Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(1):120-132. doi:10.7150/ijbs.12852 This issue Cite
Research Paper
Photodynamic-therapy Activates Immune Response by disrupting Immunity Homeostasis of Tumor Cells, which Generates Vaccine for Cancer Therapy
Yuanhong Zheng1, Guifang Yin1, Vanminh Le1, Anle Zhang1, Siyu Chen2, ![]() , Xin Liang1,
, Xin Liang1, ![]() , Jianwen Liu1,
, Jianwen Liu1, ![]()
1. State Key Laboratory of Bioreactor Engineering & Shanghai Key Laboratory of New Drug Design, School of pharmacy, East China University of Science and Technology, #268, 130 Meilong Road, Shanghai 200237, PR China
2. Department of Oncology, Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, 200092, P.R. China
Received 2015-6-1; Accepted 2015-10-21; Published 2016-1-1
Abstract

Photodynamic therapy (PDT), a regulatory approved cancer treatment, is reported to be capable of causing immunogenic apoptosis. The current data reveal PDT can cause the dysregulation of “eat me” and “don't eat me” signal by generating reactive oxygen species (ROS) -mediated endoplasmic reticulum (ER) stress. This dysregulation probably contribute to the increased uptake of PDT-killed Lewis lung carcinoma (LLC) cells by homologous dendritic cells (DCs), accompanied by phenotypic maturation (CD80high, CD86high, and CD40high) and functional stimulation (NOhigh, IL-10absent) of dendritic cells as well as subsequent T-cell responses. Morevover, C57BL/6 mice vaccinated with dendritic cells (DCs) pulsed with PDT-treated LLCs (PDT-DCs) or PDT-treated LLCs alone (PDT-LLCs) exhibited potent immunity against LLC tumors. In the current study, the PDT-induced immune response was characterized as a process related with the dysregulation of “eat me” signal and “don't eat me” signal, revealing the possibility for developing PDT into an antitumor vaccination strategy for personalized cancer immunotherapy.
Keywords: DAMPs, Immunogenic apoptosis, Antitumor vaccination, Hypericin, CD47
Introduction
Apoptosis had been considered to be immunologically silent, but recent studies distinguish between non-immunogenic and immunogenic apoptosis [1-4].
Immunogenic apoptosis is characterized by secretion of damage-associated molecular patterns (DAMPs) and the ability to elicit an antitumor immune response [5].
Photodynamic therapy (PDT) is a regulatory approved modality for the treatment of many malignant carcinomas, which is able to cause immunogenic apoptosis [5-7]. It inflicts trauma in the targeted lesion through oxidative stress triggered by photosensitizers [8]. Photosensitizers can be excited by visible light in the presence of oxygen. Hypericin is a naturally occurring photosensitizer synthesized by Hypericum plants (St. John's wort) and it's considered to be a valuable photosensitizer for PDT due to its potent photosensitizing characteristics [9], minimal dark toxicity and high singlet oxygen quantum yield [7, 10, 11]. Hypericin mainly locates to the membranes of the endoplasmic reticulum and Golgi apparatus [12, 13]. For endoplasmic reticulum (ER) stress and reactive oxygen species (ROS) production are important features of immunogenic apoptosis [14], while hypericin mediated photodynamic therapy just fit the criterion for ROS-based ER stress, this ROS-based anticancer therapy may also cause surface exposure or secretion of damage-associated molecular patterns (DAMPs) which are also called “eat me” signal and activate the host immune system [15].
DAMPs are molecules present in cells under normal conditions, and exposed on the surface or released from cells upon the stimulus of damage or physical or chemical insult/stress [11] [16]. Calreticulin (CRT), heat shock protein 70 and heat shock protein 90 (HSP70 and HSP90) are three main DAMPs identified on plasma membrane [11]. DAMPs have various effects on dendritic cells (DCs), such as maturation, activation and antigen processing/presentation. Once exposed or released from cells, DAMPs are able to interact with membrane bound or vesicular pattern-recognition receptors (PRRs) [15] [11]. At the same time, tumor cells undergoing immunogenic apoptosis downregulate the expression of “don't eat me” signals, such as surface integrin-associated protein (CD47), a cell surface molecule in the immunoglobulin superfamily. It has been demonstrated that CD47 is involved in diverse physiologic processes including cell migration, T cell and dendritic cell (DC) activation, and axon development [17].
Nowadays, there is an increasing demand for preclinical modeling of the interaction of dendritic cells with the immune system and cancer cells. In addition, an increasing number of clinical trials had been performed using DCs as experimental adjuvant treatments in cancer [18].
In this study, we report that hypericin mediated PDT (hyp-PDT) induced ROS-based oxidative stress can cause dysregulation of DAMPs and CD47 on the surface of tumor cells. This dysregulation leads to the disruption of immune homeostasis sustained by the “eat me signal” and “don't eat me signal”. Furthermore, the interaction between PDT-treated tumor cells and the immune system was affected, triggering tumor-specific immune response. Correspondingly, due to the significant immune response in vivo, both PDT-treated LLCs (PDT-LLC) and DCs co-cultured with PDT-LLCs (PDT-DC) showed potent vaccination efficacy in mouse model. Taken together, these results make it possible to prepare cancer vaccines using in vitro PDT of cell cultures and could facilitate the design of more effective immunotherapeutic strategies.
Methods and materials
Antibodies and reagents
Hypericin purchased from Yuanye bio-technology Co., Ltd (Shanghai, China) was used without any further purification. A stock solution (1mM) was made in Dimethyl Sulphoxide (DMSO), and then stored in the dark at 4◦C. Reduced L-glutathione (GSH) was purchased from Sigma-Aldrich (San Luis, USA). Antibodies against caspase 3/cleaved caspase3 and PARP/cleaved PARP were purchased from GeneTex, Inc. (California, USA). Anti-β-actin antibody was from Santa Cruz Biotechnology (California, USA). Anti-CRT, anti-HSP70 and anti-HSP90 antibodies were purchased from Epitomics Inc. (California, USA). Secondary antibody conjugated with FITC was purchased from Bioworld Technology, Inc. (Atlanta, USA). FITC conjugated-antibodies against CD47, CD80, CD86, CD40, CD4 and CD8 were from eBioscience, Inc. (California, USA), APC conjugated-antibodies against Foxp3, as well as PE conjugated-antibodies against CD25 and IFN-γ. Appropriate isotype control antibodies were also from eBioscience.
