Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Regulation of Epithelial Sodium...
Cyclic GMP/PKGII pathway
Extracellular ligand pathway
Relieving external sodium...
Moiety specificity for CPT-cGMP...
CPT-cGMP ligand docking to ENaC
Self-Inhibition as a Therapeutic...
Conclusions and Perspectives
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(4):359-366. doi:10.7150/ijbs.13764 This issue Cite
Review
CPT-cGMP Is A New Ligand of Epithelial Sodium Channels
Hong-Long Ji1 ![]() , Hong-Guang Nie2, Yongchang Chang3, Qizhou Lian4, Shan-Lu Liu5
, Hong-Guang Nie2, Yongchang Chang3, Qizhou Lian4, Shan-Lu Liu5
1. Department of Cellular and Molecular Biology, University of Texas Health Science Center at Tyler, Tyler, Texas 75708, USA.
2. Institute of Metabolic Disease Research and Drug Development, China Medical University, Shenyang 110001, China.
3. Barrow Neurological Institute, St Joseph's Hospital and Medical Center, Phoenix, Arizona, 85013, USA.
4. Department of Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China.
5. Department of Molecular Microbiology and Immunology, Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Received 2015-9-5; Accepted 2015-11-11; Published 2016-1-28
Abstract

Epithelial sodium channels (ENaC) are localized at the apical membrane of the epithelium, and are responsible for salt and fluid reabsorption. Renal ENaC takes up salt, thereby controlling salt content in serum. Loss-of-function ENaC mutations lead to low blood pressure due to salt-wasting, while gain-of-function mutations cause impaired sodium excretion and subsequent hypertension as well as hypokalemia. ENaC activity is regulated by intracellular and extracellular signals, including hormones, neurotransmitters, protein kinases, and small compounds. Cyclic nucleotides are broadly involved in stimulating protein kinase A and protein kinase G signaling pathways, and, surprisingly, also appear to have a role in regulating ENaC. Increasing evidence suggests that the cGMP analog, CPT-cGMP, activates αβγ-ENaC activity reversibly through an extracellular pathway in a dose-dependent manner. Furthermore, the parachlorophenylthio moiety and ribose 2'-hydroxy group of CPT-cGMP are essential for facilitating the opening of ENaC channels by this compound. Serving as an extracellular ligand, CPT-cGMP eliminates sodium self-inhibition, which is a novel mechanism for stimulating salt reabsorption in parallel to the traditional NO/cGMP/PKG signal pathway. In conclusion, ENaC may be a druggable target for CPT-cGMP, leading to treatments for kidney malfunctions in salt reabsorption.
Keywords: amiloride-sensitive sodium channel, cyclic guanosine nucleotides, molecular docking, lung edema.
Introduction
Cyclic guanosine monophosphates (cGMP) are important cyclic nucleotides, playing a critical role in signal transduction in many different organisms1. Signal transduction in eukaryotic cells is essential for moderating the transmission of information through the cell membrane, and as a second messenger, cGMP regulates numerous essential processes in cells by amplifying external signals, for example, from hormones and neurotransmitters. Cytosolic cGMP binds to cyclic nucleotide-gated ion channels1, 2 and phosphorylates protein kinase G (PKG) directly3-5.
Amiloride-sensitive epithelial sodium channels (ENaC) behave as ligand-gated channels, similar to several other members of the ENaC/degenerin family6. In the late part of the renal distal convoluted tubule, sodium is predominately reabsorbed via the electrogenic amiloride-sensitive ENaC. In the connecting tube, and the collecting duct, ENaC is the only pathway for retaining salt, and ENaC controls renal sodium excretion and provides the driving force for potassium secretion through the renal outer medullary potassium channel7. Loss-of-function ENaC genetic variants lead to low blood pressure, while gain-of-function mutants have been identified in Liddle's syndrome, which is characterized by increased ENaC abundance, augmented opening time, and consequent salt retention, and eventually volume-expended hypertension8. An autosomal recessive form of pseudohypoaldosteronism type 1 is caused by mutations in ENaC, with usually severe and persisting multiorgan symptoms 9.
