Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The standard paradigm for...
Evidence Oncogenic Ras is Not...
The Ras/Inflammation...
Regulation of Ras Activity by...
Post-translational Modifications...
Significance of Ras Activity...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(3):338-346. doi:10.7150/ijbs.15020 This issue Cite
Review
The Significance of Ras Activity in Pancreatic Cancer Initiation
Craig D. Logsdon1,2 ![]() , Weiqin Lu1
, Weiqin Lu1
1. Department of GI Medical Oncology, University of Texas MD Anderson Cancer Center, Houston TX 77030, USA
2. Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston TX 77030, USA
Published 2016-1-28
Abstract
The genetic landscape of pancreatic cancer shows nearly ubiquitous mutations of K-RAS. However, oncogenic K-Rasmt alone is not sufficient to lead to pancreatic ductal adenocarcinoma (PDAC) in either human or in genetically modified adult mouse models. Many stimulants, such as high fat diet, CCK, LPS, PGE2 and others, have physiological effects at low concentrations that are mediated in part through modest increases in K-Ras activity. However, at high concentrations, they induce inflammation that, in the presence of oncogenic K-Ras expression, substantially accelerates PDAC formation. The mechanism involves increased activity of oncogenic K-Rasmt. Unlike what has been proposed in the standard paradigm for the role of Ras in oncogenesis, oncogenic K-Rasmt is now known to not be constitutively active. Rather, it can be activated by standard mechanisms similar to wild-type K-Ras, but its activity is sustained for a prolonged period. Furthermore, if the level of K-Ras activity exceeds a threshold at which it begins to generate its own activators, then a feed-forward loop is formed between K-Ras activity and inflammation and pathological processes including oncogenesis are initiated. Oncogenic K-Rasmt activation, a key event in PDAC initiation and development, is subject to complex regulatory mechanisms. Reagents which inhibit inflammation, such as the Cox2 inhibitor celecoxib, block the feed-forward loop and prevent induction of PDAC in models with endogenous oncogenic K-Rasmt. Increased understanding of the role of activating and inhibitory mechanisms on oncogenic K-Rasmt activity is of paramount importance for the development of preventive and therapeutic strategies to fight against this lethal disease.
Keywords: K-RAS
Introduction
Mutant K-Ras is arguably the most studied oncogene. This is due to the extreme clinical relevance of the Ras family of small GTPases (H-Ras, K-Ras, and N-Ras), as they are the most commonly mutated oncogenes in human cancer, being present in 20% to 30% of all human tumors and K-Ras is mutated in up to 90% in pancreatic cancer [1]. Ras was first recognized in retroviruses in 1983 [2] , and a recent search for Ras in PubMed identified a more than 55,792 existing publications. Therefore, clearly there is a lot known about this molecule. Nevertheless, recent observations have shed a new light on the mechanisms involved in Ras mediated oncogenesis that emphasize the importance of Ras activity. Some of the new information is not yet well disseminated. Thus, the goal of this review is to explain the observations that led to the need to develop a revised model, to describe the new model, and to explain the implications of these ideas on how we think about Ras initiated cancer and the possibilities of preventive measures. Before describing the new model, it will be useful to review the older one.
The standard paradigm for activation of wild-type and oncogenic Ras
Ras is activated by binding GTP
Like other members of the small guanine nucleotide binding family, when the guanine nucleotide binding site on Ras is occupied by GTP, it is active (“on”) (Figure 1) [3]. This initial step of loading GTP is not spontaneous, but rather requires the interaction of Ras with activating molecules. The best known and most common means of increasing GTP loading of Ras is by interaction with a guanine exchange factor (GEF). There are at least 9 GEFs in humans that are themselves activated by numerous extracellular signaling molecules [4, 5]. Thus, Ras is generally not active until called upon by external cellular signals, but once activated it can influence multiple cellular functions.
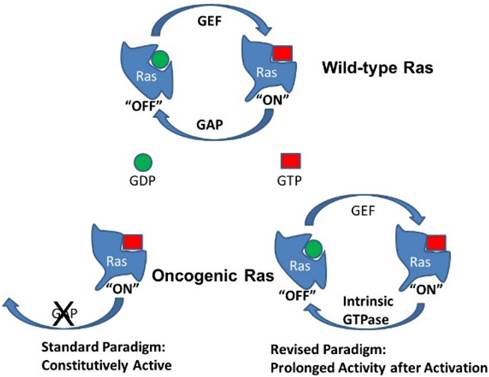
Activation of Ras by GTP loading. Wild-type K-Ras is typically bound with GDP and thus inactive (“off”). Activation of guanine exchange factors (GEFs) by interactions with receptors leads to the loading of GTP in place of GDP and K-Ras changes to the active conformation (“on”). K-Ras has intrinsic GTPase activity that will convert GTP back to GDP and turn the system back to its inactive state. However, the GTPase activity of native K-Ras is low and is greatly accelerated by interactions with GTPase activating proteins (GAPs). Oncogenic mutations in K-Ras disrupt interactions with GAPs. In the standard paradigm, this leads to constitutively active K-Ras. In the revised paradigm, this leads to prolonged signaling from oncogenic K-Ras after activation by GEFs or other mechanisms.