Cell lines and Cells culture
Lewis lung carcinomas cells (LLCs), originated as a spontaneous carcinoma of the lung in a C57BL/6 mouse, were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Lewis lung carcinoma cells stably expressing luciferase (LLC-Luc) were purchased from Shanghai innovation biotechnology Co., Ltd. (Shanghai, China). The cell lines were authenticated before receipt by means of short tandem repeat (STR) analysis and were passaged in our laboratory for fewer than 6 months. These cells were cultured in RPMI 1640 medium (Gibco, New York, USA), supplemented with 10% heat-inactivated fetal bovine serum (Gibco, New York, USA), penicillin (100 U/ml) and streptomycin (100 U/ml) (Invitrogen, California, USA). Cells were incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Mice
6 to 8 week male C57BL/6 mice were purchased from Shanghai Laboratory Animal Resource Center (Shanghai, China) and were maintained in a pathogen-free environment that was temperature- and humidity-controlled. Food and water were available ad libitum. Mice were euthanized to harvest primary cells or to end in vivo experiments at indicated time points. All of the animal experiments were carried out according to the guidelines for animal care of Ministry of Science and Technology of the PRC. Ethical approval was given by of the Administrative Panel on Laboratory Animal Care of the Shanghai Putuo Hospital.
DC generation
DCs were generated from C57BL/6 mouse bone marrow progenitor cells according to the procedure previously reported [19]. 80% of the cell population stained positive for CD11c as measured by using flow cytometry.
PDT treatment
For photosensitization, cells were seeded into 6-well plates (Corning, New York, USA) and incubated overnight in complete growth media for cell attachment.
For the experiment, cells were incubated in the dark at 37 °C with or without certain concentrations of hypericin. After 16 hours of incubation, the medium was changed with complete RPMI 1,640 medium. Cells were irradiated under light emitted from a 100-watt quartz-halogen lamp and harvested at indicated time points following irradiation. Light intensity was measured by a photo radiometer (Delta Ohm, Padua, Italy).
Assessment of apoptosis
LLC cells were exposed to PDT treatment and collected 1 hour after irradiation. Cell death was assessed by using an AnnexinV-FITC/PI apoptosis detection kit (Invitrogen, California, USA) as described by the manufacturer's instruction. Samples were analyzed by FACScan (BD Bioscience, California, USA). Data were further analyzed using Flowjo software.
Western blotting
Proteins were extracted and separated in SDS-PAGE gels, and western blot were performed as previously described [20].β-actin was used as the loading control.
Flow cytometric analysis of cell surface proteins
Cells were harvested at the indicated time points following PDT treatment, washed twice with PBS, then fixed in PBS containing 0.25% paraformaldehyde (PFA) for 5 min, washed with cold PBS twice and incubated with primary antibody for 30 min. The cells were washed and incubated with the FITC-conjugated monoclonal or polyclonal secondary antibody for 30 min. Both primary antibody and secondary antibody were diluted in cold blocking buffer (2% FBS in PBS). Each sample was then analyzed by FACScan (BD Bioscience) to identify cell surface HSP70, HSP90, and CRT. Secondary antibody alone was used as the control. Dead cells and cell aggregates were gated out based on light scatter measurements. Subsequently, single parameter histograms and contour maps were drawn. Data were analyzed using Flowjo software and presented as histograms.
For phagocytosis, DCs were stained with a DiO cell membrane green fluorescent probe (Beyotime, Shanghai, China). Tumor cells were subjected to hyp-PDT treatment. Immature DCs (day 6) were co-cultured with tumor cells at a DC/tumor cell ratio of 1:5 for 24h. The cells were fixed in 0.25% paraformaldehyde for 20 minutes, washed in PBS for 20 minutes, and analyzed by flow cytometry.
Immunofluorescence
For surface immunofluorescence analysis, LLC cells were fixed in 0.25% paraformaldehyde, incubated with anti-CRT, anti-HSP70 and anti-HSP90 antibodies, and then with the secondary antibody conjugated with FITC. Fluorescence was imaged with a Nikon A1R laser scanning confocal microscope (Nikon, Tokyo, JP) and NIS-Elements D3.2 software.
Analysis of murine DC maturation, NO, and cytokines
LLC cells exposed to high dose of hyp-PDT treatment with or without GSH were co-incubated with imDCs (day 6) at a ratio of 1:5 (imDCs: LLCs) for 24 h. ImDCs (day 6) were stimulated with 100 ng/ml of Escherichia coli-derived LPS (Sigma-Aldrich, San Luis, USA) for 24 h, and were regarded as positive controls for DC maturation. The cells were detached, washed and stained with the following antibodies: FITC-anti-CD86, anti-CD80 and anti-CD40 according to the manufacturer's instructions. FITC-conjugated armenian hamster IgG and FITC-conjugated rat IgG2a antibodies were used as isotype controls. The DC cells were analyzed using a FACScan (BD Bioscience) flow cytometer. The DCs were gated according to the forward (FSC) and side scatter (SSC) properties and 50,000 viable DCs were acquired for each experiment. The co-culture supernatant derived from these imDCs and LLCs co-incubation experiments was collected for further analysis. NO in the supernatant was quantified by Griess assay [21]. Immunoreactive levels of mouse IL-10 in the media was measured by using mouse IL10 elisa kit (Abcam, Cambridge, UK). The samples were prepared according to the manufacturers' instructions and analyzed on EnSpire® Multimode Plate Reader (PerkinElmer, Massachusetts, USA).
Evaluation of tumor-specific T cell and regulatory T cell (Treg) activated by DCs in vitro
Unpulsed or tumor-loaded DCs were co-cultured with autologous T cells at a ratio of 1:10 on days 0 and 7 of culture in the presence of IL-2 (25-50 IU/mL; Peprotech, Rocky Hill, USA). A week after the last stimulation with DCs, cells were harvested and stained with FITC-anti-CD8 antibody for 30min. After washed with cold PBS, cells were fixed with 2% formaldehyde and permeabilized with 0.5% saponin. They were then incubated with PE-anti-IFN-γ for 30min. PE-conjugated rat IgG2a was used as the isotype control. Gating was performed on CD8+ T cells and the percentage of IFN-γ+ cells was calculated. Additionally, cells were stained for Foxp3, CD25 and CD4 to identify the regulatory T cell (Treg) population according to the manufacturer (Bioscience). A FACScan flow cytometer (BD Bioscience, California, USA) was used for these experiments and the data were analyzed with Flowjo software and presented as dot plots.
