. All quantitative data were means ± SEM of three independent experiments in triplicates. The levels of miR-125b and miR-130a were normalized to those of actin mRNA.
NF-κB/p65-DICER-miRs axis regulates TNF-α production
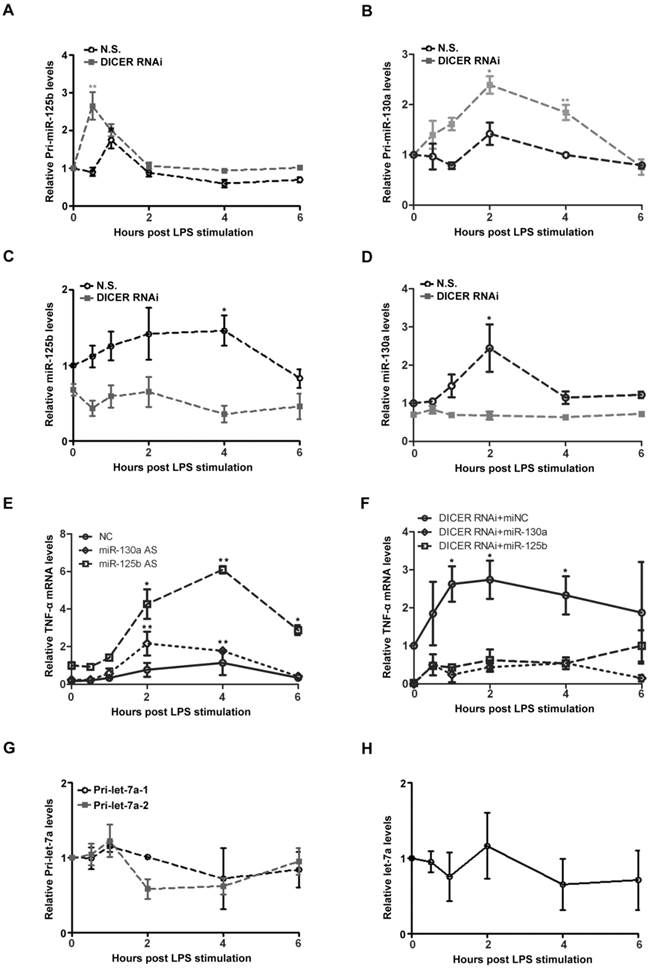
We next attempt to study the underlying mechanism of NF-κB/p65-DICER-miRNAs axis in TNF-α production. In response to LPS treatment, the levels of pri-miR-125b and pri-miR-130a were rapidly increased in Huh7 cells (Figures 4A and 4B). Interestingly, the up-regulation of these pri-miRNAs was further enhanced when DICER was knocked down. In contrast to the changes of the pri-miRNAs, we found that the levels of the mature miR-125b and miR-130a were decreased when DICER was knocked down (Figures 4C and 4D). These results clearly showed that DICER played an important role in conversion of the pri-miR-125b and pri-miR-130a to their mature forms, and that DICER down-regulation led to an accumulation of the immature forms accompanied by the reduction of their mature forms. To provide further evidence that DICER expression driven by NF-κB indeed affects TNF-α levels, we examined the effects of miR-125b and miR-130a on TNF-α expression. The LPS-induced TNF-α mRNA levels were greatly increased when miR-125b or miR-130a were down-regulated (Figure 4E). Consistently, expressing miR-125b and miR-130a reversed the increase in TNF-α mRNA levels caused by DICER down-regulation (Figure 4F). These results supported an explanation that increases of miR-130a and miR-125b by DICER activity in the later phase of LPS treatment provides a mechanism in controlling the overproduction of TNF-α. Since TLR-induced gene expression is generally mediated by NF-κB [31], we further examined the levels of both primary and mature forms of let-7a, a miRNA known to target IL-6 [32]. Neither primary nor mature form of let-7a was affected (Figure 4G and 4H), suggesting that regulation of IL-6 production was different from that of TNF-α. Taken together, the above results revealed a regulatory mechanism involving NF-κB/p65, DICER, miR-125b and miR-130a that controls TNF-α homeostatic production in response to NF-κB activation in hepatocytes.