Five ENaC subunits have been cloned to date, namely α-, β-, γ-, δ-, and ε-ENaC. Among them, the β- and γ-subunits regulate the channel activity of the 'self-conducting' α-, δ-, and ε-ENaC subunits when heterologously expressed in oocytes and cell lines 10-12. Luminal impermeable reagents and hormones have been confirmed to regulate ENaC in lung, kidney, and colon, and ENaC is regulated by a spectrum of protein kinases, such as PKG and protein kinase A (PKA) 13-15. However, our understanding of the cellular and molecular mechanisms by which cGMP regulates ENaC is incomplete.
Regulation of Epithelial Sodium Channels by CPT-cGMP
Cyclic GMP is one of the most prominent nucleotides in eukaryotic cells. Guanylyl cyclases elevate cell cGMP levels, which regulate complex signaling cascades through immediate downstream effectors, cGMP-dependent protein kinases, and cyclic nucleotide-gated ion channels 1. CPT-cGMP is a derivative analog of parental cGMP, and has long been used for studying the NO/cGMP/PKG cascade 16, 17. Regulation of epithelial sodium channels by CPT-cGMP and its cAMP analog is summarized in Table 1. Species- and culture-dependence of ENaC properties, and the strategies used for elevating cell cGMP content, including nitric oxide (NO) donors and PKG isoform-specific cGMP analogs, may contribute to these divergent observations.
Regulation of sodium channel activity by nucleotides.
| Nucleotides | Concentration | Models | Effects | Ref. |
|---|---|---|---|---|
| CPT-cGMP* | 1 μM | Xenopus 2F3 cells | Increase in Isc | 62 |
| 400 μM | Rat ATII cells | No change in Isc | 63 | |
| 2 mM | Rat bronchioalveolar epithelial cells | Increase in Isc/ 22Na influx | 64, 65 | |
| 100 μM | Rat ATII cells | Increase in whole cell activity | 66 | |
| 4 mM | Human A549 cells | Increase in whole cell activity | 67 | |
| 10 μM | Frog urinary bladder epithelium | Increase in single channel activity | 68, 69 | |
| 0.1 - 1000 μM | Heterologous human αβγ ENaC in oocytes | Increase in whole cell activity | 16 | |
| 0.5 mM | Human lung lobes | Increase in alveolar fluid clearance | 24 | |
| 0.2 mM | Mouse pleural tissues | Increase in Isc | 15 | |
| 1 mM | Rat ATII cells | Decrease in single channel activity | 56 | |
| 100 μM | Human A549 cells | Decrease in whole cell activity | 70 | |
| CPT-cAMP | 100 μM | MDCK cells | Increase in Isc | |
| 40 μM | Liddle's disease lymphocytes | Increase in whole cell activity | 71 | |
| 200 μM | Heterologous human αβγ ENaC in oocytes | Increase in whole cell activity | 14, 21, 72 | |
| 100 µM | BeWo cells | Increase in whole cell activity | 73 |
*Abbreviations: ATII, alveolar type II epithelial; MDCK, Madin-Darby canine kidney; Isc, short-circuit current; CPT-cAMP, 8-pCPT-cAMP-Na; CPT-cGMP, 8-pCPT-cGMP-Na.
Cyclic GMP/PKGII pathway
To date, three PKG isoforms have been isolated, namely, PKGI-α, PKGI-β, and PKGII 18. Accumulating evidence from genetically engineered animals suggests that cardiovascular phenotypes are predominant with the PKGI knockout, while PKGII deficiency leads to dysfunction in epithelial tissues [19, 20]. Our results using Ussing chamber and voltage clamp techniques showed that 8-pCPT-cGMP increased the amiloride-sensitive short-circuit current across H441 cell monolayers and also increased heterologously expressed αβγδ-ENaC activity in a dose-dependent manner, most probably through the stimulation of PKGII enzymatic activity and subsequent activation of channel function 15 (Figure 1). A carton briefly showed the interaction of cGMP and ENaC (Figure 2). Recent studies showed that PKG-interacting proteins mediate cellular targeting of PKG isoforms by interacting with their leucine zipper domains, and that protein recognition is mediated through surface charge interactions 19.