Evaluation of tumor-specific T cell and regulatory T cell (Treg) population in vivo
Seven days after tumor rechallenge in all groups of mice, splenocytes were harvested and stimulated with PDT-treated tumor (20:1) and IL-2 (20 U/ml) for 5 days. IFN-γ-producing CD8+ tumor-specific T cell and regulatory T cell (Treg) population were evaluated as described in subsection 2.10.
In-vivo analysis of immune system priming in mice
A total of 35 male C57BL/6 mice were divided into 5 groups (each containing 7 mice): control, F/T-LLC, F/T-DC, PDT-LLC, PDT-DC.
Normal LLC cells were exposed to the highest dose of hyp-PDT (hypericin, 0.25μM; light dose, 1.85J/cm2). Following a 24-hour-recovery, 1×106 cells in 200μL PBS were injected subcutaneously into the left flank of 6-week-old male C57BL/6 mice. Immunization was done twice within 10 days. Control mice were injected with 200μL PBS. Seven days after the second immunization, mice were rechallenged with 5×105 live untreated LLC-Luc cells in the right flank. The 'communication' between the tumor antigenic memory and the adaptive immune system was studied by analyzing the tumor sizes at the rechallenge site. Hence, following the rechallenge, the mice were monitored with in-vivo imaging system for the presence of tumors at the rechallenge site every 4~5 days. The mice were anesthetized with phenobarbital sodium (40mg/kg). D-luciferin solution (150 mg/kg) was injected i.p following anaesthesia (no movement in response to tweezers pinch). Bioluminescence imaging expose times ranged from 1~2min. The imaging continued until the tumors in the control mice became unmanageable, that is, either necrotic or capable of causing movement problems (about 20~30 days).
CTL assay
Seven days after the second vaccination, splenocytes were harvested from all group of mice. CTL assays were performed as previously reported [22].
Statistical analysis
All experiments were repeated no less than three times and the results were presented as the mean of those independent experiments. All data were described as (mean±SD). The Spearman test was used for correlation analyses. Two-tailed Student's T-tests were used to assess differences between the treated and control groups. Statistical analyse was performed by GraphPad prism5 software to evaluate the significance of the difference between groups, shown as * p<0.05; ** p<0.01; *** p<0.001[20].
Results
Oxidative stress induced by PDT causes cancer cells to undergo apoptosis
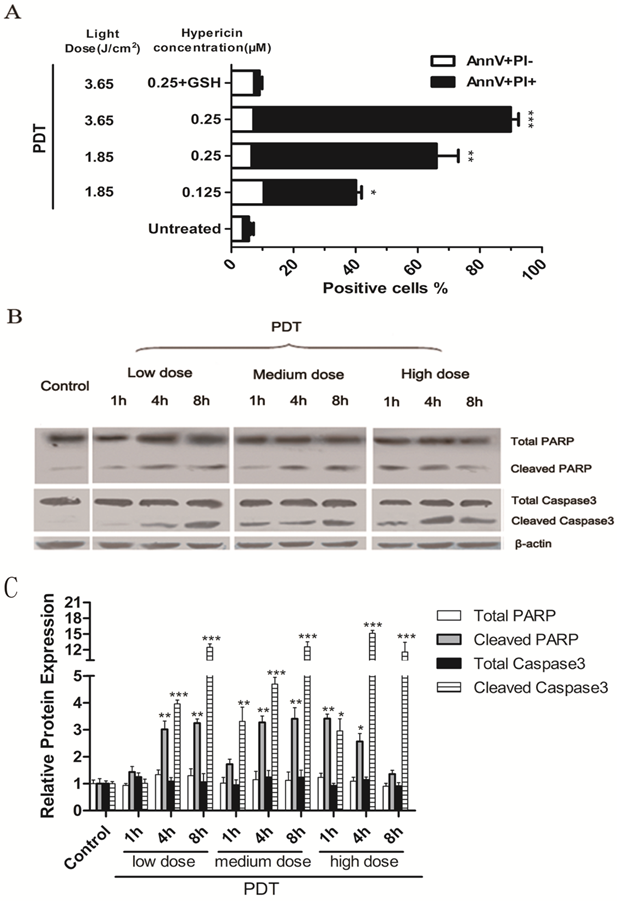
The percentage of apoptotic PDT-treated LLCs was determined by PI and Annexin V staining. LLC cells were incubated with 125nM or 250nM hypericin for 16 hours in the dark and were irradiated with different light doses (3.65 J/cm2 or 1.85 J/cm2). Cells were harvested 1 hour after irradiation. As the results shown in Fig.1A, PDT induced apoptosis in LLC cells in a light dose- and a hypericin concentration-dependent manner. Moreover, PDT treatment also induced the main hallmarks of apoptosis, including caspase-3 and PARP cleavage in LLC cells (Fig.1, B). All results mentioned above could be reversed by the exogenous antioxidant, GSH. Treatment with hypericin or light alone had little toxic effect on LLC cells (data not shown). It suggests that PDT-induced apoptosis is caused by oxidative stress triggered by hyp-PDT. The dose of hyp-PDT that killed more than 50% of tumor cells (hypericin, 0.25μM; light dose, 1.85J/cm2) was used for subsequent experiments to investigate whether cancer cells dying in response to PDT associated (hyp-PDT based; unless otherwise mentioned) oxidative stress can activate mouse immature DCs (imDCs).
LLC cells undergo apoptosis upon hyp-PDT treatment. Cells were incubated with 125nM or 250nM hypericin for 16 h and were irradiated at a fluency of either 1.85 or 3.65 J/cm2. (A) Percentage of early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptotic cells were determined by flow cytometry 1 hour after PDT. Data are presented as percent survival; the values are means of 3 independent experiments (five replicate determinations for each) ± SD (*p<0.05, **p<0.01, ***p<0.001, vs. untreated). (B) LLC cells showed caspase 3 and PARP cleavage after Hyp-PDT. LLC cells were treated with the low (hyp, 125nM; Light dose, 1.85J/cm2), medium (hyp, 250nM; Light dose, 1.85J/cm2), high (hyp, 250nM; Light dose, 3.65J/cm2) PDT dose or left untreated (Control), and whole-cell lysates were harvested 1hour post PDT and analyzed by western blot. (C) Quantification of 3 independent Western blots of each protein level represented as fold increase relative to control. The relative expression of each band = (density of each band/density of actin band). Mean ± SD was from three independent experiments. *p<0.05, **p< 0.01, ***p<0.001versus control.