MiRs were upregulated to inhibit TNF-α production induced by p65/DICER signaling. Pri-miR-125b (A), pri-miR-130a (B), miR-125b (C) and miR-130a (D) levels in Huh7 cells transfected with N.S. or DICER RNAi for 72h and stimulated with LPS were determined and normalized to actin mRNA levels. *P<0.05, **P<0.01vs 0h. (E) TNF-α mRNA levels in Huh7 cells transfected with negative control (NC), miR-125b or miR-130a AS for 72h and treated with LPS were determined and normalized to actin mRNA levels. *P<0.05, **P<0.01 vs 0h. (F) TNF-α mRNA levels in Huh7 cells transfected with DICER RNAi in addition to miNC, miR-125b or miR130a for 72h and stimulated with LPS were determined and normalized to actin mRNA levels. *P<0.05 vs 0h. Pri-let-7a-1, pri-let-7a-2 (G) and mature let-7a (H) levels in Huh7 cells stimulated with LPS were determined and normalized to actin mRNA levels.
NF-κB/p65-DICER-miRNAs axis regulates TNF-α production in LPS induced sepsis shock
Since LPS induced-septic shock is mostly caused by NF-κB-dependent TNF-α production [8], we next examined the in vivo roles of hepatic DICER in a mouse LPS-induced liver septic shock model. Similar as the results obtained from cultured cells, the expression levels of TNF-α (Figure 5A and 5B), miR-125b and miR-130a (Figure 5C and 5D) in mice were all greatly increased in response to LPS treatment. To study the roles of DICER in liver in LPS-induced septic shock, we generated hepatocyte-specific Dicer-deficient mice by breeding Dicerflox/flox mice (DicerF/F) with mice expressing Cre recombinase under control by either the albumin promoter (Alb-Cre) [33] or inducible Mx1 promoter (Mx1-Cre) [34], resulting in DicerF/FAlbCre or DicerF/FMx1Cre mice. Efficient deletion of Dicer in liver was confirmed by quantitative RT-PCR in these mice (Supplementary Figure 5A-C). The DicerF/FAlbCre and DicerF/FMx1Cre mice could survive to adulthood without obvious abnormalities [35]. We found that administration of LPS could markedly elevated TNF-α mRNA in the livers and TNF-α protein in sera of DicerF/FAlbCre and DicerF/FMx1Cre mice, with levels significantly higher than in the control DicerF/F mice (Figure 5E-G). Agreeing well, the mature hepatic miR-125b levels were decreased (Supplementary Figure 5D). Notably, although the serum TNF-α levels were not affected, we did observe a significant decrease in miR-125b in the livers of the DicerF/+AlbCre mice after LPS treatment (Supplementary Figure 5E and 5F). Collectively, TNF-α production in Dicer mutant mice was much higher and last longer compared with wild type mice after treated with LPS.
Dicer expression was important for hepatic TNF-α expression in mice. (A) Dicer mRNA levels in the purified hepatocytes of DicerF/F mice treated with saline (Ctrl) or LPS for 1h. Each dot represented the measurement of an animal (n=5). **P<0.01 vs Ctrl. (B) Immunoblots of total proteins isolated from the purified hepatocytes of DicerF/F mice treated with LPS for 4h. MiR-125b (C) and miR-130a (D) levels in the purified hepatocytes of DicerF/F mice treated with saline (Ctrl) or LPS for 1h. Each dot represented measurement of an animal (n=5). **P<0.01 vs Ctrl. The mRNA levels of hepatic TNF-α in DicerF/FAlbCre (E) or DicerF/FMx1Cre (F) mice treated with LPS were measured, normalized to those of hepatic actin (n=5). **P<0.01 vs DicerF/F. (G) Serum TNF-α levels of mice after LPS treatment were measured by ELISA (n=5). *P<0.05, **P<0.01 vs DicerF/F. (H) Serum TNF-α levels in liposome-treated DicerF/FAlbCre or DicerF/F mice treated with LPS for 1h (n=6). **P<0.01 vs PBS, *P<0.05 vs DicerF/F or PBS (DicerF/FAlbCre).