Ras is inactivated by GTP hydrolysis, which is impaired in oncogenic Ras mutants
Physiologically, wild-type Ras activity is transient. Ras molecules have intrinsic GTPase activity that will hydrolyze GTP to GDP, thus returning Ras to the inactive (“off”) state and shutting off the signal. However, the GTPase activity of unmodified wild-type Ras is relatively low and is greatly stimulated by association with GTPase-activating proteins (GAPs) [6]. Interference with Ras/GAP interactions therefore prolong the GTP bound state and increase Ras signaling. The mutant form of Ras most often associated with cancer acquires point mutations of residues 12 or 61 that impair the interaction of Ras with common GAPs. Importantly, these mutations do not alter the interactions with GEFs and do not affect the intrinsic GTPase activity of Ras itself. Nevertheless, in the standard paradigm, the prolongation of Ras activity by these mutations is suggested to be indefinite, so that oncogenic Ras remains permanently activated (Fig 1). In contrast, in the revised model being presented, at physiologically relevant levels of expression, oncogenic mutant Ras is not constantly active but shows a prolonged activity state after interactions with GEFs. This seemingly small difference has large biological ramifications.
Ras has multiple down-stream effects
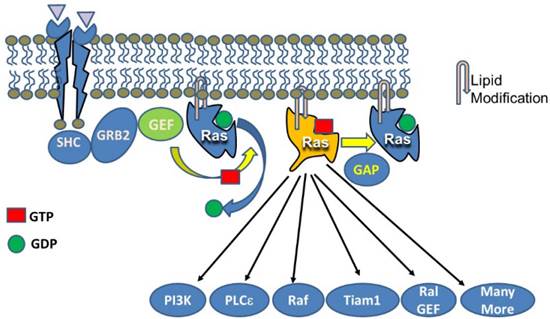
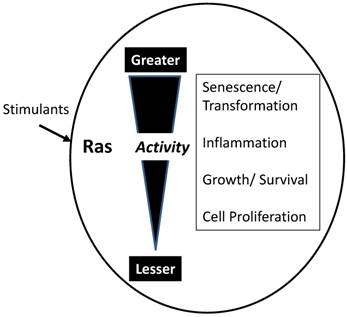
As indicated in Figure 2, GTP-bound active Ras is able to interact with a large number of effectors leading to changes in numerous cellular processes. However, the actual consequences of activating Ras depend on the overall activity level (Figure 3). While each molecule of Ras is a binary switch, the cells have many copies of Ras and the total Ras activity is determined by the number of active molecules. Thus, ultimately, Ras activity is more like a rheostat than a binary switch. Importantly, physiological Ras signaling occurs when a very low percentage of Ras molecules are occupied by GTP. This is illustrated by studies in cell lines with high levels of EGF receptors which found that a maximal concentration of EGF led to approximately occupation of 50% of total Ras with GTP [7]. In the pancreas, K-Ras activity is necessary for several normal cell functions [8]. Ras activity is elevated by physiologic levels of secretagogues, such as cholecystokinin, that are released during a meal. This level of activity correlates with increased rates of protein synthesis. At higher, but still physiological Ras activity levels, acinar cell proliferation is stimulated. In rodents, the addition of a trypsin inhibitor to the diet increases the release of endogenous cholecystokinin to a high physiologic level and the pancreas adapts to this increased signaling by hypertrophy and hyperplasia [9]. High Ras activity is also associated with alterations of the differentiation program of the cells. Acinar-ductal metaplasia occurs with high levels of Ras activity [10]. At very high levels, observed in pathological conditions such as inflammation, Ras activity can lead to senescence, cell death, or in the absence of tumor suppressors, to transformation. Therefore, when examining diagrams of Ras signaling (such as Figure 2) it is important to remember that the diagrams do not typically indicate the relative sensitivities of the different effectors.