Hyp-PDT-treated cancer cells expose DAMPs on the surface
We next analyzed the surface exposure of CRT and heat shock proteins (HSP90 and HSP70) following oxidative stress induced by PDT treatment (hypericin, 0.25μM; light dose, 1.85J/cm2). GSH is an essential antioxidant that is oxidized during oxidative stress to form a GSSG disulfide. In our experiments, exogenous glutathione (GSH) was used to protect cancer cells from apoptosis caused by PDT induced oxidative stress. Flow cytometry results show that hyp-PDT caused an enhanced surface exposure of CRT, HSP70 and HSP90 on cancer cells (Fig. 2A). A significant surface upregulation of surface-associated DAMPs was detectable as early as 30 min after hyp-PDT; maximal fluorescence was observed at 1 hour post PDT treatment. Although the upregulation declined overtime, it was still evident 4 hours later. This increase of DAMPs caused by hyp-PDT was partly diminished in the presence of GSH, thus revealing its dependence on oxidative stress. (Fig. 2B)
Expression of DAMPs (CRT、HSP70 and HSP90) on the cell surface following PDT treatment. (A) Representative histograms of single experiment showing the expression of CRT, HSP70, and HSP90 after PDT treatment. Gray solid histogram represents isotype control. (B) Quantitative analysis of CRT, HSP70 and HSP90 expression on cells subjected to PDT treatment at indicated time points (1hour and 4 hours) after irradiation. The fluorescence intensity of DAMPs was normalized by that of the isotype control, respectively. The expression of the indicated markers is shown as a fold change of the mean fluorescence intensity (MFI) when compared with untreated cells. (C) Confocal microscopy images of cells treated with hyp-PDT and stained for HSP70, HSP90, and CRT. (D) Representative histograms of single experiment showing the expression of CD47 at 1 and 4 hours post PDT. The gray solid histogram represents isotype control. (E) The fluorescence intensity of CD47 was normalized by that of the isotype control. The expression of CD47 is shown as a fold change in the MFI when compared with untreated cells. The summary of a total of 5 independent experiments is shown. #, *p < 0.05; ##, **p<0.01; ###, *** p<0.001.
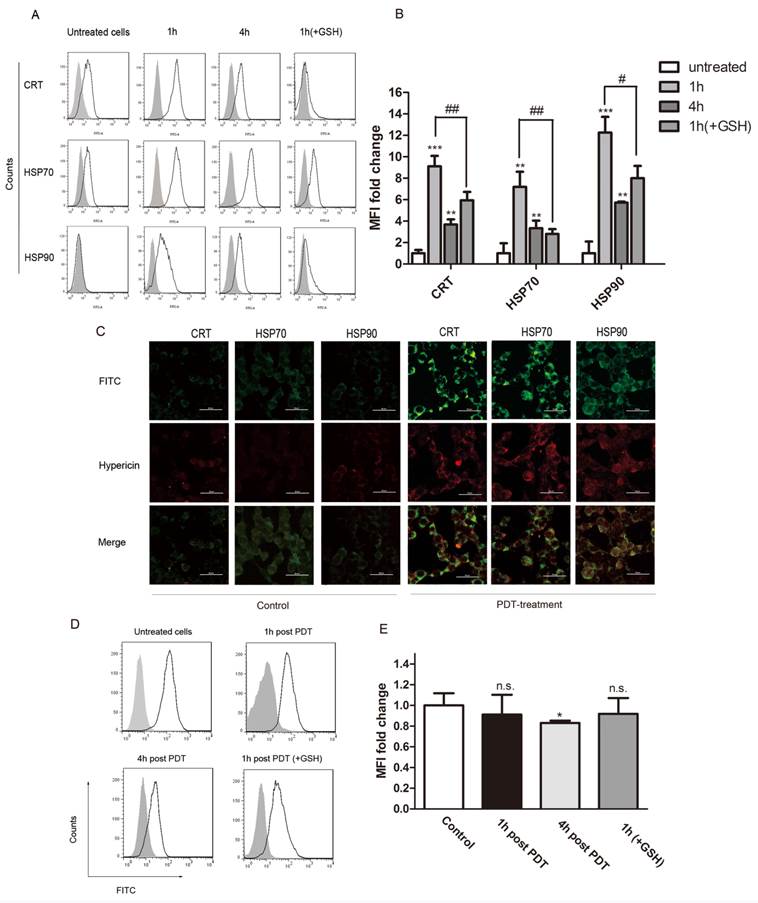
CRT, HSP70 and HSP90 on hyp-PDT-treated cells were also detected by immunofluorescence staining. Consistent with the result from flow cytometry, prominent expression of CRT, HSP70 and HSP90 could be seen on the cell surface of LLCs after PDT treatment (Fig. 2C, right). No significant DAMP upregulation was detectable with light-only (Fig. 2C, left) and hypericin-only controls (data not shown).
Cell surface CD47 is downregulated on LLC cells treated by PDT
DAMPs will be translocated to the cell surface upon immunogenic apoptosis. They are members of so-called “eat me” or “find me” signals [11]. In contrast, CD47, a "don't eat me" signal, is previously reported to be involved in mechanism by which tumor cells escape immunosurveillance [23].
Flow cytometry was utilized to determine the CD47 expression of cells undergoing PDT treatment. There was prominent surface expression of CD47 on untreated LLC cells. The CD47-associated-fluorescence decreased slightly by 1hour post PDT. The decreased level got significant at 4 hours post PDT (Fig 2D, E). As such, both “eat me” / “find me” signals and "don't eat me" signal were affected by hyp-PDT, indicating that the status of the immune system might be altered in the tested system.
LLC cells subjected to hyp-PDT undergo phagocytic interactions with imDCs
In vitro phagocytosis assays were performed on LLC cells exposed to hyp-PDT with or without exogenous GSH which was previously demonstrated to diminish the dyregulation of DAMPs. In this experiment, DC cells were labeled with DiO dye, while tumor cells were red fluorescent due to the inner existence of hypericin. Thus, the rate of phagocytosis of LLC tumor cells by DCs could be detected by flow cytometry. The results show that LLC cells subjected to hyp-PDT underwent phagocytic interactions with imDCs. The phagocytosis had already occurred to a slight extent by 12 hours since co-incubation (data not shown). After 24 hours, the rate of phagocytosis went up to over 40%. Furthermore, LLC cells pretreated with GSH were also subjected to hyp-PDT. These cells showed less stimulated effect on immatured DCs (Fig. 3A).