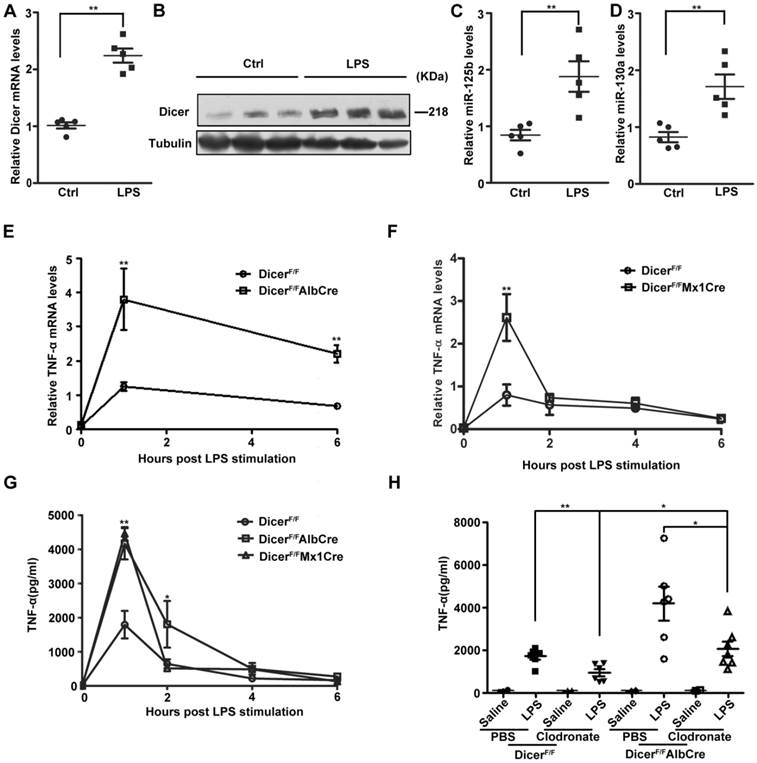
We then depleted macrophages by liposome-encapsulated clodronate [36, 37] and examined LPS-induced TNF-α production. Although depletion of macrophages (Supplementary Figure 6A) significantly reduced serum TNF-α levels in DicerF/F and DicerF/FAlbCre mice, but the levels in DicerF/FAlbCre mice still remained much higher than in DicerF/F mice after LPS injection (Figure 5H). In contrast to the observations that the TNF-α production in hepatocytes from DicerF/FAlbCre mice was much greater than that from DicerF/F mice (Supplementary Figure 6B), the LPS-induced TNF-α production in the peritoneal macrophages isolated from these mice was not different (Supplementary Figure 6C).
The serum transaminase activity between these mice was not different (Supplementary Figure 6D and 6E), suggesting the impact of overproduction of TNF- α was systemic rather than liver-specific. Therefore, DICER upregulation in hepatocytes prevented TNF-α overproduction during LPS treatment, avoiding the systemic effects mediated by TNF-α and other cytokines, which could cause tissue injury, disseminated intravascular coagulation, shock and death [34, 38].
