Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(11):1272-1280. doi:10.7150/ijbs.12108 This issue Cite
Research Paper
AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms
Yu-Lu Dai, Su-Ling Huang, Ying Leng ![]()
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zu Chong Zhi Road 555, Shanghai 201203, China
Received 2015-3-12; Accepted 2015-8-12; Published 2015-9-14
Abstract

The role of AMP-activated protein kinase (AMPK) in pancreatic β-cell apoptosis is still controversial, and the reasons for the discrepancies have not been clarified. In the current study, we observed the effects of two well-known AMPK activators 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) and metformin, on apoptosis in rat insulinoma INS-1E cells, and further explored their possible mechanisms. Both AICAR and metformin protected INS-1E cells from palmitate-induced apoptosis, as reflected by decreases in both cleaved caspase 3 protein expression and caspase 3/7 activity, and these protective effects were abrogated by AMPK inhibitor compound C. The protective action of AICAR was probably mediated by the suppression of triacylglycerol accumulation, increase in Akt phosphorylation and decrease in p38 MAPK phosphorylation, while metformin might exert its protective effect on INS-1E cells by decreases in both JNK and p38 MAPK phosphorylation. All these regulations were dependent on AMPK activation. However, under standard culture condition, AICAR increased JNK phosphorylation and promoted INS-1E cell apoptosis in an AMPK-dependent manner, whereas metformin showed no effect on apoptosis. Our study revealed that AMPK activators AICAR and metformin exhibited different effects on INS-1E cell apoptosis under different culture conditions, which might be largely attributed to different downstream mediators. Our results provided new and informative clues for better understanding of the role of AMPK in β-cell apoptosis.
Keywords: AICAR, Metformin, AMPK, INS-1E cells, apoptosis
Introduction
As cellular energy sensor, AMPK exerts pleiotropic benefits to control metabolic homeostasis by regulating glucose and lipid metabolism in insulin-sensitive tissues including liver, skeletal muscle and adipose tissue [1]. However, its role in β-cells still remains controversial and the major dispute arises in its role in β-cell apoptosis, which could be largely attributed to differences in cell lines and culture conditions [2]. AICAR, a specific AMPK activator, was reported to inhibit high glucose-induced apoptosis in INS-1E cells [3], but could also synergize with glucose to induce apoptosis in MIN6N8 cells [4]. The seemingly contradictory results suggested that effect of AICAR on β-cell apoptosis under the same culture condition might be cell line specific. Therefore, for studies designed to explore the mechanisms of effects of AMPK activators on β-cell apoptosis, it seems more rational to carry out the studies in the same cell line. However, multiple investigations showed that AMPK activators exhibited different effects on apoptosis in the same cell line under different culture conditions. For instance, AICAR could protect β-cells against palmitate-induced apoptosis, but could also induce apoptosis under standard culture condition in MIN6 cells [5-7]. This was also the case for metformin in this cell line, and regulation of apoptosis were dependent on activation of AMPK by metformin [5, 8]. Similarly, AICAR was demonstrated to inhibit palmitate-induced apoptosis in INS-1E cells, but also promote apoptosis under standard culture condition [9, 10]. However, whether the antiapoptotic or proapoptotic effect of AICAR in this cell line required activation of AMPK is unclear. In addition, effects of metformin on apoptosis under palmitate-challenged and standard culture conditions in INS-1E cells have not been defined.
In pathological state, β-cell apoptosis is primarily initiated by chronic exposure to elevated concentrations of glucose and saturated fatty acids, which is currently defined as 'glucolipotoxicity' and could be induced by chronic exposure of β-cells to palmitate [11-13]. The precise biochemical basis of glucolipotoxicity is still under extensive investigation. Emerging evidences indicate that abnormal lipid partitioning consequent to dysregulated nutrition metabolism is considered to be fundamental. In overt diabetes state, chronic exposure of excessive glucose leads to accumulation of malonyl-CoA, an allosteric inhibitor of carnitine palmitoyltransferase 1 (CPT1). CPT1 is the rate-limiting enzyme of fatty acid oxidation. As a consequence, superfluous fatty acids incorporate into lipids such as triacylglycerol (TG), ceramide, diacylglycerol and other metabolic intermediates, most of which are detrimental to β-cells [12, 14-16]. In liver and muscle, activation of AMPK increases fatty acid oxidation and inhibits lipogenesis through phosphorylating its substrate acetyl coenzyme A carboxylase (ACC), leading to decreased lipids deposition [1]. However, it remains unclear whether AMPK activators could also exert such lipid-lowering effect in palmitate-challenged β-cells. Moreover, effects of AMPK activators on β-cell lipid deposition were infrequently covered in studies on their protective effect on palmitate-induced apoptosis.