The immune functional status of DCs after interacting with PDT-LLCs. The phagocytosis of LLC cells treated with hyp-PDT by immature dendritic cells (imDCs) (DiO) was assessed by flow cytometry. Dot plots of the representative experiments are shown in (A, left). The percentages of phagocytosis at 24 hours (pretreated with GSH or not) are shown relative to the control (the 24-hour-co-incubation of normal LLC cells and imDCs) (A, right). (B) Confocal fluorescence images show phagocytic interactions between PDT-treated LLC cells (red) and imDCs (green). (C~E) Correlation between the expression of DAMPs and the rate of phagocytosis at 24 hours. (F) Correlation between the expression of CD47 and the rate of phagocytosis at 24 hours. Summary of 8 independent experiments is shown. (G, H) Cytokine and respiratory burst patterns exhibited by DCs undergone different treatments. LLC cells were left untreated (CNTR), freeze/thawed (F/T), or treated with a high PDT dose. They were then co-incubated with imDCs (CNTR-DCs、F/T-DCs、PDT-DCs). As a positive control, imDCs were stimulated with LPS (LPS-DCs) or 24 h. The conditioned co-culture media (CCM) derived from co-incubation experiments were collected and analyzed for the concentrations of nitrite (solubilized form of nitric oxide or NO) (G) and IL-10 (H). Values have been shown for three independent experiments. #,*p < 0.05; ##, **p<0.01; ###, *** p<0.001, versus CNTR-DCs.

Spearman rank test analysis showed that the rate of phagocytosis closely correlated with the intensity of CRT and HSP70 expression and, to a lesser degree, with the intensity of HSP90 expression (Fig. 3C~E). In contrast, CD47, the "don't eat me" signal, showed a negative correlation with the rate of phagocytosis (Fig. 3F).
PDT-associated-oxidative stress has significant impact on the functional status of DCs
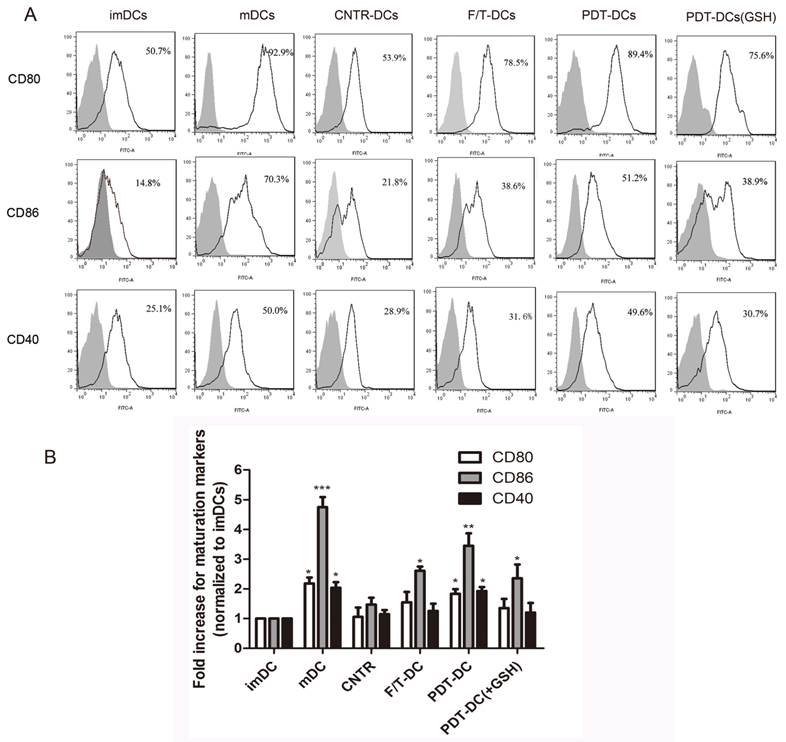
The ability of DCs to activate the immune response depends on their maturation status and the expression of co-stimulatory molecules. The prepared PDT-DCs were characterized by their phenotypic maturation. The hyp-PDT treated dying LLC cells induced an elevated expression of co-stimulatory molecules CD80/86 and CD40. They were markedly different from imDCs and DCs co-cultured with freeze-thawed LLC cells (F/T-DCs). The expression of these surface molecules on PDT-DCs was similar to mDCs (stimulated by lipopolysaccharide (LPS)). GSH also played a reversal role in this process (Fig. 4A, B).
PDT-treated dying LLC cells induce phenotypic maturation of DCs. (A) Representative histograms of single experiment showing the expression of CD80, CD86, and CD40 after the indicated treatment. On day 6, immature DCs were collected and cultured with LLC cells undergone different treatments (untreated, CNTR; freeze/thawed, F/T; or treated with a high PDT dose) for 24 hours. As a positive control, imDCs were stimulated with LPS for 24 h. The results are shown as “mDC”. After co-incubation/stimulation, DCs were immunostained by directly conjugated antibodies and analyzed for CD80、CD86 and CD40. The gray solid histogram represents the isotype control. (B)The expression of CD80, CD86, and CD40 is shown as a fold change of mean fluorescence intensity (MFI) when compared with “imDCs”. Values have been shown for three independent experiments with 5 replicate determinations of each. *p<0.05, **p< 0.01, ***p<0.001, versus “imDC”.
To further identify the functional status of DCs, we evaluated the generation of nitric oxide (NO) as a marker for respiratory burst. The co-incubation of hyp-PDT treated LLC cells and imDCs induced large amount of nitric oxide production at a level more significant than imDCs exposed to LPS (Fig. 3G). Moreover, ELISA assay shows that the formation of PDT-DCs produced little IL10, an immunosuppressive cytokine (Fig. 3H).
From the above results, the functional status of DCs changed significantly, enabling their interaction with PDT-treated tumor cells and more T cell activation. Of note, freeze-thawed LLC cells did not stimulate DC maturation. It rules out the possibility that accidental necrosis might contribute to the increase DC maturation. Moreover, F/T-DCs stimulated an abundant production of IL-10. These findings suggest that cells killed by photodynamice therapy might be quite different from the ones undergone accidental necrosis in inducing immunity response.