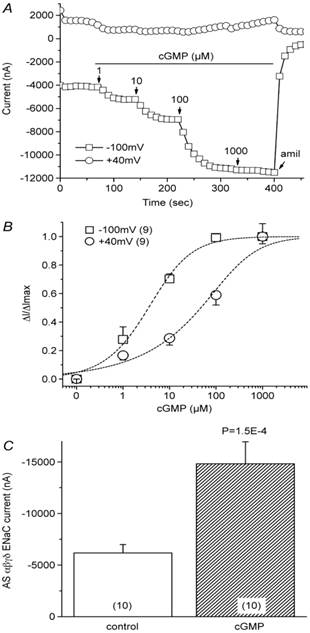
Stimulation of heterologously expressed human ENaCs in Xenopus oocytes by cGMP. A: Representative current trace recorded at +40 mV and -100 mV. Oocytes were perfused with 8-pCPT-cGMP ranging from 1 μM to 1 mM, as indicated by arrows. Amiloride was added to the bath before the recordings were terminated. B: Dose-response curve. The average cGMP-activated current fraction in the presence of cGMP (ΔI) was normalized to the maximal cGMP-elevated current (ΔImax) and plotted as a function of cGMP concentration. Dashed lines and the EC50 value were derived by fitting the raw data with the Hill equation. n=9. C: Average amiloride-sensitive (AS) αβγδ ENaC currents before (control) and after cGMP perfusion. Holding potential, -100 mV. n=10. Figure reprinted from 15, and used with permission.
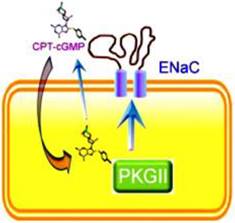
Extracellular and intracellular mechanisms for CPT-cGMP or CPT-cAMP to activate ENaC. These cell permeable compounds, when applied in vivo or in vitro, will stimulates ENaC activity by both acting as an external ligand to release self-inhibition and as a cytosolic signal molecule to phosphorylate cGKII that subsequently activates ENaC.
Extracellular ligand pathway
In sharp contrast to the native ENaC, heterologously expressed αβγ-ENaC in oocytes has been considered to be cAMP/PKA-independent, particularly for acute regulation. The cell-permeable PKGII isoform-specific activator, 8-pCPT-cGMP, acutely stimulates human, rat, and mouse αβγ-ENaC reversibly. Furthermore, intraoocyte domains of ENaC subunits and traditional soluble protein kinases may not be targets of this compound 20. A critical extracellular domain has recently been identified as the 8-CPT-cAMP binding site 21, indicating that 8-CPT-cAMP is an extracellular ligand for ENaC 6, 21. Moreover, 8-CPT-cAMP can stimulate δβγ-ENaC up to approximately 3-fold, and αβγ-ENaC by 2-fold 14. Co-expression of δ-ENaC with αβγ channels conferred CPT-cAMP-mediated activation with an EC50 (concentration for 50% of maximal effect) value of 30 μM, similar to that for αβγ channels (49 μM) 22.
Relieving external sodium self-inhibition
External sodium self-inhibition is an intrinsic feature of ENaC. A rapidly increase in extracellular sodium ions to a physiological concentration (150 mM for mammals and 100mM for amphibians) generates a maximal peak current in seconds, and then the permeability of ENaC to Na+ ions is gradually reduced to a relatively stable level with a current level of approximately half of the maximal value. This phenomenon is called extracellular sodium self-inhibition of ENaC activity. It differs from the down-regulation of ENaC activity by slowly accumulating intracellular Na+ content in a feedback manner. External Na+ self-inhibition is a crucial mechanism to limit overwhelming salt absorption to prevent a quick raise in epithelial cell volume and blood pressure. It maintains salt and fluid homeostasis at the luminal surface of the respiratory system to keep normal cilia beating in the airways, hyperpolarize apical membrane, and finely adjust fluid volume to host leukocytes and physiological regulators as the forefront battle against noxious aspirated insults 22.