Previous investigations have shown that multiple signalling pathways are involved in palmitate-induced β-cell apoptosis. Exposure of β-cells to palmitate impairs phosphatidylinositol 3-kinase (PI3K)/Akt signalling pathway, and there is evidence that activation of PI3K/Akt signalling improves β-cell function and prevents palmitate-induced apoptosis [17, 18]. Moreover, chronic exposure to palmitate activates mitogen-activated protein kinases (MAPKs) signalling, c-jun N-terminal kinase (JNK) and p38 MAPK in particular. JNK and p38 MAPK are activated via various cellular stress and trigger apoptosis in several cell lines [19]. This is also the case in β-cells that activation of JNK and p38 MAPK has been proposed to be involved in palmitate-induced apoptosis [20], and their specific inhibitors could prevent apoptosis [6, 21].
In the present study, we compared the effects of AICAR and metformin on apoptosis in rat insulinoma INS-1E cells, and further investigated the underlying mechanisms including lipid metabolism and alterations in Akt and MAPK signalling. These results provided additional insights to resolve the controversies related to the effects of AMPK on β-cell apoptosis.
Materials and Methods
Chemicals and reagents
AICAR and metformin were purchased from Toronto Research Chemicals (ON, Canada) and Sigma-Aldrich (MO, USA) respectively. Compound C and JNK-IN-8 were from Millipore (MA, USA) and Selleck (China) respectively. Palmitate/BSA solution was prepared as previously described [22]. In brief, 100 mM palmitate (Sigma-Aldrich) stock was dissolved in 5% (w/v) fatty acid free BSA (PAA Laboratories, QLD, Australia) in a 55℃ water bath and filtered. The palmitate and BSA solutions were cooled to room temperature and diluted in serum-free medium to concentrations of 0.25 mM and 0.25%, respectively.
Cell culture
Rat insulinoma INS-1E cells (a kind gift from Dr L.G. Luo, Brown University Medical School, USA) (passage 11-25) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (11.1 mM glucose), supplemented with 10% (v/v) FBS (Hyclone, UT, USA), 1 mM sodium pyruvate, 10 mM HEPES, 50 μM β-mercaptoethanol, 100 units/ml penicillin and 100 μg/ml streptomycin at 37℃ and 5% CO2. For experiments, cells were incubated in the presence of 0.25 mM palmitate (palmitate-challenged condition) or in the absence of palmitate (standard culture condition) for indicated time.
Caspase 3/7 activity assay
After culturing in black 96-well optical-bottom plates for 24 h, cells were exposed to 0.25 mM palmitate with or without compounds for 16 h. Caspase 3/7 activity was measured with Apo-ONE® Homogeneous Caspase-3/7 Assay kit (Promega, WI, USA) according to manufacturer's instruction. Briefly, equal volumes of Caspase-3/7 reagent were added to each sample and incubated for 3 h at room temperature. Fluorescence was determined at lengths of 499 nm/521 nm (excitation/emission).
Fatty acid oxidation
Fatty acid oxidation was determined by measurement of 3H2O released from [3H]-palmitatic acid. In brief, after exposure to 0.25 mM palmitate with or without compounds for 10 h, cells were further incubated in fresh medium containing 0.5 μCi/ml [3H]-palmitatic acid (PerkinElmer, MA, USA) and 1 mM carnitine at 37℃ for 2 h. The supernatant was collected and mixed with 10% activated charcoal (Sigma-Aldrich), followed by centrifugation at 13,000 rpm for 20 min. 3H2O in the aqueous phase was measured by liquid scintillation counting (Perkin Elmer Trilu, MA, USA) and normalized to protein contents.
Measurement of cellular TG content
Cells cultured in 6-well plates were exposed to 0.25 mM palmitate with or without compounds for 10 h and then harvested by trypsinization, pelleted by centrifugation, resuspended in distilled water and ultrasonized. Total TG content in the lysate was determined enzymatically with commercial kits (East Ou Jin Ma Biotech, Wenzhou, China). Absorbance was normalized to protein contents.
Western blot
Samples were prepared as previously described [23]. Equal protein samples (20 or 30 μg) were separated by SDS-PAGE, transferred to PVDF membranes, and blotted with primary antibodies against cleaved caspase 3 (17 kDa), GAPDH (37 kDa), phosphorylated AMPK (Thr172, 62 kDa), phosphorylated ACC (Ser79, 280 kDa), phosphorylated Akt (Ser473, 60 kDa), phosphorylated JNK (Thr183/Tyr185, 54 kDa for p54 JNK and 46 kDa for p46 JNK) and phosphorylated p38 MAPK (Thr180/Tyr182, 43 kDa). Subsequently, samples were incubated with anti-rabbit IgG HRP-linked antibody. Primary antibodies and anti-rabbit IgG HRP-linked antibody were purchased from Cell Signaling Technology (MA, USA) and Bio-Rad (CA, USA) respectively. The signals were detected by the ECLTM Prime Western Blotting Detection Reagent (GE healthcare, Buckinghamshire, UK) and quantified by densitometry (Bio-Rad).