In vitro activation of tumor specific T cells by DCs pulsed with PDT-treated LLC cells
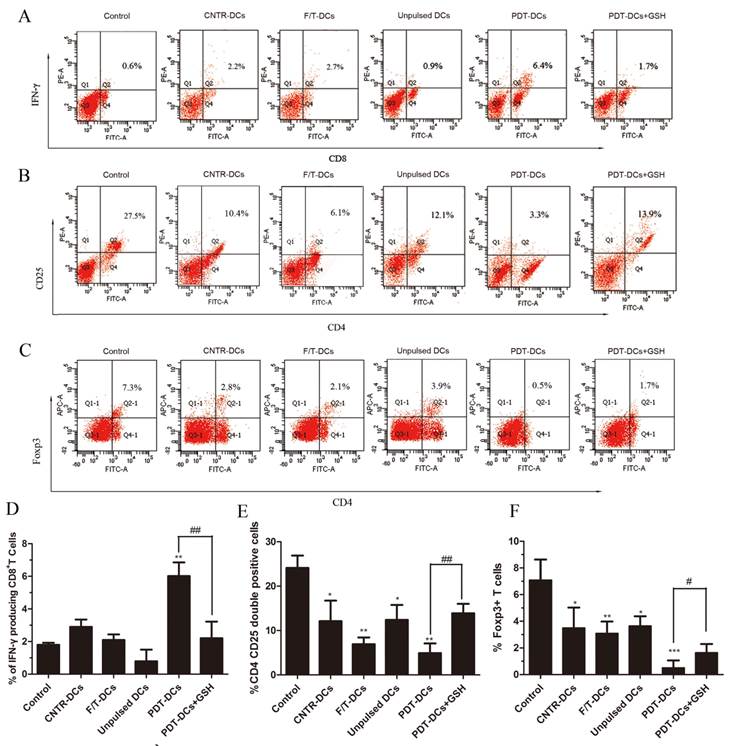
The balance of effector versus Treg cells could be influenced by the environment where the immune response is generated [24]. Thus, we evaluated the ability of tumor cell-loaded DCs to activate a tumor-specific T-cell response. LLC cells treated by PDT were co-cultured with immature DCs. As a positive control, imDCs were stimulated with LPS. These DCs were then used as stimulators of autologous T cells. The IFN-γ-producing CD8+ cell populations and the CD4+CD25+FoxP3+ Treg cell population were analyzed by flow cytometry.
As shown in Fig. 5, co-incubation of T cells and PDT-DCs resulted in a potent stimulation of IFN-γ-secreting CD8+ T cells (Fig. 5A, D) and a remarkable suppression of the CD4+CD25+Foxp3 Treg cell population (Fig. 5B, C, E and F). There is a growing realization that Treg cells play an important role in suppressing the immune response against tumor cells [25], while these cells are depleted by a high dose of PDT, therefore potentiating immunity. Antitumor responses have also been found to correlate with IFN-γproducing CD8+ T cells and CD4+CD25+FoxP3+ T cells [26, 27]. Increased IFN-γ-producing-CD8+ T cells were detected upon stimulation with PDT-DCs.
DCs pulsed with PDT-LLCs induce tumor-specific immunity. Bone marrow-derived DCs were pulsed with LLC cells undergone different treatment (untreated, CNTR; freeze/thawed, F/T; or treated with a high PDT dose) and then used for the stimulation of autologous T cells for 7 days. Spleen lymphocytes these mice without any treatment were used as the control. The number of IFN-γ producing CD8+ T cells was analyzed by flow cytometry. (A) shows representative staining of 5 independent experiments, while (D) shows a summary. The frequency of CD4+CD25+FoxP3+ cells was analyzed to determine the Treg cell population. (B), and (C) are representatives of 5 independent experiments. Data in (E) and (F) show a summary of 5 independent experiments. *p<0.05, **p< 0.01, ***p<0.001; #p<0.05, ##p<0.01 versus control.
These results suggest that PDT-DCs can induce a potent antitumor response in vitro. This immunity response was caused by PDT-associated oxidative stress, which could be inhibited by the exogenous antioxidant, GSH.
Both PDT-DC and PDT-LLC vaccination can induce immune response in vivo
To investigate the ability of PDT-treated LLC cells to activate the adaptive immune system, in vivo experiments were carried out in immunocompetent C57BL/6 mice. Firstly, mice were immunized with PBS (Control), F/T-LLCs, F/T-DCs, PDT-LLCs and PDT-DCs twice within 10 days. The immunized mice were rechallenged with live LLC cells that stably expressing luciferase (LLC-Luc). The solid tumor growth at the rechallenge site was monitored to evaluate the priming of the adaptive immune system. To different degrees, PDT-DCs and PDT-LLCs immunization inhibited the tumor growth seen in non-immunized mice (Fig. 6 A~C). F/T-DCs immunized mice showed certain vaccination efficacy in the initial stage of rechallenge. However, the rate of tumor growth became even faster than the control group. F/T-LLCs didn't show any vaccination efficacy during the experiment.
Vaccination efficacies of PDT-DCs and PDT-LLCs. Following immunization with PBS (Control), F/T-LLCs, F/T-DCs, PDT-LLCs and PDT-DCs, mice were rechallenged with live LLC-Luc cells. 7 mice were included in each group. (A) In vivo fluorescence imaging of vaccinated C57BL/6 mice at day 3, 7, 11, 15, 19 and 23 post-rechallenge (an example is shown in the photograph). (B) Tumor sizes were measured for the 23 days post-rechallenge. Data are presented as tumor growth curves. (C) Show the survival rates of mice rechallenged by live LLC-Luc cells. Splenocytes were harvested. The number of IFN-γ producing CD8+ T cells and Treg cell population were analyzed by flow cytometry. (C) is the representative graph of 7 independent experiments. Data in (D), (E) and (F) show a summary of 7 independent experiments. (H) CTL assays were performed based upon LDH. Seven days after the second DC vaccination, splenocytes were harvested from mice of each group were harvested as effectors, and were fed with LLC tumor cells as targets. CTL assays were performed using LDH assay. Values have been shown for 3 independent experiments. *p<0.05, **p< 0.01, ***p<0.001 compared with the control.