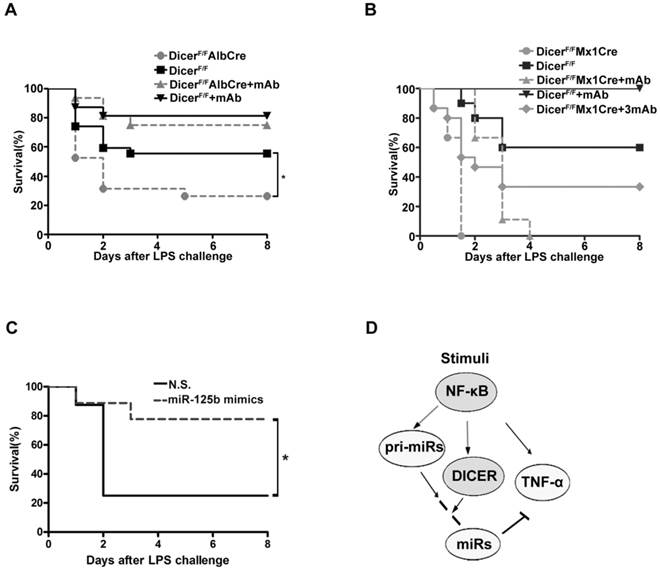
We then examined the mouse survival after injection with a lethal dose LPS. Eight days after injection, more than 50% of the DicerF/F mice still remained alive, whereas only 26% DicerF/FAlbCre mice survived the challenge (Figure 6A). Further, administration of infliximab, the anti-TNF-α antibody known to block the action of TNF-α [39], significantly protected both the DicerF/FAlbCre and DicerF/F mice from LPS-induced toxicity. Remarkably, while all of the DicerF/FMx1Cre mice succumbed to LPS 36h after the challenge, 30% of the mice survived when infliximab was given (Figure 6B). Moreover, injection of miR-125b mimics could similarly prevent mice from LPS-induced toxicity (Figure 6C). These results suggested that the higher mortality rates of the Dicer mutant mice were mainly caused by the systemic effects of hepatic TNF-α overproduction.
Knock down of DICER greatly sensitized mice to septic shock. Survival of DicerF/FAlbCre (A) or DicerF/FMx1Cre (B) mice injected intraperitoneally with saline or Infliximab (30μg/g, mAb or 90μg/g, 3mAb) 1 h before application of LPS was monitored over a period of 8 days (n=15-19). *P<0.05 vs DicerF/F. (C) Survival of DicerF/FAlbCre mice injected intravenously (i.v.) via lateral tail veins with nonsense (N.S.) or miR-125b mimics (1.5μg/g)12h before application of LPS was monitored over a period of 8 days (n=8-9). *P<0.05 vs N.S.. (D)A model depicting NF-κB-DICER-miRs regulatory circuitry in hepatic TNF-α expression. NF-κB activation induced TNF-α expression and also directed the expression of DICER and miRs to prevent overproduction of TNF-α.
Discussion
We have shown here that the NF-κB-dependent transcription of DICER is important for TNF-α homeostasis in hepatocytes. Several lines of evidence support this conclusion. First, activation of NF-κB was sufficient to stimulate DICER promoter activity and induce its expression; NF-κB activation was also necessary for LPS or TNF-α to enhance DICER promoter activity and its expression (Figures 1-3). Second, DICER, pri-miR-125b, pri-miR-130a and TNF-α expressions were induced in response to NF-κB activation; but the TNF-α expression was suppressed when the peak expression of DICER and miR-125b occurred (Figure 4). Third, after NF-κB activation, DICER processed pri-miR-125b or pri-miR-130a to miR-125b or miR-130a to suppress TNF-α production (Figure 3 and 4). Finally, inhibition of DICER expression resulted in an enhanced and prolonged production of TNF-α in cultured cells and in mice (Figure 2D, 5E, 5F and 5G). These findings led us to propose a model to illustrate how TNF-α production was stringently controlled by NF-κB signaling (Figure 6D). The LPS-induced transcription of TNF-α gene is dependent on NF-κB activation. Meanwhile, activation of NF-κB also triggers transcriptional and post-transcriptional mechanisms to suppress TNF-α production or prevent TNF-α overproduction, which involves the upregulation of DICER and pri-miRs and subsequent miRs formation. The outcome of these NF-κB-directed regulatory mechanisms is the balanced production of TNF-α, which is critical for physiological immune and inflammatory responses.
Activation of TLR4 by LPS initiates NF-κB signaling pathway, leading to the production of proinflammatory cytokines, such as TNF-α and IL-6 [40, 41]. In our study, NF-κB signaling drove a cluster of gene expression, including DICER, miR-125b, miR-130a and TNF-α, in which TNF-α expression was inhibited by DICER/miR-125b/miR-130a. An RNAi-based screening reveals that DICER is required for the rapid decay of TNF-α mRNA in mammalian cells [27]. Thus, the NF-κB/DICER pathway forms a novel negative feedback loop in TNF-α production, in which activation of a transcriptional factor (NF-κB) directs the negative feedback achieved by its products (DICER, miR-125b, miR-130a and TNF-α) at post-transcriptional levels. This feedback loop was thus an important and specific mechanism for the fine tune of TNF-α expression since inhibition of DICER expression led to an enhanced production of TNF-α, but not IL-6 or IL-8 (Figures 2E and 2F).