Statistical analysis
All data were presented as means ± SEM. and P value was analyzed by Student's t test or ANOVA. Values of P<0.05 were considered statistically significant.
Results
AICAR and metformin protect INS-1E cells from palmitate-induced apoptosis
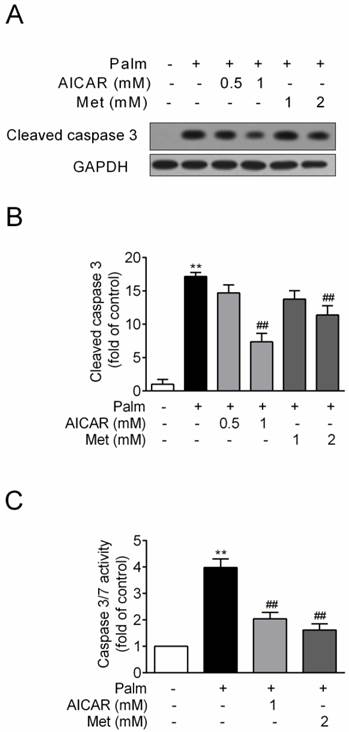
INS-1E cells were exposed to 0.25 mM palmitate with or without compounds for 16 h. Challenge of INS-1E cells with palmitate resulted in a significant increase of cleaved caspase 3 protein expression, an important biomarker of apoptosis, and this index was markedly reduced by 57% and 34% in the presence of 1 mM AICAR and 2 mM metformin respectively (P<0.01 vs palmitate-exposed cells; Fig. 1A and 1B). Meanwhile, AICAR and metformin showed similar inhibition of palmitate-induced apoptosis in terms of decreased caspase3/7 activity (P<0.01 vs palmitate-exposed cells; Fig. 1C).
Effects of AICAR and metformin on palmitate-induced INS-1E cell apoptosis. INS-1E cells were exposed to 0.25 mM palmitate with or without AICAR or metformin for 16 h, followed by evaluation of apoptosis. (A, B) Apoptosis was evaluated by immunoblotting of cleaved caspase 3 (representative immunoblots; quantification of four independent experiments). (C) Apoptosis was evaluated by measuring caspase 3/7 activity (quantification of three independent experiments). **P<0.01 vs control (cells exposed to 0.25% BSA); ##P<0.01 vs cells exposed to 0.25 mM palmitate. Palm, palmitate; Met, metformin.
AICAR and metformin prevent palmitate-induced INS-1E cell apoptosis in an AMPK-dependent manner
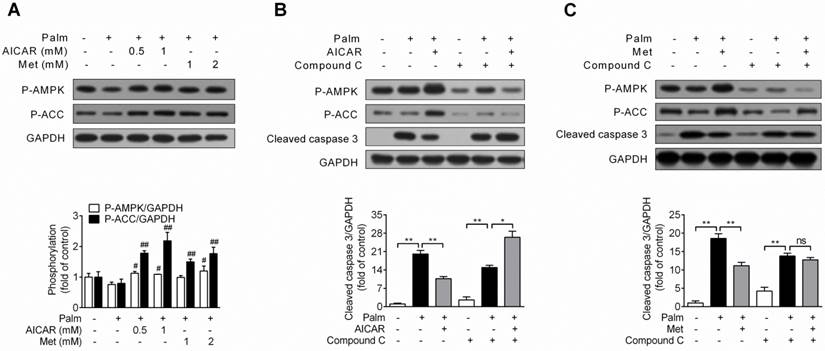
Under condition of palmitate-induced apoptosis, both AICAR and metformin increased AMPK and ACC phosphorylation (P<0.05 and P<0.01 vs palmitate-exposed cells; Fig. 2A). Furthermore, in combination with AMPK inhibitor compound C (10 μM), the protective effect of AICAR (Fig. 2B) and metformin (Fig. 2C) were abrogated, as shown by relief of decreased cleaved caspase 3 protein expression. These findings demonstrated that prevention of palmitate-induced INS-1E cell apoptosis by AICAR and metformin were dependent on their activation of AMPK.
Role of AMPK activation in prevention of palmitate-induced INS-1E cell apoptosis by AICAR and metformin. (A) Effects of AICAR or metformin on AMPK and ACC phosphorylation in INS-1E cells exposed to 0.25 mM palmitate with or without compounds for 16 h. Representative immunoblots were shown and protein contents were quantified separately from four independent experiments. (B, C) INS-1E cells were preincubated with or without compound C (10 μM) for 30 min, and then exposed to 0.25 mM palmitate with or without 1 mM AICAR (B) or 2 mM metformin (C) for 16 h, followed by immunoblotting of AMPK and ACC phosphorylation and cleaved caspase 3 (representative immunoblots; quantification of cleaved caspase 3 protein levels from four independent experiments). #P<0.05 and ##P<0.01 vs cells exposed to 0.25 mM palmitate; *P<0.05 and **P<0.01 vs corresponding columns as indicated; ns, not significant. Palm, palmitate; Met, metformin.