Activation of Ras stimulates a large number of down-stream pathways. Canonical activation of Ras occurs when a tyrosine kinase receptor (e.g. EGFR) is occupied by a ligand leading to phosphorylation of the receptor and interaction with signaling components such as SHC which then binds to GRB2 and ultimately with the GEF, SOS. SOS acts on Ras to replace GDP with GTP. GTP loading changes the conformation of Ras allowing it to interact with multiple effectors. The five most widely studied effects include PI3K, PLCε, Raf, Tiam1, and Ral GEF. However, many more effectors have been described. One thing not indicated by figures of this sort is the relative sensitivities of the various effectors.
Influence of total Ras activity on cell function. While individual molecules or Ras act as binary switches, the biological response depends on the summation of active Ras molecules. Low levels of Ras stimulants activate a small proportion of available Ras molecules. Low Ras activity is involved in maintenance of cellular homeostasis and activities such as protein synthesis. Slightly elevated total Ras activity stimulates growth, both hypertrophy and hyperplasia. High levels of Ras activity generate inflammatory mediators and inflammation. Very high levels of Ras activity typically lead to cell cycle arrest and senescence. However, if senescence mechanisms are compromised, high levels of Ras activity transform the cells leading to carcinogenesis.
Evidence Oncogenic Ras is Not Constitutively Active
Before describing the new model, we will present several lines of argument indicating that oncogenic Ras, at endogenous levels of expression, is not fully active and is not sufficient to lead to oncogenesis. A summary of these issues is listed in Table 1.
A summary of evidence that oncogenic mutant K-Ras is not constitutively active.

Lack of effectiveness in vivo
Perhaps the most obvious indication that endogenous levels of oncogenic mutant K-RAS alone is insufficient for oncogenesis is the relatively minor effect observed after expressing oncogenic K-Ras from its endogenous promoter in cells of the pancreas in genetically engineered mouse models (GEMMs). Specifically, in animals bearing a floxed copy of K-RasG12D cloned into the wild-type K-Ras gene locus crossed with mice expressing Cre from a pancreas specific developmental promoter, all of the cells of the pancreas are K-RasG12D/+. If oncogenic K-Ras were constitutively active, as suggested by the classical model, then these cells would possess extremely high Ras activity (50% activated K-Ras molecules). This would be expected to stimulate virtually all of the down-stream pathways and effectors influenced by Ras. Yet, only a tiny minority of cells in these GEMMs form pancreatic intraepithelial neoplasms (PanINs) [11]. Moreover, expression of oncogenic K-Ras at endogenous levels in adult pancreatic acinar cells has almost no discernable effect [12, 13]. The lack of visible pathological changes associated with expression of endogenous levels of K-RasG12D is accompanied by a complete lack of activation of signaling pathways down-stream of Ras, such as the Raf/MEK/Erk pathway, AKT, or Cox2 [13]. These observations are not consistent with 50% of the cellular K-Ras being GTP bound and active.
Requirement for Inflammatory Stimulus
Several previous studies have indicated that an inflammatory insult can initiate carcinogenesis in pancreatic cells expressing endogenous levels of oncogenic mutant K-Ras [12-14]. However, the mechanisms involved in this effect have been unclear. Because GEMMs in which oncogenic K-Ras is expressed during development apparently show greater sensitivity to the expression of oncogenic mutant K-Ras compared to adult cells, a common interpretation of this observation has been that dedifferentiated cells are more susceptible to Ras oncogenesis. Thus, the ability of inflammation to cause acinar-ductal metaplasia has been suggested as the important action of inflammation to sensitize the pancreas to oncogenic K-Ras. However, there are several lines of evidence against this interpretation. Inflammatory insults which are less severe than supraphysiological caerulein treatments typically employed to induce pancreatitis in mouse models are also able to increase the activity of endogenous levels of oncogenic K-Ras and lead to transformation and PDAC. For example K-Ras GTP loading was increased by elevated levels of Cox2 expression, or feeding of camostat or a high-fat diet [13, 15]. None of these treatments caused ADM on their own, yet each led to PDAC in the presence of oncogenic K-Ras. Furthermore, direct expression of high levels of oncogenic K-Ras, which leads to high levels of K-Ras activity without the need for inflammatory treatments, transforms adult acinar cells with high efficiency [13]. Therefore, Ras activity at a sufficient level can efficiently transform even adult cells.
Common Presence of Oncogenic Ras in Humans
Another finding inconsistent with oncogenic K-Ras being fully activated comes from the observation that most people develop oncogenic mutant Ras in the pancreas and other organs as they age, but they do not necessarily develop cancer [16-19]. Examination of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma indicated that PanIN frequency in healthy individuals was dependent on age and reached greater than 70% between ages 50 and 60 [20]. Other studies have found that at last 74% of PanINs harbor K-RAS mutations [21]. Therefore, if oncogenic Ras were fully active, one might expect a higher rate of pancreatic cancer than is actually present.