DICER is the key enzyme of miRNA maturation and ubiquitously expressed. Recent study in epithelial cells uncovered several NF-κB/p65-dependent miRNAs, which expression in the mature form is not increased although the up-regulation of their primary transcripts was found [19, 42], indicating that post-transcriptional mechanism is also critical in regulation of miRNA expression. Since NF-κB signaling upregulated DICER, pri-miR-125b, pri-miR-130a, miR-125b and miR-130a levels (Figure 4A-D), it is possible that NF-κB induced miR-125b and miR-130a expression by activating transcription of pri-miR-125b and pri-miR-130a which were subsequently converted to miR-125b and miR-130a by DICER. Our results also showed that the expression of IL-6, pri-let7 and let-7, a miRNA targeting IL-6, was not regulated by the NF-κB/DICER pathway. These results suggested that post-transcriptional regulation of miRNAs by DICER might require the coordination of transcriptional upregulation of both pri-miRNA transcripts and DICER.
It should be pointed out that NF-κB-dependent transcription of DICER was not observed in the RAW264.7 cell, a mouse macrophage cell line, following LPS stimulation although the production of TNF-α was highly induced (Supplementary Figure 3A and 3B). That the LPS-induced upregulation of DICER found in Huh7 cells and MEF (non-hematopoietic cells) could not be detected in macrophages suggests that the NF-κB/DICER/miRs pathway was a cell type specific mechanism involved in regulation of TNF-α expression. Since LPS-induced NF-κB activation lasted much longer in these non-hematopoietic cells than in RAW264.7 cells, the long-lasting activation of NF-κB may greatly affect DICER expression. Indeed, the duration of NF-κB activation contributes to the selectivity of the transcriptional response [43]. Alternatively, LPS-induced long-lasting NF-κB activation in the non-hematopoietic cells appears to be that TNF-α induced by LPS acts in an autocrine fashion to maintain the long-term NF-κB activation [44].
TNF-α is mainly produced by macrophages. But, its expression has been found in a broad variety of cells, including lymphoid cells, endothelial cells, fibroblasts, glia and hepatocytes [45]. The production of TNF-α by non-hematopoietic cells have an important role in physiology and pathology of many tissues, including liver [46]. Our Kupffer depletion experiments showed a reduction in serum TNF-α level by 50% in mice given LPS, indicating the presence of a non-Kupffer cell source in mice (Figure 5H). We also found that TNF-α protein level was increased in LPS-treated isolated hepatocytes (Supplementary Figure 6B). Our findings consistent with many studies provide evidence that hepatocytes could be considered not only as targets of TNF-α, but also as potentially active participants of the local and systemic cytokine network [47, 48]. In this context, the transcriptional control of DICER expression in hepatocytes is critical for the homeostatic regulation of TNF-α production in response to NF-κB activation.
Collectively, our results suggested that a novel axis involving NF-κB, DICER, miR-125b and miR-130a that provides important homeostatic regulation in TNF-α production by hepatocytes in addition to Kupffer cells and other macrophages (Figure 6D). Down-regulation of DICER affects TNF-α, but not IL-6 or IL-8, in response to NF-κB activation shown here, indicating that temporal and spatial expression of a protein may influence DICER effect on its expression. In term of the importance of TNF-α in septic shock, our findings might present a new hepatocyte-possessing mechanism to limit TNF-α production therapeutically.