Effects of AICAR and Metformin on fatty acid oxidation and TG accumulation in palmitate-challenged INS-1E cells
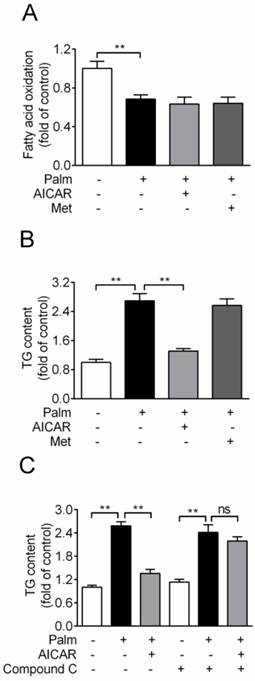
Based on the principle of glucolipotoxicity, effects of AICAR and metformin on fatty acid oxidation and TG content were detected. Chronic exposure of INS-1E cells to 0.25 mM palmitate resulted in a ~30% reduction of fatty acid oxidation, which was not rescued by an incubation with 1 mM AICAR or 2 mM metformin (Fig. 3A). In addition, cellular TG content increased 2.7-fold with palmitate incubation for 16 h. AICAR significantly inhibited TG accumulation, whereas metformin had no effect (P<0.01 vs palmitate-exposed cells; Fig. 3B). Furthermore, the lipid-lowering effect of AICAR was completely abrogated in the presence of compound C (Fig. 3C). This indicated that AICAR might inhibit palmitate-induced TG accumulation through activation of AMPK.
Effects of AICAR and metformin on fatty acid oxidation and TG accumulation in INS-1E cells exposed to palmitate. (A) Fatty acid oxidation was determined after INS-1E cells were exposed to 0.25 mM palmitate with or without 1 mM AICAR or 2 mM metformin for 10 h and further incubated with [3H]-palmitate for 2 h. Fatty acid oxidation was determined by liquid scintillation counting of 3H2O released into the aqueous phase. (B) Cellular TG content was evaluated after INS-1E cells were exposed to 0.25 mM palmitate with or without 1 mM AICAR or 2 mM metformin. (C) INS-1E cells were pretreated with compound C (10 μM) for 30 min and then exposed to 0.25 mM palmitate with or without 1 mM AICAR for 10 h, followed by measurement of TG content. Three or four independent experiments were performed. **P<0.01 vs corresponding columns as indicated; ns, not significant. Palm, palmitate; Met, metformin.
Signalling mechanisms involved in AICAR and metformin inhibition of palmitate-induced INS-1E cell apoptosis
Since impairment of PI3K/Akt signalling pathway and activation of JNK and p38 MAPK are involved in palmitate-induced β-cell apoptosis, effects of AICAR and metformin on Akt, JNK and p38 MAPK phosphorylation were first investigated under palmitate-challenged condition. After incubation with 0.25 mM palmitate for 16 h, Akt phosphorylation decreased, JNK phosphorylation increased and p38 MAPK phosphorylation remained unchanged. Under this stress condition, AICAR could largely reverse the inhibitory effect of palmitate on Akt phosphorylation and significantly decrease p38 MAPK phosphorylation. However, an unexpected additional increase in JNK phosphorylation was observed. On the other hand, metformin scarcely affected Akt phosphorylation, but significantly decreased both JNK and p38 MAPK phosphorylation (P<0.05 and P<0.01 vs palmitate-exposed cells; Fig. 4A).
Effects of AICAR and metformin on Akt, JNK and p38 MAPK phosphorylation in palmitate-challenged INS-1E cells. (A) INS-1E cells were exposed to 0.25 mM palmitate with or without 1 mM AICAR or 2 mM metformin for 16 h, followed by immunoblotting of Akt, JNK and p38 MAPK phosphorylation. (B, C) INS-1E cells were preincubated with or without compound C (10 μM) for 30 min, and then exposed to 0.25 mM palmitate with or without 1 mM AICAR (B) or 2 mM metformin (C) for 16 h, followed by immunoblotting of Akt (only for AICAR), JNK and p38 MAPK phosphorylation. Representative immunoblots were shown and protein contents were quantified separately from three or four independent experiments. *P<0.05 and **P<0.01 vs control (cells exposed to 0.25% BSA) or corresponding columns as indicated; #P<0.05 and ##P<0.01 vs cells exposed to 0.25 mM palmitate. Palm, palmitate; Met, metformin.