Ras Activity, but Not Ras Mutation, is Necessary for Oncogenesis
Ras activity can be elevated to levels capable of cellular transformation without oncogenic mutation. For example, in hepatocellular carcinoma (HCC) Ras itself is not mutated. Rather, in HCC Ras activity is elevated by alterations of regulatory molecules [22]. For example, nearly all samples of human HCC were observed to have decreased levels of at least one Ras GAP (RASAL1, DAB2IP, or NF1) [23]. The major GAP, RASAL1, has been shown to be decreased in HCC by the silencing of the PITX transcription factor [24]. Decreased levels of GAPs result in prolonged Ras signaling, similarly to the oncogenic mutations of Ras itself.
Evidence for Lack of GTP Occupancy Under Basal Conditions
The most direct evidence that oncogenic K-Ras is not constitutively active comes from studies where acinar cells were isolated from GEMMs in which K-RasG12D was knocked-in using a highly efficient adult acinar cell specific Cre. In these acinar cells, which are K-RasG12D/+, analysis of GTP occupancy of K-Ras using a standard Raf pull-down assay indicated that the percent of K-Ras occupied under basal conditions was only a few percent [25]. This is significantly less than the 50% predicted by the standard model.
Oncogenic Ras Activity is Influenced by Stimulants
In the revised paradigm, oncogenic Ras requires interactions with GEFs to become active. Experimentally, oncogenic K-Ras was shown to be activated by stimulants such as EGF [13]. It has also been shown that expression of a dominant negative GEF can reduce Ras activity in cells expressing oncogenic Ras [26]. These data directly contradict the notion that oncogenic mutant K-Ras is always active. These data also provide an explanation for the observation that Ras stimulants can accelerate and initiate carcinogenesis in cells bearing endogenous levels of oncogenic K-Rasmt. Importantly, acinar cells with endogenous levels of oncogenic K-Ras were found to respond to standard stimuli with prolonged Ras activity [13]. This is what would be predicted from the known interference of the oncogenic mutations with Ras interactions with GAP proteins. This prolongation of the Ras activity allows for the development of a feed-forward cycle of Ras stimulation that is critical for oncogenesis.
The Ras/Inflammation Feed-Forward Model of Pancreatic Cancer Initiation
The most straight-forward interpretation of all available data leads to a revised model for the functioning of oncogenic Ras that has the following components:
Oncogenic mutant K-Ras is not constitutively activity
Rather than being constantly occupied by GTP, mutant K-Ras requires activation similarly to wild-type K-Ras, but maintains prolonged activity after stimulation by classic mechanisms (Figure 4).
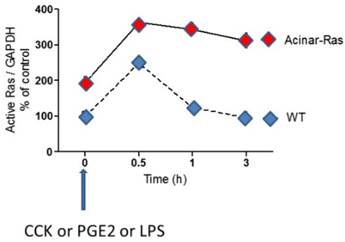
K-Ras-GTP loading is increased by stimulants and mutant oncogenic K-Ras prolongs the activity. Murine pancreatic acinar cells isolated from mice that express endogenous levels of oncogenic mutant K-Ras in all acinar cells, and those isolated from wild-type mice, were stimulated with a variety of stimulants (CCK, PGE2 and LPS). For each of these stimulants it was observed that Ras was activated by treating the cells (indicated by analysis of GTP binding) and that the activity of Ras was prolonged in the cells expressing oncogenic mutant K-Ras. [13]
High Ras activity is necessary for oncogenesis
Endogenous levels of mutant K-Ras expression do not transform cells. Yet, high levels of mutant K-Ras expression, for example those obtained using strong promoters, are highly efficient at cellular transformation [10]. This is because high levels of mutant K-Ras generate high levels of Ras activity, while endogenous levels have only a small effect on basal Ras activity. Endogenous levels of expression of oncogenic mutant K-Ras, while having little effect on basal levels of Ras activity, magnify the response of the cells to stimulants.
There is a threshold beyond which Ras activity becomes self-sustaining
Treatment of cells bearing endogenous levels of oncogenic mutant K-Ras with high levels of Ras activators causes Ras activity to reach a threshold at which it generates its own stimulants. Ras can activate down-stream pathways, including NFκB, Cox2, Stat3, and ROS, that generate direct and indirect Ras activators. This critical threshold is not reached during normal physiological functions, even with endogenous levels of mutant Ras present, as GEMMs expressing K-RasG12D in adult pancreatic acinar cells are unaffected by normal living [13]. However, higher levels of stimulation, such as those generated by a high fat diet [15] or feeding with a trypsin inhibitor [13] are sufficient to reach this threshold. Importantly, genetic expression of high levels of oncogenic K-Ras generate sufficient activity to fully transform adult pancreatic acinar cells without the need for external stimulants. The levels of activity (percentage of GTP bound K-Ras) observed in mice with transgenic overexpression of K-Ras are similar to those observed in transformed pancreatic cells from either human or mouse. Therefore, the level of expression of oncogenic Ras is important, because it affects Ras activity. The mechanisms that explain the high activity with high levels of expression and low activity with low levels of expression are not completely clear. This is primarily a significant consideration in GEMMs, where levels of expression of oncogenic Ras may be artificially high.