Materials and Methods
Mice
DicerF/F (Jax: 006366), Alb-Cre (Jax: 003574) and Mx1-Cre (Jax: 002527) mice were from the Jackson Laboratories. DicerF/F mice were crossed with Alb-Cre or Mx1-Cre transgenic mice to generate hepatocyte-specific Dicer-deficient mice (DicerF/FAlbCre mice) and DicerF/FMx1Cre mice (efficient deletion in both the liver and hematopoietic cells). DicerF/FAlbCre mice were identified as described previously [49]. Deletion of folxed alleles in Mx1-Cre mice was performed as described previously[34]. DicerF/F littermates without the Cre transgene were served as WT controls. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the Institute of Neuroscience, Shanghai, China.
Constructs
All plasmid sequences are available upon request. We created human DICER-luciferase reporter constructs by cloning the promoter fragments into the pGL3-Basic luciferase reporter vector (Promega) between KpnI and HindIII. Specific primers for DICER promoter's deletion constructs were listed in Supplementary Table S1. DICER-RNAi plasmids were generated according to the previous report [50].
Cell Culture, Transfection and Reagents
HEK293 cells, human hepatoma Huh7 cells, mouse embryonic fibroblasts (MEFs) and murine macrophage cell line RAW264.7 were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) in a 5% CO2 atmosphere at 37℃. HeLa cells were cultured in DMEM (Invitrogen) supplemented with 4% FBS (Thermo Fisher Scientific) in a 5% CO2 atmosphere at 37℃. Primary hepatocytes were prepared as described[51]. Transfections of HEK293, Huh7 cells or HeLa cells with siRNAs (GenePharma, Shanghai; listed in Supplementary Table S1) or plasmids were carried out with lipofectamine 2000 (Invitrogen) or FuGene6 (Roche, in HeLa cells) according to the manufacturer's protocol. The cells were incubated for 72 h to down-regulate p65 or DICER. The knockdown effect was determined by quantitative RT-PCR and Western blot analysis. Ammonium pyrrolidine dithiocarbamate (APDC, 100 ng/ml), BAY 11-7082 (BAY, 30 μM), BMS-345541 (BMS, 1 μM) and LPS (1 μg/ml) were purchased from Sigma, and TNF-α (10 ng/ml) from R&D. All other reagents were purchased from Shanghai Sangon Biological Engineering Technology & Services Co, Ltd (Shanghai, China) unless otherwise specified.
Luciferase Reporter Assay
For the relative luciferase reporter assay, 1.5×105 cells/well seeded into 12-well plates were transiently cotransfected with 1 µg of the luciferase constructs plus 0.25 µg of galactosidase encoding plasmid DNA in each well. In cotransfection, 1 µg of expression vector for p65 (p65-GFP) or 2 µg of siRNAs against p65 was also added and the total amount of DNA or siRNA added to each well was equalized using the empty vector or nonsense siRNA. We prepared cell extracts 48h after cotransfection. The luciferase activity was analyzed using the luciferase assay system (Promega).
Reverse Transcription and Quantitative PCR
Total RNA was isolated using Trizol (Invitrogen) and cDNA was prepared by reverse transcription (MBI Fermentas) followed by real-time PCR analysis (SYBR green; TaKaRa) on a Rotor-Gene Q 2plex HRM analyzer (QIAGEN). Primers used were listed in Supplementary Table S1.
Western Blot
Immunoblot analysis was performed by probing target proteins with antibodies listed in Supplementary Table S2. The quantitation of protein band density was done using ImageQuant 5.2 software (GE Healthcare).
ChIP Assay
ChIP assays in Huh7 cells were performed using a ChIP Assay Kit (Upstate) following the manufacturer's instructions. The cell extracts were subjected to immunoprecipitation using IgG, anti-p65 or anti-p50 antibodies (Santa Cruz). The DNA in the precipitates was extracted and then amplified by qPCR or PCR using primers in Supplementary Table S1. All the PCR products have been confirmed by sequencing analysis.
Kupffer Cell Depletion
Clodronate-containing liposomes were prepared as described[52]. Kupffer cells were depleted in mice via intravenous administration of 100 ul liposome-encapsulated clodronate (Sigma) per 25 g body weight 48 h before LPS administration. Control mice were injected with liposome-encapsulated phosphate-buffered saline (PBS). Kupffer cell elimination was confirmed by the measurement of F4/80 (a macrophage maker) mRNA levels using qRT-PCR [53].