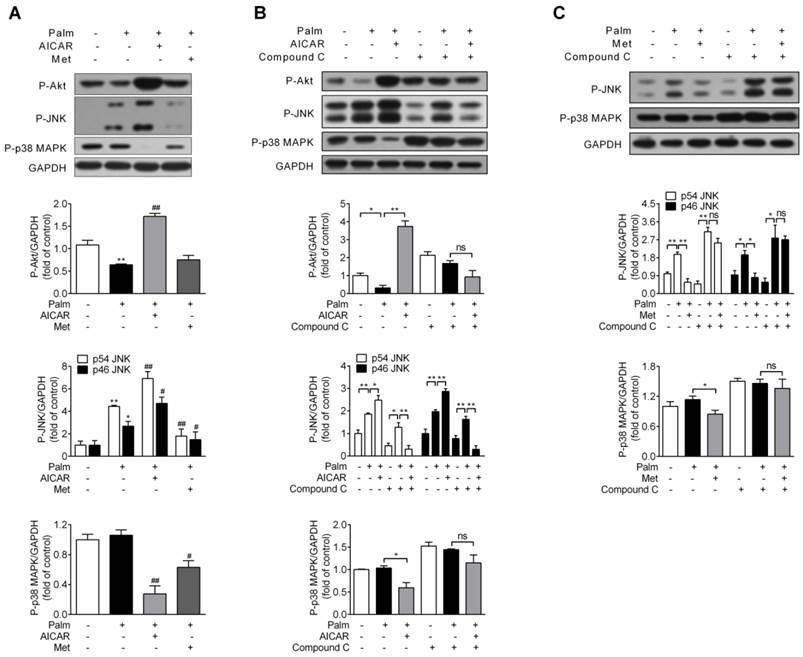
Given that both AICAR and metformin could protect against palmitate-induced apoptosis in an AMPK-dependent manner, we studied whether regulations on Akt, JNK and p38 MAPK by AICAR and metformin were downstream of AMPK activation. After application of compound C, AICAR-mediated increase of Akt and JNK phosphorylation and decrease of p38 MAPK phosphorylation were abolished (Fig 4B). Moreover, suppression of both JNK and p38 MAPK phosphorylation by metformin was also reversed by application of compound C (Fig 4C). This indicated that regulations of these kinases by AICAR or metformin were all dependent on their activation of AMPK signalling pathway.
AMPK-activated JNK contributes to AICAR-induced INS-1E cell apoptosis under standard culture condition
Considering the possibility that activation of JNK by AICAR might cause cell apoptosis under standard culture condition, INS-1E cells were incubated with 1 mM AICAR or 2 mM metformin for 16 h. Assessment of JNK phosphorylation and cleaved caspase 3 protein expression was performed. Under this condition, chronic treatment of AICAR indeed increased JNK phosphorylation, whereas metformin decreased JNK phosphorylation (P<0.01 vs control) (Fig. 5A). Simultaneously, we observed a moderate increase in the cleaved form of caspase 3 (P<0.05 vs control) after treatment with AICAR, but metformin had no effect on it (Fig. 5B). This demonstrated that AICAR, but not metformin, would trigger cell apoptosis under standard culture condition.
Effects of AICAR and metformin on INS-1E cell apoptosis under standard culture condition. (A, B) Under standard culture condition, INS-1E cells were stimulated with 1 mM AICAR or 2 mM metformin for 16 h, followed by immunoblotting of JNK phosphorylation (A) and cleaved caspase 3 (B). 0.25 mM palmitate was set as positive control. (C) INS-1E cells were preincubated with or without JNK-IN-8 (0.3 μM) for 30 min, and then exposed to 1 mM AICAR for 16 h, followed by immunoblotting of JNK phosphorylation and cleaved caspase 3. (D) INS-1E cells were preincubated with or without compound C (10 μM) for 30 min, and then exposed to 1 mM AICAR for 16 h, followed by immunoblotting of AMPK, ACC and JNK phosphorylation and cleaved caspase 3. Representative immunoblots were shown and protein contents of cleaved caspase 3 and P-JNK were quantified. Three or four independent experiments were performed. *P<0.05 and **P<0.01 vs control (cells not exposed to palmitate) or corresponding columns as indicated; ns, not significant. Palm, palmitate; Met, metformin.