Regulation of Ras Activity by External Stimuli
Because oncogenic mutations are not sufficient to activate Ras, and Ras activity is the key to oncogenesis, it is useful to consider in more detail some of the mechanisms through which various regulators affect Ras activity.
Inflammatory Stimuli
That inflammation is a risk factor and a driver of PDAC has been known for many years and multiple studies have been conducted to identify the specific factors, networks, and cells involved. This has led to the identification of a large number of potential factors that can be involved (for a review see 24818722). However, it was not previously appreciated that mutant oncogenic K-Ras is not constitutively active and is the key target of these factors. It was also not recognized that high K-Ras activity itself generate several inflammatory mediators. With the current knowledge it becomes clear that any stimulus that activates K-Ras is capable of being an initiator of PDAC. Stimuli capable of activating oncogenic K-Ras may originate from stressed pancreatic cells, from infiltrated inflammatory cells, from adipose tissues, from ingested or inhaled chemicals or other sources. If oncogenic mutant K-Ras is present and activated, then transformation is possible.
Receptor Tyrosine Kinases
The original and best known mechanism for Ras activation involves activation by guanine nucleotide exchange factors (GEFs). In the canonical pathway, phosphorylation of a receptor tyrosine kinase (e.g. EGFR) leads to binding of the adapter protein GRB2 to phosphorylated tyrosine and subsequently recruits the GEF SOS. This GEF stimulates the release of GDP from Ras and enhances loading with GTP, thereby activating Ras and initiating down-stream signaling [3]. In the standard paradigm, because oncogenic mutant Ras proposed to be fully activated, it would not be predicted that growth factors would influence pancreatic cancer cells. In the revised paradigm, growth factors are one of the potential triggers of oncogenesis in cells expressing endogenous levels of oncogenic mutant Ras.
G protein coupled receptors (GPCR)
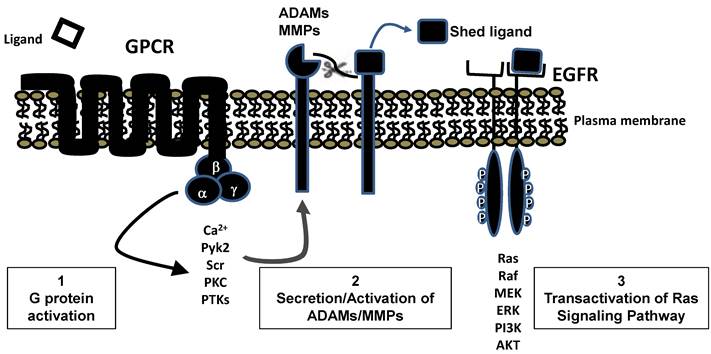
Ras can also be activated by GPCRs [27]. Because GPCRs do not directly interact with any known GEFs, there activation of Ras occurs by an indirect route. Typically this involves the release of membrane bound EGFR ligands, by secretion of proteases such as the matrix metalloproteinases, which can then activate EGFR (Figure 5). Mammalian ligands that bind EGFR include EGF, amphiregulin, betacellulin, epigen, epiregulin, heparin-binding EGF-like growth factor, and Transforming Growth Factor α [28] . These ligands are all type I transmembrane proteins inserted into the plasma membrane that can be cleaved by proteases to release mature growth factors. Thus, ultimately, the GPCRs also often activate Ras through the functioning of GEFs The importance of EGFR in transducing signals to Ras from multiple GPCR receptors may help explain the requirement for EGFR expression for transformation of acinar cells in some GEMMs [29, 30] . It would seem highly unlikely that EGFR would be required for transformation if high levels of oncogenic Ras were expressed in the acinar cells, as at high levels of expression no external stimulus is necessary for Ras activity to reach the feed-forward loop threshold. However, this specific study has yet to be done.
GPCR transactivation of EGFR leading to Ras stimulation. Stimulation of many GPCRs leads, through standard second messenger pathways, to secretion or activation of ADAMs or MMPs. Activity of these proteases frees bound EGFR ligands which interact with EGFR and generate typical tyrosine kinase signaling pathways including activation of RAS.