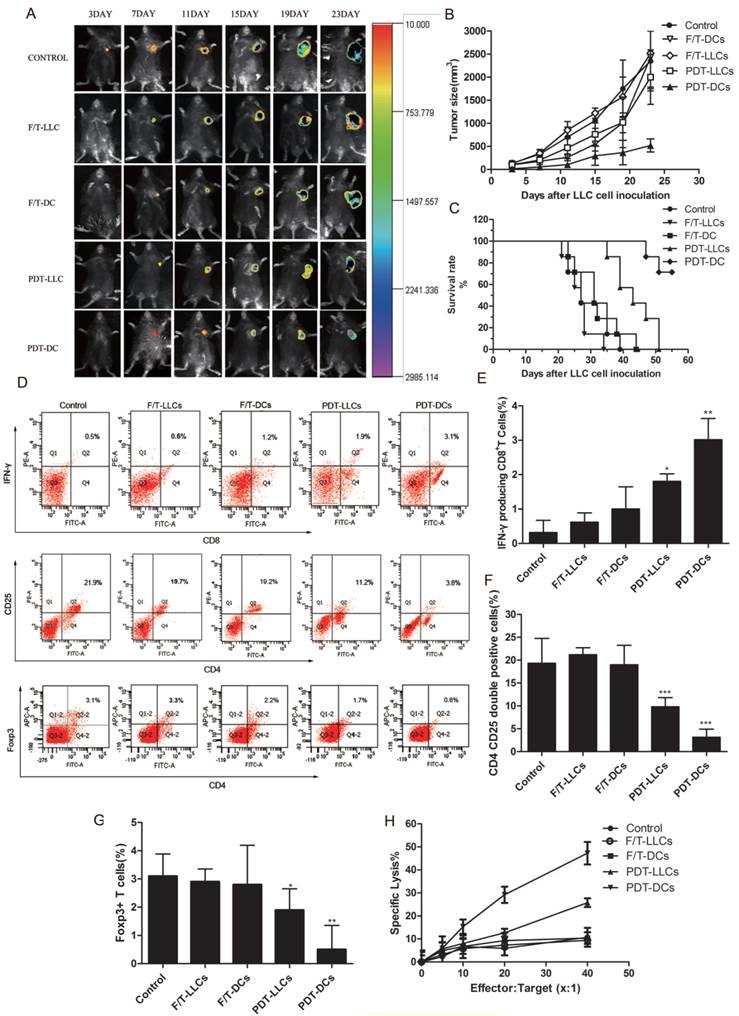
Next, to further investigate the in vivo immune responses induced by PDT-DCs and PDT-LLCs vaccination, we examined the CTL responses in tumor-bearing mice of each group. As shown in Fig. 6H, the PDT-DCs vaccination group exhibited significant CTL activity (more than 50% cell death) against LLC targets, while PDT-LLCs group showed approximately 20% cell death. Moreover, splenocytes were isolated before tumor rechallenge. The IFN-γ-producing CD8+ T cell population and the CD4+CD25+FoxP3+ Treg cell population were analyzed by flow cytometry similar to the aforementioned in vitro experiments. Results showed that both PDT-DC and PDT-LLC vaccination caused efficient tumor-specific CTL response, potent stimulation of IFN-γ-secreting CD8+ T cells (Fig. 6D, upper), and the remarkable suppression of the CD4+CD25+FoxP3+ Treg cell population (Fig. 6D, bottom), just to different extents.
Taken together, these results strongly suggest that PDT-DCs or PDT-LLCs prepared by hyp-PDT show potent antitumor responses against LLC-Luc cells rechallenge.
Discussion
In the current study, we observed the level of “eat me” signal and “don't eat me” signal changed in an opposite way after PDT treatment. GSH, which can diminish the dysregulation of these immune-associated antigen, can also reduce the PDT-induced immune response. Combined with the correlation analysis results, we speculate that the dysregulation of “eat me” signal and “don't eat me” signals may contribute to undermining the ability of LLC cells to escape immune attack. This imbalance of the immune homeostasis involved a change of DCs immune status, the release of immune-related cytokines (NOhigh, IL-10absent), and the stimulation of tumor-specific IFN-γ-producing CD8+ T cells, as well as lower numbers of Tregs.
In addition, in vivo experiments were performed to examine the vaccination potential of PDT-DCs and PDT-LLCs against LLC solid tumors. Compared with untreated mice, the results show that tumor growth was obviously inhibited in mice vaccinated with PDT-DCs and PDT-LLCs respectively, demonstrating that PDT-DCs and PDT-LLCs are potential to be used as an effective cancer vaccine. These results are inconsistent with that in a study by Nam-Chul Jung and his colleague [19]. In their study, they reported that PDT-DCs could induce an effective antitumor response. However, PDT tumor lysates alone had little or no effect on the growth of tumors. As for this phenomenon, we suppose that, DH-I-180-3, the photosensitizer they utilized is not mainly located in ER. In other words, the extent of ER-stress induced by DH-I-180-3 is not as strong as hypericin does.
What's needed to stated, our work is only on one specific cancer type and one cell line. Cancer itself is so heterogenous even within the same cancer that it is hard to extrapolate that this treatment option will be effective with all cancers.
It is worth mentioning that DC had been considered as the main antigen-presenting cell for many years, while macrophages had been believed to specialize in degrading antigens rather than presenting them. For this reason, we initially chose to focus on the functional status of dendritic cells and its' capacity to induce tumor cell-specific T cells. Recent reports have shown that macrophages could be effective at antigen presentation and are powerful initiators of the CD8+T cell response [28], while CD47 and DAMPs also participate in the process [29, 30]. Furthermore, anti-CD47 antibody was reported to be capable of inhibiting the growth of various blood cancers and solid tumors [31, 32]. From this sense, we suspect that both DCs and macrophages play important roles in the elicitation of a DAMP-driven immune response. The combination of PDT with anti-CD47 antibody may strengthen the antitumor efficacy of PDT for further undermining the balance of immune escape. Further studies are necessary to prove this theory.
Many studies have been reported that a cytoprotective effect may occur in some cancer cell types during the process of PDT treatment [33]. The cytoprotective mechanisms enable cancer cells to avoid the cytotoxic effects of PDT. This phenomenon is mainly due to the high levels of antioxidant molecules and ROS enzyme, as well as proteins regulate PDT-induced apoptosis or participate in the repair of lesions induced by oxidative stress [34]. As mentioned above, exogenous antioxidant, GSH, can reverse the antitumor immunity effect induced by hyp-PDT. Consequently, we hypothesize that the cytoprotective effect may have some impact on the tumor-specific immune response in some cell lines. Additional studies are needed.
PDT treatment kills tumors through direct cytotoxic effects [35], vascular damage [36], and the induction of an immunity response [11]. However, PDT is not an effective treatment for disseminated tumors, which results in its' limited clinical efficacy [19]. Therefore, it is necessary to develop new approaches that either enhances the efficacy of PDT or uses it in conjunction with other treatment modalities.