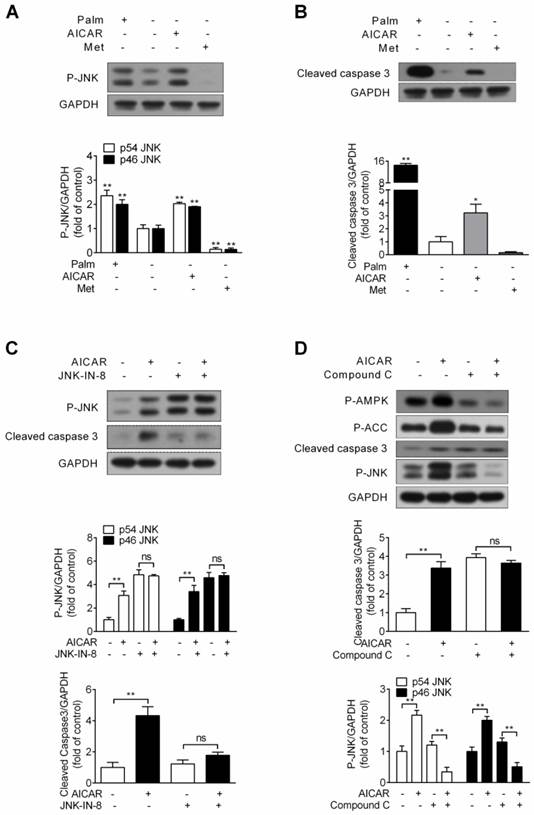
The JNK inhibitor JNK-IN-8 was then applied to identify the role of AICAR-activated JNK in mediating cell apoptosis. As Figure 5C showed, AICAR failed to increase JNK phosphorylation and cleaved caspase 3 protein expression in the presence of JNK-IN-8, implicating that JNK activation contributed to AICAR-induced apoptosis. Moreover, AICAR markedly increased AMPK and ACC phosphorylation, and AICAR was incapable promoting cell apoptosis and increasing JNK phosphorylation in the presence of compound C (Fig. 5D). This revealed that AMPK activation was responsible for AICAR-mediated cell apoptosis and JNK activation.
Discussion
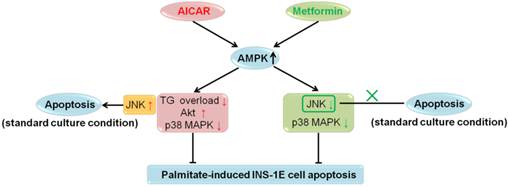
In recent years, extensive in vitro and in vivo studies on the role of AMPK in β-cell apoptosis were carried out; however, no definite conclusions have been drawn [2]. In the current investigation, we studied the effects of AICAR and metformin on apoptosis under both palmitate-challenged and standard culture conditions in rat insulinoma INS-1E cells. We showed that both AICAR and metformin could exert AMPK-dependent protection against palmitate-induced apoptosis via different downstream mechanisms. AICAR might prevent apoptosis by reversing TG overload, activating Akt and inhibiting p38 MAPK. By contrast, metformin protected INS-1E cells probably through suppression of JNK and p38 MAPK. All these regulations were dependent on activation of AMPK signalling pathway. On the other hand, under standard culture condition, AICAR would induce apoptosis through AMPK-mediated JNK activation; while metformin did not induce apoptosis (Fig. 6). Therefore, we speculated that different culture conditions and differences in downstream mediators were the primary causes for the differential regulations of apoptosis by AICAR and metformin in INS-1E cells. This speculation provided a new insight into the current understanding of the controversies regarding the role of AMPK in β-cell apoptosis.
Summary for regulations of AICAR and metformin on INS-1E cell apoptosis under palmitate-challenged and standard culture conditions.
According to the basis of glucolipotoxicity, accumulation of detrimental lipids is an instrumental initiator of β-cell apoptosis. In β-cells, overexpression of lipogenic transcription factor sterol regulatory element binding protein-1c (SREBP-1c) could reproduce the phenotypes of glucolipotoxicity including excess TG deposition and cell apoptosis [9, 24]. Moreover, progressive TG accumulation contributes to the onset of β-cell failure in Zucker diabetic fatty (ZDF) rat, a typical type 2 diabetes model characterized with β-cell apoptosis [25-27]. AICAR and metformin have been claimed to decrease hepatic lipid accumulation by inducing fatty acid oxidation and suppressing lipid synthesis [28-30]. Similarly, previous findings suggested that both TG overload and apoptosis could be reversed by AICAR in β-cells with SREBP-1c overexpression [9, 24]. Besides, AICAR and troglitazone might preserve β-cell function in ZDF rats probably through reducing lipids deposition [31, 32], further highlighting the significance of the hypolipidemic action of AMPK activators in protecting β-cells. Therefore, effects of AICAR and metformin on lipid contents were determined in the context of palmitate-induced apoptosis, and herein TG was set as a paradigm of deposited lipids. We found that AICAR could reverse palmitate-induced TG accumulation, and this effect mainly relied on activation of AMPK and inhibition of ACC, similar to its effect on peripheral tissues. However, metformin had no effect on TG overload, as supported by findings that metformin protected human islets from lipotoxicity without changing TG levels [33]. The reasons for the different regulations of AICAR and metformin on TG accumulation are unknown and require further investigation.