Post-translational Modifications Regulating Ras Activity
While GEFs are the best known and most common mechanism for Ras activation, GEFs are not the only means of activating Ras proteins. Post translational modifications can also activate Ras. Like all members of the RAS superfamily, K-RAS function is tightly regulated by post-translational modifications. Several post-translational modifications of Ras have been reported to influence its activity [31]. In the standard paradigm oncogenic mutant Ras is fully active and these mechanisms are not considered important. However, in the revised paradigm, these post-translational modifications may be extremely significant. All of these modifications and their influences have been studies using wild-type Ras. We know very little about the influence these modifications have on oncogenic mutant K-Ras. The different modifications include:
Farnesylation
K-RAS is normally farnesylated on a C-terminal cysteine and one splice form (K-RAS4A) is subsequently palmitoylated [31] . These modifications regulate K-RAS function by increasing its association with the plasma membrane. The positioning of Ras at the inner side of the plasma membrane is essential for its activation and for some of its activities. Because of the importance of this cellular localization, interference with farnesylation was predicted to inhibit Ras activation and provide therapeutic benefit. Thus, several drugs were developed to inhibit this process. Unfortunately, attempts to block these modifications for therapeutic purposes has, as yet, not been successful. Nevertheless, efforts to improve therapeutics aimed at this important post-translational modification continue and may eventually become useful [32]. Farnesylation also influences the ability of Ras to interact with different specific effectors in different cellular compartments including golgi, ER, and mitochondria. For example, protein kinase C (PKC) is reported to facilitate the translocation of K-RAS from plasma membrane to mitochondria through regulating a farnesyl-electrostatic switch [33]. Localization of oncogenic K-Ras to the mitochondrial inner member was reported to induce a rapid suppression of respiratory chain complex-I and a decrease mitochondrial transmembrane potential by affecting the cyclosporin-sensitive permeability transition pore, therefore, causing metabolic switch from mitochondrial oxidative phosphorylation to the aerobic glycolysis and ROS stress in cancer cells, and promoting tumor development [34]. However, more investigations will be required to elucidate the specific roles of Ras activity in these different compartments.
Ubiquitination
Mono-ubiquitination of lysine 147 of K-Ras inhibits GAP-mediated hydrolysis and therefore increases Ras activation [35]. This is yet another mechanism that would be expected to prolong Ras signaling. Interestingly, mono-ubiquitination of H-Ras at Lys-117 also increases its activity, but in this case the mechanism appears to be an enhancement of GTP-GDP exchange [36]. Di-ubiquitination of H-Ras and N-Ras, on the other hand, promotes endosomal localization or retention and thereby restricts the ability of these molecules to signal to ERK [37]. Thus, ubiquitination can have multiple effects on Ras signaling depending on the specific Ras form and the specific sites modified.
Acetylation
Mutant K-RAS that is acetylated has been found to be unable to re-load GTP in an efficient manner, resulting in attenuated transforming activity [38]. The mechanism of these inhibitory effects has been demonstrated to involve acetylation of lysine 104 which interferes with GEF-induced nucleotide exchange. The deacetylases HDAC6 and SIRT2 were identified as being involved in this process in cancer cells. Inhibition of either of these enzymes dramatically reduced the growth of cancer cell lines expressing oncogenic mutant K-RAS [38]. These results support the concept that K-Ras must be continually reactivated in order to maintain its transforming abilities and suggest that targeting of HDAC6 and/or SIRT2 may represent a new therapeutic strategy to treat cancers expressing mutant forms of K-RAS.
Modification of the redox-sensitive C118
As mentioned above, Ras can be activated independently of GEFs by modifications of the thiol residue of cysteine 118 (C118). It has been proposed that redox activation of Ras may be a central mechanism of oxidative stress signaling [39, 40]. Oxidative stress acts on redox-sensitive RAS molecules to perturb GTPase nucleotide-binding interactions that result in the enhancement of the guanine nucleotide exchange of small GTPases [41]. Wild type K-Ras is subject to ROS or RNS regulation for the enhanced activity through forming the oxygenated GDP adduct (putatively assigned as 5-oxo-GDP) or S-nitrosylated cysteine at C118 of the redox-sensitive NKC118D motif of K-Ras RAS molecules, including Hras, Nras and K-Ras, possess a redox sensitive NKC118D motif and are subject to reactive oxygen species (ROS) and reactive nitrogen species (RNS) regulation [39, 40, 42]. The action of redox agents on these redox-sensitive GTPases is similar to that of guanine nucleotide exchange factors (GEFs) in that they perturb GTPase nucleotide-binding interactions that result in the enhancement of the guanine nucleotide exchange of small GTPases [41]. Wild type K-Ras is subject to ROS or RNS regulation for the enhanced activity through forming the oxygenated GDP adduct (putatively assigned as 5-oxo-GDP) or S-nitrosylated cysteine at C118 of the redox-sensitive NKC118D motif of K-Ras [39, 40, 43]. It is as yet unknown whether this mechanism plays a part in the functioning of oncogenic mutant K-Ras. However, levels of ROS in transformed cells are especially elevated.