Our previous studies showed that external CPT-cAMP stimulated human, but not rat and murine, αβγ-ENaC in a dose-dependent and external sodium concentration-dependent fashion 14. ENaC mutations that abolished self-inhibition (βΔV348 and γH233R) almost completely eliminated CPT-cAMP mediated activation. In contrast, mutations that both enhanced self-inhibition and elevated CPT-cAMP sensitivity increased the stimulating effects of the compound. Our above data confirmed that CPT-cAMP acts as a ligand to regulate heterologous ENaC by relieving self-inhibition. Edelheit et al 23 studied alanine mutations in 17 conserved charged residues of ENaC and found that these residues are involved in conformational changes that lead to channel constriction and to the sodium self-inhibition response upon sodium ion flooding. Similarly, our recent data showed that elimination of self-inhibition of αβγ-ENaC may be a novel mechanism for CPT-cGMP to stimulate salt reabsorption in the human epithelium (Figure 3) 24.
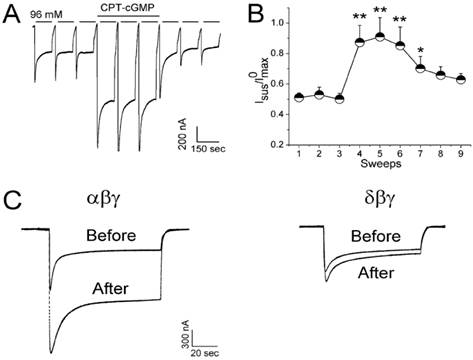
CPT-cGMP modifies self-inhibition of αβγ ENaC in oocytes. Self-inhibition was initiated by fast switching the low sodium bath solution (1 mM sodium) to regular ND96 medium (96 mM sodium). A: Whole-cell current trace digitized at -60 mV. CPT-cGMP (0.2 mM) was added after the first three sweeps and washed out for the last three sweeps. B: Normalized and averaged currents; *P < 0.05 and **P < 0.01 versus the first sweep. C: Comparison of CPT-cGMP on self-inhibition of αβγ and δβγ ENaC channels. Paired traces were recorded before and after application of CPT-cGMP, n=9. Figure reprinted from 24, and used with permission.
Moiety specificity for CPT-cGMP serving as an ENaC ligand
CPT-cGMP, an analog of CPT-cAMP, was capable of activating ENaC in the identical manner in cell-free outside-out patches 16. Both point mutations of the putative consensus PKG phosphorylation sites and truncation of entire cytosolic NH2- and COOH-terminal tails of ENaC did not alter the response to CPT-cGMP. The ENaC activity was activated to the same extent by CPT-cGMP in cells in which PKGII expression was knocked down using small interfering RNA. As we demonstrated, the parachlorophenylthio moiety and ribose 2'-hydroxy group of CPT-cGMP are crucial for activating ENaC 16, and CPT-cGMP may serve as a novel ENaC ligand in addition to activating the PKG signal. The strict requirement for both parachlorophenylthio and the ribose 2'-hydroxy groups helps to clarify the basis for the inconsistent results observed with the regulation of ENaC by a variety of cGMP analogs (Figure 4).
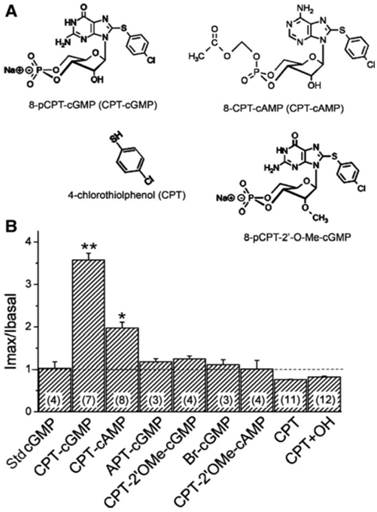
Domain-dependence of CPT-cGMP and CPT-cAMP analogs. A: Modified positions are shown for 8-pCPT-cGMP and 8-CPT-cAMP. B: The responses of human αβγ-ENaC to these compounds (0.2 mM) were compared: 8-pCPTcGMP, (CPT-cGMP); 8-CPT-cAMP, (CPT-cAMP); 2-aminophenylthio-cGMP, (APT-cGMP); 8-pCPT-methylated ribose 2'-hydroxy (2'-O-Me)-cGMP (CPT-2'OMe-cGMP); Sp-8-Br-cGMP, (Br-cGMP); 8-CPT-2'-O-Me-cAMP, (CPT-2'OMe-cAMP), 4-chlorothiolphenol, (CPT); a mixture of CPT and KOH, (CPT+OH). The normalized currents before and after application of these compounds were compared with that of the standard cGMP molecule (Std cGMP). **P < 0.01, *P < 0.05. Numbers in parentheses are the number of oocytes examined for each group. Figure reprinted from 16, and used with permission.