Since metformin did not affect TG deposition, other mechanisms might be involved in mediating its antiapoptotic action. As mentioned above, PI3K/Akt and MAPK signalling pathways are involved in palmitate-induced β-cell apoptosis, and there is preliminary evidence that inhibition of JNK was implicated in metformin-mediated prevention of ER stress-induced β-cell apoptosis [34]. Based on this, we studied the effect of metformin on JNK phosphorylation in palmitate-challenged INS-1E cells, and found that metformin markedly inhibited JNK phosphorylation. Unexpectedly, AICAR showed an additional increase in JNK phosphorylation in the presence of palmitate. Moreover, both AICAR and metformin could inhibit p38 MAPK phosphorylation. However, AICAR significantly increased Akt phosphorylation, whereas metformin scarcely affected it. Therefore, different regulations of TG overload and phosphorylation of JNK and Akt obviously indicated that AICAR and metformin prevented palmitate-induced INS-1E cell apoptosis via different downstream mechanisms. According to previous investigations, whether AICAR and metformin could regulate Akt, JNK or p38 MAPK through activation of AMPK remained unclear. Nevertheless, our study clearly showed that effects of AICAR or metformin on Akt (only for AICAR), JNK and p38 MAPK were dependent on their activation of AMPK signalling, further suggesting their involvement in AMPK-dependent prevention of palmitate-induced apoptosis by AICAR or metformin.
Sustained activation of JNK triggers apoptosis in response to multiple types of stress and has been proposed to mediate apoptosis in β-cells chronically exposed to palmitate [19, 20], which was successfully achieved in our cell preparations. In our study, an additional activation of JNK by AICAR was observed in the presence of palmitate, raising the possibility that AICAR-induced JNK activation could cause INS-1E cell apoptosis. In the present study, we observed that AICAR stimulated JNK and triggered cell apoptosis under standard culture condition, which was blunted by application of JNK inhibitor JNK-IN-8. Besides, cell apoptosis and JNK activation were both abrogated as AMPK signalling was inhibited by compound C. These results unveiled a conceivable sequence from AMPK, over JNK to caspase 3, which was responsible for AICAR-induced apoptosis under this condition. The same mechanism has been demonstrated to mediate AICAR-triggered apoptosis in MIN6 cells [6]. Additionally, AICAR was reported to cause apoptosis in rat and mouse primary β-cells in an AMPK-dependent manner, and this proapoptotic action was absent in β-cells isolated from AMPKα2-deficient mice [21]. As for metformin, it exerted remarkable suppression on JNK and did not cause apoptosis. Based on the causal role of JNK activation in AICAR-triggered apoptosis, inhibition of JNK was likely a key contributor in the non-apoptotic effect of metformin.
Given that JNK activation contributed to AICAR-induced INS-1E cell apoptosis under standard culture condition, AICAR should have synergized with palmitate to cause apoptosis. However, AICAR-mediated reversal of TG overload, activation of Akt and inhibition of p38 MAPK under palmitate-challenged condition were sufficient to compensate for the impairment caused by JNK activation. Thus, an antiapoptotic action was ultimately presented. Nevertheless, TG was not overloaded under standard culture condition (without exposure of palmitate), and thus the protective part contributed by reversion of palmitate-induced TG accumulation was gone. Therefore, the detrimental role of JNK activation might occur, thereby triggering apoptosis.
In conclusion, the present study demonstrated that different regulations of AICAR and metformin on INS-1E cell apoptosis were caused by differences in culture conditions and downstream mediators. Our work should be informative for future investigations focusing on the effects of AMPK activators on apoptosis in other β-cell lines or primary β-cells.
Abbreviations
ACC, acetyl coenzyme A carboxylase; AICAR, 5-aminoimidazole-4-carboxamide ribonucleoside; AMPK, AMP-activated protein kinase; BSA, bovine serum albumin; CPT1, carnitine palmitoyltransferase 1; ER, endoplasmic reticulum; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; JNK, c-jun N-terminal kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; RPMI, Roswell Park Memorial Institute; SREBP-1c, sterol regulatory element binding protein-1c; TG, triacylglycerol; ZDF, Zucker diabetic fatty.
Acknowledgements
This study was supported by the National Nature Science Foundation of China (grant numbers 81202570).