Ras dimerization
Ras dimers first were suggested by radiation inactivation experiments performed on H-Ras-transformed cells where Ras proteins with a molecular weight suggesting dimers were observed [44]. More recently, the existence of N-Ras, H-Ras, or K-Ras dimers have been reported under various experimental conditions [45-47]. The importance of Ras dimerization for the pathology of oncogenic Ras signals is currently unknown. However, Ras dimerization could offer molecular explanations for several observations that are currently not understood. For example, at this time the role of wild-type K-Ras in cancer development and progression is not understood. Some studies suggest a dominant negative effect of wild-type K-Ras [48]. Although not yet proven, it may be that heterodimers of wild-type and mutant K-Ras behave much differently than homodimers of mutant K-Ras. This may also explain why high levels of oncogenic mutant K-Ras have spontaneous high activity while endogenous levels do not. Furthermore, the existence of functional dimerization of heterologous Ras molecules would greatly increase the potential complexity of Ras signaling.
Significance of Ras Activity Model to Cancer Prevention
One of the most significant contributions of the Ras Activity Paradigm is the insight it has given to understanding cancer risk factors and preventative measures. Activation of oncogenic Ras likely explains the mechanism behind the effects of known risk factors for development of PDAC, including inflammation, chronic pancreatitis, and oxidative stress. Furthermore, the understanding that oncogenic K-Ras needs to be activated to cause cancer means that inhibition of K-Ras activation is a reasonable preventative strategy. At least in terms of cancer initiation, it may not be necessary to directly inhibit Ras activity but simply to prevent it from reaching pathological levels. The preventative effects of drugs that reduce cancer risk, such as nonsteroidal anti-inflammatory agents (eg, aspirin [49]) and various antioxidants [50], may well be due to their ability to inhibit activation of K-Ras. Knowing that oncogenic mutant Ras is still regulated makes it a much more amenable target for intervention. However, the information in this review applies to the earliest stages of PDAC initiation. It is unclear what the role of oncogenic mutant Ras is in more highly developed PDAC. Furthermore, in order for K-Ras to transform the cells, other important obstacles such as senescence must be overcome. Nonetheless, the new model for the role of oncogenic K-Ras in pancreatic cancer initiation should help in our understanding of the disease beginnings and provide new ideas for ways to prevent and treat PDAC.
Acknowledgements
This work was in part supported by grants from National Institutes of Health DK052067 to CDL. Further support came from the University of Texas MD Anderson Cancer Center Sheikh Ahmed Bin Zayed Al Nahyan Center for Pancreatic Cancer Research to W. Lu and the Lockton Foundation Endowment for CDL.
Competing Interests
The authors have declared that no competing interest exists.
References
1. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047-53
2. Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2-27
3. Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178-87
4. Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563-74
5. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842-57
6. Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul. 2014;55:1-14
7. Osterop AP, Medema RH, vd Zon GC, Bos JL, Moller W, Maassen JA. Epidermal-growth-factor receptors generate Ras.GTP more efficiently than insulin receptors. Eur J Biochem. 1993;212:477-82
8. Nicke B, Tseng MJ, Fenrich M, Logsdon CD. Adenovirus-mediated gene transfer of RasN17 inhibits specific CCK actions on pancreatic acinar cells. Am J Physiol. 1999;276:G499-506
9. Holtz BJ, Lodewyk KB, Sebolt-Leopold JS, Ernst SA, Williams JA. ERK activation is required for CCK-mediated pancreatic adaptive growth in mice. Am J Physiol Gastrointest Liver Physiol. 2014;307:G700-10
10. Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J. et al. Ras Activity Levels Control the Development of Pancreatic Diseases. Gastroenterology. 2009
11. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437-50
12. Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L. et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291-302
13. Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S. et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519-28
14. Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic K-Ras. Biochem Biophys Res Commun. 2009;382:561-5
15. Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB. et al. A High-Fat Diet Activates Oncogenic K-Ras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology. 2013
16. Yan L, McFaul C, Howes N, Leslie J, Lancaster G, Wong T. et al. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology. 2005;128:2124-30