LPS-induced Endotoxin Shock
At the age of 12 weeks, DicerF/FAlbCre, DicerF/FMx1Cre and DicerF/F mice weighing 20-25 g were intraperitoneally injected (i.p.) with NaCl (0.9%) or Infliximab (mAb, 30 μg/g; 3mAb, 90 μg/g) 1 h before application of LPS (10 μg/g). At different time points after injection, blood was taken from the animals and serum was prepared for further analysis. Alanine transaminase (ALT) and aspartate aminotransferase (AST) activities in serum were measured using the ALT and AST Reagent kit (Nanjing Jiancheng Bio-engineering Institute). Animals were continuously monitored for LPS-induced lethality for a period of 8 days after LPS challenge (n=15-19 per treatment group).
Cytokine Level Assays
Cytokine concentration in serum was determined by enzyme-linked immunoabsorbent assay (ELISA) at the indicated time points. ELISA assays measuring the levels of TNF-α were carried out with the Mouse TNF-α ELISA kit (R&D systems).
In Vivo Transfection
At the age of 12 weeks, DicerF/FAlbCre mice weighing 20-25 g were injected intravenously (i.v.) via lateral tail veins with nonsense or miR-125b mimics (1.5μg/g) 12 h before application of LPS (10 μg/g) using in vivo-jetPEI™-Gal (Polyplus-transfection, New York) according to the manufacturer's protocol. Animals were continuously monitored for LPS-induced lethality for a period of 8 days after LPS challenge (n=8-9 per treatment group).
Statistics
All data were presented as mean ± SEM of three independent experiments. Comparisons between groups for statistical significance were performed with Student's t test, analysis of variance (ANOVA), or the log-rank test. Results were considered significant difference at *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001.
Supplementary Material
Supplementary Method, Supplementary Tables and Figures.
Acknowledgements
We thank C. Wang for MEF, ZH. Zheng for DICER constructs, FL.Yang and ZJ. Fan for technical assistance. This work was supported by grants from 973 Program (2011CB00400), the Ministry of education innovation team development plan (IRT13101) and the National Natural Science Foundation of China (81130081, 30571836 and 31300963).
Author contributions
Y.G. and HL.Y. conducted the experiments and wrote the manuscript. JF.W., and KL.S. assisted experimentally. Liu Cao and YZ.W. supervised the project and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634-5
2. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. The New England journal of medicine. 2003;348:138-50
3. Feldmann M. Translating molecular insights in autoimmunity into effective therapy. Annual review of immunology. 2009;27:1-27
4. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of neuroinflammation. 2008;5:45
5. Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Current directions in autoimmunity. 2010;11:27-60
6. Stamou P, Kontoyiannis DL. Posttranscriptional regulation of TNF mRNA: a paradigm of signal-dependent mRNA utilization and its relevance to pathology. Current directions in autoimmunity. 2010;11:61-79
7. Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell research. 2011;21:223-44
8. Lin WJ, Yeh WC. Implication of Toll-like receptor and tumor necrosis factor alpha signaling in septic shock. Shock. 2005;24:206-9
9. Neilson JR, Sharp PA. Small RNA regulators of gene expression. Cell. 2008;134:899-902
10. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
11. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature reviews Genetics. 2004;5:522-31
12. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350-5
13. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259-69
14. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annual review of medicine. 2009;60:167-79
15. Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nature immunology. 2008;9:839-45
16. Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B. et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082-9
17. Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-55
18. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nature reviews Molecular cell biology. 2009;10:126-39
19. Zhou R, Hu G, Gong AY, Chen XM. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic acids research. 2010;38:3222-32
20. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12481-6
21. Levy C, Khaled M, Robinson KC, Veguilla RA, Chen PH, Yokoyama S. et al. Lineage-specific transcriptional regulation of DICER by MITF in melanocytes. Cell. 2010;141:994-1005
22. Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL. et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986-90