Contribution Statement
YLD, SLH and YL designed the study. YLD and SLH acquired, analysed and interpreted the data. YLD wrote the manuscript. SLH and YL reviewed/edited the manuscript. All authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407-16
2. Fu A, Eberhard CE, Screaton RA. Role of AMPK in pancreatic beta cell function. Mol Cell Endocrinol. 2013;366:127-34
3. Nyblom HK, Sargsyan E, Bergsten P. AMP-activated protein kinase agonist dose dependently improves function and reduces apoptosis in glucotoxic β-cells without changing triglyceride levels. J Mol Endocrinol. 2008;41:187-94
4. Kim WH, Lee JW, Suh YH, Lee HJ, Lee SH, Oh YK. et al. AICAR potentiates ROS production induced by chronic high glucose: roles of AMPK in pancreatic β-cell apoptosis. Cell Signal. 2007;19:791-805
5. Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X. et al. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10:268-77
6. Kefas BA, Cai Y, Ling Z, Heimberg H, Hue L, Pipeleers D. et al. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol. 2003;30:151-61
7. Cai Y, Martens GA, Hinke SA, Heimberg H, Pipeleers D, Van de Casteele M. Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic Biol Med. 2007;42:64-78
8. Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H. et al. Metformin-induced stimulation of AMP-activated protein kinase in β-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol. 2004;68:409-16
9. Li J, Liu X, Ran X, Chen J, Li X, Wu W. et al. Sterol regulatory element-binding protein-1c knockdown protected INS-1E cells from lipotoxicity. Diabetes Obes Metab. 2010;12:35-46
10. Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M. et al. AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27:1706-23
11. Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev. 2008;29:351-66
12. El-Assaad W, Joly E, Barbeau A, Sladek R, Buteau J, Maestre I. et al. Glucolipotoxicity alters lipid partitioning and causes mitochondrial dysfunction, cholesterol, and ceramide deposition and reactive oxygen species production in INS832/13 β-cells. Endocrinology. 2010;151:3061-73
13. Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2010;1801:289-98
14. Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in β-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405-13
15. Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116:1802-12
16. Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M. et al. A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J. 2011;435:267-76
17. Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β-cells (INS-1). J Biol Chem. 2002;277:49676-84
18. Elghazi L, Rachdi L, Weiss AJ, Cras-Meneur C, Bernal-Mizrachi E. Regulation of beta-cell mass and function by the Akt/protein kinase B signalling pathway. Diabetes Obes Metab. 2007;9(Suppl 2):147-57
19. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838-49
20. Natalicchio A, Labarbuta R, Tortosa F, Biondi G, Marrano N, Peschechera A. et al. Exendin-4 protects pancreatic beta cells from palmitate-induced apoptosis by interfering with GPR40 and the MKK4/7 stress kinase signalling pathway. Diabetologia. 2013;56:2456-66
21. Kefas BA, Heimberg H, Vaulont S, Meisse D, Hue L, Pipeleers D. et al. AICA-riboside induces apoptosis of pancreatic beta cells through stimulation of AMP-activated protein kinase. Diabetologia. 2003;46:250-4
22. Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology. 2006;147:3398-407
23. Huang SL, Yu RT, Gong J, Feng Y, Dai YL, Hu F. et al. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia. 2012;55:1469-81
24. Diraison F, Parton L, Ferre P, Foufelle F, Briscoe CP, Leclerc I. et al. Over-expression of sterol-regulatory-element-binding protein-1c (SREBP1c) in rat pancreatic islets induces lipogenesis and decreases glucose-stimulated insulin release: modulation by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR). Biochem J. 2004;378:769-78
25. Unger RH. How obesity causes diabetes in Zucker diabetic fatty rats. Trends Endocrin Met. 1997;8:276-82
26. Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. Faseb J. 2001;15:312-21
27. Tiano JP, Delghingaro-Augusto V, Le May C, Liu S, Kaw MK, Khuder SS. et al. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents beta cell failure in rodent models of type 2 diabetes. J Clin Invest. 2011;121:3331-42
28. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167-74
29. Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H. et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898-905
30. Henriksen BS, Curtis ME, Fillmore N, Cardon BR, Thomson DM, Hancock CR. The effects of chronic AMPK activation on hepatic triglyceride accumulation and glycerol 3-phosphate acyltransferase activity with high fat feeding. Diabetol Metab Syndr. 2013;5:29
31. Higa M, Zhou YT, Ravazzola M, Baetens D, Orci L, Unger RH. Troglitazone prevents mitochondrial alterations, β cell destruction, and diabetes in obese prediabetic rats. Proc Natl Acad Sci U S A. 1999;96:11513-8
32. Yu X, McCorkle S, Wang M, Lee Y, Li J, Saha AK. et al. Leptinomimetic effects of the AMP kinase activator AICAR in leptin-resistant rats: prevention of diabetes and ectopic lipid deposition. Diabetologia. 2004;47:2012-21
33. Lupi R, Del Guerra S, Fierabracci V, Marselli L, Novelli M, Patane G. et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes. 2002;51(Suppl 1):S134-7
34. Jung TW, Lee MW, Lee YJ, Kim SM. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem Biophys Res Commun. 2012;417:147-52
Author contact
![]() Corresponding author: Professor Ying Leng, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zu Chong Zhi Road 555, Shanghai 201203, China, Tel: + 86 21 50806059, Fax: + 86 21 50807088, E-mail: ylengac.cn
Corresponding author: Professor Ying Leng, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zu Chong Zhi Road 555, Shanghai 201203, China, Tel: + 86 21 50806059, Fax: + 86 21 50807088, E-mail: ylengac.cn