17. Lu X, Xu T, Qian J, Wen X, Wu D. Detecting K-ras and p53 gene mutation from stool and pancreatic juice for diagnosis of early pancreatic cancer. Chin Med J (Engl). 2002;115:1632-6
18. Yakubovskaya MS, Spiegelman V, Luo FC, Malaev S, Salnev A, Zborovskaya I. et al. High frequency of K-ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int J Cancer. 1995;63:810-4
19. Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T. et al. Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology. 1996;110:227-31
20. Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol. 2003;16:996-1006
21. Shi C, Hong SM, Lim P, Kamiyama H, Khan M, Anders RA. et al. K-RAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Mol Cancer Res. 2009;7:230-6
22. Delire B, Starkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Invest. 2015;45:609-23
23. Calvisi DF, Ladu S, Conner EA, Seo D, Hsieh JT, Factor VM. et al. Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. J Hepatol. 2011;54:311-9
24. Kolfschoten IG, van Leeuwen B, Berns K, Mullenders J, Beijersbergen RL, Bernards R. et al. A genetic screen identifies PITX1 as a suppressor of RAS activity and tumorigenicity. Cell. 2005;121:849-58
25. Huang H, Daniluk J, Liu Y, Chu J, Li Z, Ji B. et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2013
26. Bossu P, Vanoni M, Wanke V, Cesaroni MP, Tropea F, Melillo G. et al. A dominant negative RAS-specific guanine nucleotide exchange factor reverses neoplastic phenotype in K-ras transformed mouse fibroblasts. Oncogene. 2000;19:2147-54
27. Almendro V, Garcia-Recio S, Gascon P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr Drug Targets. 2010;11:1169-80
28. Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2-13
29. Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK. et al. EGF receptor is required for K-RAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304-17
30. Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318-30
31. Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39-51
32. Holstein SA, Hohl RJ. Is there a future for prenyltransferase inhibitors in cancer therapy? Curr Opin Pharmacol. 2012;12:704-9
33. Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A. et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Molecular cell. 2006;21:481-93
34. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y. et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399-412
35. Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER. et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2011;4:ra13
36. Baker R, Wilkerson EM, Sumita K, Isom DG, Sasaki AT, Dohlman HG. et al. Differences in the regulation of K-Ras and H-Ras isoforms by monoubiquitination. J Biol Chem. 2013;288:36856-62
37. Pfleger CM. Ubiquitin on ras: warden or partner in crime? Sci Signal. 2011;4:pe12
38. Yang MH, Laurent G, Bause AS, Spang R, German N, Haigis MC. et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol Cancer Res. 2013;11:1072-7
39. Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195-8
40. Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD. et al. Redox regulation of cell signalling. Nature. 1996;381:380-1
41. Heo J. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxidants & redox signaling. 2011;14:689-724
42. Ferro E, Goitre L, Retta SF, Trabalzini L. The Interplay between ROS and Ras GTPases: Physiological and Pathological Implications. J Signal Transduct. 2012:365769
43. Heo J, Campbell SL. Superoxide anion radical modulates the activity of Ras and Ras-related GTPases by a radical-based mechanism similar to that of nitric oxide. The Journal of biological chemistry. 2005;280:12438-45
44. Santos E, Nebreda AR, Bryan T, Kempner ES. Oligomeric structure of p21 ras proteins as determined by radiation inactivation. J Biol Chem. 1988;263:9853-8
45. Guldenhaupt J, Rudack T, Bachler P, Mann D, Triola G, Waldmann H. et al. N-Ras forms dimers at POPC membranes. Biophys J. 2012;103:1585-93
46. Lin WC, Iversen L, Tu HL, Rhodes C, Christensen SM, Iwig JS. et al. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci U S A. 2014;111:2996-3001
47. Thompson H. US National Cancer Institute's new Ras project targets an old foe. Nat Med. 2013;19:949-50
48. Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G, Li J. et al. Wildtype K-Ras2 can inhibit lung carcinogenesis in mice. Nature genetics. 2001;29:25-33
49. Cui XJ, He Q, Zhang JM, Fan HJ, Wen ZF, Qin YR. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas. 2014;43:135-40
50. Stan SD, Singh SV, Brand RE. Chemoprevention strategies for pancreatic cancer. Nature reviews Gastroenterology & hepatology. 2010;7:347-56
Author contact
![]() Corresponding author: Craig D. Logsdon, Ph.D., 1515 Holcombe Blvd. Box 1906, Houston TX 77030. Phone: 713 563-3585; EMAIL: clogsdonorg
Corresponding author: Craig D. Logsdon, Ph.D., 1515 Holcombe Blvd. Box 1906, Houston TX 77030. Phone: 713 563-3585; EMAIL: clogsdonorg