23. Warren HS. Editorial: Mouse models to study sepsis syndrome in humans. J Leukoc Biol. 2009;86:199-201
24. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387-98
25. He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell research. 2011;21:159-68
26. Osawa Y, Nagaki M, Banno Y, Brenner DA, Asano T, Nozawa Y. et al. Tumor necrosis factor alpha-induced interleukin-8 production via NF-kappaB and phosphatidylinositol 3-kinase/Akt pathways inhibits cell apoptosis in human hepatocytes. Infect Immun. 2002;70:6294-301
27. Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J. et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623-34
28. Li H, Chen X, Guan L, Qi Q, Shu G, Jiang Q. et al. MiRNA-181a regulates adipogenesis by targeting tumor necrosis factor-alpha (TNF-alpha) in the porcine model. PLoS One. 2013;8:e71568
29. Liu M, Wang Z, Yang S, Zhang W, He S, Hu C. et al. TNF-alpha is a novel target of miR-19a. Int J Oncol. 2011;38:1013-22
30. Zhang J, Wu H, Li P, Zhao Y, Liu M, Tang H. NF-kappaB-modulated miR-130a targets TNF-alpha in cervical cancer cells. J Transl Med. 2014;12:155
31. Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81-96
32. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693-706
33. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM. et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. The Journal of biological chemistry. 1999;274:305-15
34. Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell. 2009;136:249-60
35. Hand NJ, Master ZR, Le Lay J, Friedman JR. Hepatic function is preserved in the absence of mature microRNAs. Hepatology. 2009;49:618-26
36. Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, van Rooijen N, Medof ME. et al. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology. 2009;49:1326-34
37. Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83-93
38. Sass G, Heinlein S, Agli A, Bang R, Schumann J, Tiegs G. Cytokine expression in three mouse models of experimental hepatitis. Cytokine. 2002;19:115-20
39. Grattendick KJ, Nakashima JM, Feng L, Giri SN, Margolin SB. Effects of three anti-TNF-alpha drugs: etanercept, infliximab and pirfenidone on release of TNF-alpha in medium and TNF-alpha associated with the cell in vitro. Int Immunopharmacol. 2008;8:679-87
40. Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S. et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annual review of immunology. 2006;24:353-89
41. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783-801
42. Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS pathogens. 2009;5:e1000681
43. Sen R, Smale ST. Selectivity of the NF-{kappa}B response. Cold Spring Harbor perspectives in biology. 2010;2:a000257
44. Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857-61
45. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45-65
46. Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-kappaB: effects on liver homeostasis and beyond. Endocrine reviews. 2007;28:365-86
47. Garcia-Lazaro JF, Thieringer F, Luth S, Czochra P, Meyer E, Renteria IB. et al. Hepatic over-expression of TGF-beta1 promotes LPS-induced inflammatory cytokine secretion by liver cells and endotoxemic shock. Immunol Lett. 2005;101:217-22
48. Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM. et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:1548-53
49. Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A. 2005;102:10898-903
50. Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673-7
51. Yin Z, Ellis EC, Nowak G. Isolation of mouse hepatocytes for transplantation: a comparison between antegrade and retrograde liver perfusion. Cell Transplant. 2007;16:859-65
52. van Rooijen N, van Kesteren-Hendrikx E. "In vivo" depletion of macrophages by liposome-mediated "suicide". Methods Enzymol. 2003;373:3-16
53. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ. et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59:347-57
Author contact
![]() Corresponding authors: Liu Cao, Key Laboratory of Medical Cell Biology, China Medical University, 77 Pu-He Road, Shenyang, 110122, P.R.China. Email: caoliucmu.edu.cn or Yizheng Wang, Institute of Neuroscience, SIBS, 320 Yue-Yang Road, Shanghai, 200031, P. R. China. Email: yzwangac.cn
Corresponding authors: Liu Cao, Key Laboratory of Medical Cell Biology, China Medical University, 77 Pu-He Road, Shenyang, 110122, P.R.China. Email: caoliucmu.edu.cn or Yizheng Wang, Institute of Neuroscience, SIBS, 320 Yue-Yang Road, Shanghai, 200031, P. R. China. Email: yzwangac.cn