In conclusion, we characterized the process of tumor-specific immune response underlying the membrane dyregulation of DAMPs and CD47. It imply that the immune response may be partly due to the breakdown of the balance between DAMPs and CD47, which lay the foundation of combination between gene therapy and photodynamic therapy. In a more general sense, photodynamic therapy may be a potential inducer of 'immunogenic cancer cell death' and may also have the potential to be developed into antitumor vaccination strategies for personalized therapy.
Abbreviations
PDT, photodynamic therapy; Hyp-PDT, hypericin mediated PDT; LLCs, lewis lung carcinoma cells; ROS, reactive oxygen species; DAMPs, damage-associated molecular patterns; CD47, integrin-associated protein; CRT, calreticulin; HSP, heat shock protein; DCs, dendritic cells; mDCs, mature DC cells; imDCs, immature DC cells; PDT-LLCs, PDT-treated LLCs; PDT-DCs, DCs co-cultured with PDT-LLCs; DMSO, dimethyl sulfoxide; PI, propidium iodide; nM, nmol/L; μM, μmol/L; CTL, Cytotoxic T-Lymphocyte.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81502540), Fundamental Research Fund for the Central Universities of China (No. 222201514333), Shanghai Committee of Science and Technology (No. 13140902300), Nano Science and Technology Special Funding of Shanghai Committee of Science and Technology (No.11nm0503700), the Shanghai Committee of Science and Technology [grant 11DZ2260600] and the Fundamental Research Funds for the Central Universities.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798-804
2. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell death and differentiation. 2012;19:107-20
3. Zitvogel L, Kroemer G. The immune response against dying tumor cells: avoid disaster, achieve cure. Cell Death Differ. 2008;15:1-2
4. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nature reviews Immunology. 2009;9:353-63
5. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T. et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. The EMBO journal. 2012;31:1062-79
6. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. The EMBO journal. 2009;28:578-90
7. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer immunology, immunotherapy: CII. 2012;61:215-21
8. Lin A, Hahn SM. Photodynamic therapy: a light in the darkness? Clin Cancer Res. 2009;15:4252-3
9. Karioti A, Bilia AR. Hypericins as potential leads for new therapeutics. Int J Mol Sci. 2010;11:562-94
10. Galeotti N, Vivoli E, Bilia AR, Vincieri FF, Ghelardini C. St. John's Wort reduces neuropathic pain through a hypericin-mediated inhibition of the protein kinase Cgamma and epsilon activity. Biochemical pharmacology. 2010;79:1327-36
11. Garg AD, Nowis D, Golab J, Agostinis P. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis. 2010;15:1050-71
12. Ritz R, Roser F, Radomski N, Strauss WS, Tatagiba M, Gharabaghi A. Subcellular colocalization of hypericin with respect to endoplasmic reticulum and Golgi apparatus in glioblastoma cells. Anticancer research. 2008;28:2033-8
13. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP. et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell death and differentiation. 2012;19:1880-91
14. Ullrich E, Bonmort M, Mignot G, Kroemer G, Zitvogel L. Tumor stress, cell death and the ensuing immune response. Cell death and differentiation. 2008;15:21-8
15. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochimica et biophysica acta. 2010;1805:53-71
16. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nature reviews Cancer. 2012;12:860-75
17. Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Current opinion in immunology. 2012;24:225-32
18. Imhof M, Karas I, Gomez I, Eger A. Interaction of tumor cells with the immune system: implications for dendritic cell therapy and cancer progression. Drug discovery today. 2013;18:35-42
19. Jung NC, Kim HJ, Kang MS, Lee JH, Song JY, Seo HG. et al. Photodynamic therapy-mediated DC immunotherapy is highly effective for the inhibition of established solid tumors. Cancer letters. 2012;324:58-65
20. Zheng Y, Le V, Cheng Z, Xie S, Li H, Tian J. et al. Development of rapid and highly sensitive HSPA1A promoter-driven luciferase reporter system for assessing oxidative stress associated with low-dose photodynamic therapy. Cell stress & chaperones. 2013;18:203-13
21. Becker AJ, Uckert S, Tsikas D, Noack H, Stief CG, Frolich JC. et al. Determination of nitric oxide metabolites by means of the Griess assay and gas chromatography-mass spectrometry in the cavernous and systemic blood of healthy males and patients with erectile dysfunction during different functional conditions of the penis. Urological research. 2000;28:364-9
22. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. 2012;188:1592-9
23. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271-85
24. Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer immunology, immunotherapy: CII. 2005;54:307-14
25. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nature reviews Cancer. 2006;6:535-45
26. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G. et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer research. 2010;70:855-8
27. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H. et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012;189:558-66
28. Zhou F, Xing D, Chen WR. Regulation of HSP70 on activating macrophages using PDT-induced apoptotic cells. Int J Cancer. 2009;125:1380-9
29. Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK. et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88-91
30. Martins I, Kepp O, Galluzzi L, Senovilla L, Schlemmer F, Adjemian S. et al. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Annals of the New York Academy of Sciences. 2010;1209:77-82
31. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB. et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11103-8
32. Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends in immunology. 2010;31:212-9
33. Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochimica et biophysica acta. 2007;1776:86-107
34. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250-81
35. Buytaert E, Matroule JY, Durinck S, Close P, Kocanova S, Vandenheede JR. et al. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene. 2008;27:1916-29
36. Chen B, Roskams T, Xu Y, Agostinis P, de Witte PA. Photodynamic therapy with hypericin induces vascular damage and apoptosis in the RIF-1 mouse tumor model. Int J Cancer. 2002;98:284-90
Author contact
![]() Corresponding authors: Jianwen Liu, Ph.D., Professor; Xin Liang, Ph.D.; Professor; Siyu Chen, Ph.D., Professor. Tel. /fax: +86 21 64252044; +86 21 52665957; +86-21-25077642. E-mail address: liujianedu.cn (J.W. Liu); xin.liangedu.cn (X. L.); siyu.chenedu.cn (S.Y. Chen).
Corresponding authors: Jianwen Liu, Ph.D., Professor; Xin Liang, Ph.D.; Professor; Siyu Chen, Ph.D., Professor. Tel. /fax: +86 21 64252044; +86 21 52665957; +86-21-25077642. E-mail address: liujianedu.cn (J.W. Liu); xin.liangedu.cn (X. L.); siyu.chenedu.cn (S.Y. Chen).