CPT-cGMP ligand docking to ENaC
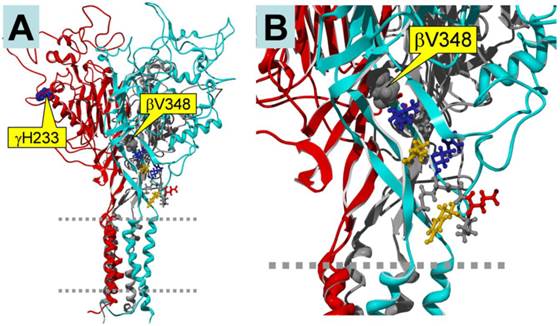
In our previous experiment, we constructed mutants abolishing (βV348 and γH233R), or augmenting (αY458A and γM432G), ENaC self-inhibition 14, 24. The mutations eliminating self-inhibition resulted in a loss of response to CPT-cGMP, whereas those enhancing self-inhibition facilitated the stimulatory effects of this compound. Figure 5 shows the potential binding sites for the CPT-cGMP ligand in ENaC domains that are crucial for self-inhibition. βV348 is located in the center of the palm region of the subunit, and γH233 is located in the vicinity of the putative binding site for protons. These domains potentially directly or allosterically interact with CPT-cGMP.
Homology model of trimeric human αβγ ENaC. The subunits are color-coded as follows: α, grey (back, center); β, cyan (front, right); γ, red (front, left). A: Side view of the entire trimer. Two dashed grey lines indicate the plasma membrane. The locations of the mutations γH233 and βV348 are marked by yellow boxes. B: Close-up view, showing the location of βV348. Note that βV348 is located in the center of the palm region of the subunit, and its downstream residues extend to the wrist region of the subunit that seems to be important in the coupling between extracellular domains to the pore region. Thus, deletion of βV348 may change the position and orientation of the downstream residues. γH233 is located in the vicinity of the putative binding site for protons. Since 8-CPT-cGMP is much larger than a proton, its binding site may include the H233 residue.
Self-Inhibition as a Therapeutic Mechanism
Reagents able to release self-inhibition
Previous results suggested that external Zn2+ rapidly and reversibly activates ENaC in a dose-dependent manner by relieving the channel from sodium self-inhibition25. In addition to Zn, extracellular chloride 26, temperature27, 28, halogenated gases 27-29, sulfhydryl group-modifying reagents (e.g., p-chloromercuriphenylsulfonate, pCMPS and p-chloromercuribenzoate, pCMB) 30, p-chloromercuribenzoate benzimidazolylguanidine 30-33, protons 26, cupper 34, and proteases 28, 35, 36 are also proposed to modulate ENaC-gating by relieving sodium self-inhibition. A classic earlier review summarized most of these reagents27,37,38. Non-cleaved channels have a low intrinsic open probability that may reflect enhanced channel inhibition by external sodium, and cleavage at a minimum of two sites within the α- or γ-ENaC subunits is required to activate the channel, presumably by releasing inhibitory fragments 39. The extent of ENaC proteolysis is dependent on channel residency time at the plasma membrane, as well as on the balance between levels of expression of proteases that activate epithelial sodium channels and inhibitors of these proteases. For example, furin cleavage of ENaC subunits activates the channels by relieving sodium self-inhibition, and this activation requires that the α-ENaC subunit be cleaved twice35.
Specific domains/sites changing self-inhibition
His(88) and Asp(516) of the γ subunit play a role in the Zn2+-regulating sodium self-inhibition mentioned above. Recent studies showed that palmitoylation of the γ subunit activates ENaC by increasing the open probability of the channels 40. ENaC mutants with the mutations γC33A, γC41A, or γC33A/C41A have significantly enhanced sodium self-inhibition and reduced open probability compared with wild type ENaCs, suggesting that ENaC palmitoylation is an important post-translational mechanism of channel regulation. Exon 11 within the human α, β, and γ ENaC genes encodes structurally homologous yet functionally diverse domains, and exon 11 in the α-subunit encodes a module that regulates channel gating 41.
In contrast to the other mutations, γL511Q largely eliminated the sodium self-inhibition response, reflecting a down-regulation of ENaC open probability by extracellular sodium 42. γL511Q is a gain-of-function human ENaC variant and it enhances ENaC activity by increasing channel open probability and dampens channel regulation by extracellular sodium and proteases 42.
Therapeutic effects of targeting self-inhibition
Renal handling of sodium and water is abnormal in chronic kidney diseases. Filtrated sodium is reabsorbed from the glomerular filtrate, and potassium is secreted through a tight epithelium in the kidney. Sodium crosses the apical membrane and enters the epithelial cell through the ion-selective ENaC 43. ENaC is responsible for the reabsorption of sodium through the apical membrane of the connecting tubule and the collecting duct, and plays a key role in controlling sodium balance, extracellular fluid volume, and blood pressure 44. Regulated epithelial sodium channel proteolysis has been observed in rat kidney and in human airway epithelia. Pseudohypoaldosteronism type 1 is a monogenic disorder of mineralocorticoid resistance characterized by salt-wasting, hyperkalemia, high aldosterone levels, and failure to thrive 45. An autosomal recessive form of pseudohypoaldosteronism type 1 is caused by mutations in ENaC, and is usually associated with severe and persisting multiorgan symptoms. The relief of self-inhibition by CPT-cAMP contributes to the acute effects, in addition to the well-known cAMP-PKA signal pathway 14. In fact, the clinical relevance of the abnormal regulation of ENaC by CPT-cAMP has been implicated in the upregulation of ENaC in autosomal recessive pseudohypoaldosteronism type 1 46.
Another distressed transapical sodium transport occurs in injured lungs, for example, acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (please see classical reviewes 47-51). Apical ENaC contributes to up to 60% of transepithelial sodium transport in mammalian lungs under physiological conditions. This critical process is sensitive to aspirated pollutants, allergens, pathogens, and bacterial endotoxins. In addition to increased leaking through alveolar microvascular endothelium (indirect ALI), lung edema ususally results from reduced edema fluid resolution via ENaC (direct ALI). ENaC is a promising target for developing new therapeutical strategies to alleviate lung edema, at least for the phenotype of direct ALI 52, 53.
CPT-cGMP and self-inhibition
Human serum cGMP level is 6 nM and 3-time greater in human bronchoalveolar lavage 54. It appears that cGMP may serve as an autocrine and paracrine to regulate ENaC function. However, the effective dose for CPT-cGMP and CPT-cAMP to blunt self-inhibition is micromolar, suggesting an uncertain physiological role for cGMP and analogs. A large dose of cGMP compound (1 mg/kg) was administered to patients as reported by two clinical trials 55, and numerous preclinical studies (from 100 µM to 2 mM) 56-58. It is therefore feasible to apply aerosolized nucleotides to mitigate edematous lung injury. We have demonstrated that CPT-cGMP up-regulates ENaC via two mechanisms: release self-inhibition externally and activates ENaC through cGMP/cGKII pathway intracellularly 24. Thus, these compounds could regulate sodium absorption via either or both mechanisms in a cell permeability-dependent manner. Administration of cAMP could be a potent pharmaceutical treatment for edematous lung injury 59, and cGMP may have similar potential. cGMP increased in murine and rat lungs both in vivo and in vitro following NO application 60, and increased cGMP may augment the cGMP-sensitive pathway for lung fluid removal from alveolar sacs 61. Our previous study demonstrated for the first time that PKGII is an ENaC activator in non-ciliated bronchial secretory cells 15. Accordingly, upregulation of the rate-limiting ENaCs in respiratory epithelial cells by specific PKGII activators may be a potent clinicopharmaceutical strategy for alleviating airspace flooding in fatal edematous lung diseases. The observation of our previous study that specific moieties of 8-pCPT-cGMP are required for activating ENaC may provide pivotal information for developing potential ENaC channel openers structurally related to 8-pCPT-cGMP, which would be extremely useful for treating diseases associated with lower ENaC function. We postulate that when the tight epithelial layer is damaged, for example, in injured lungs, even though the mixture of extracellular matrix proteins, including collagens, albumin, and fibrins, will seal the epithelial cell-free alveolar surface, the potency of ENaC stimulator will be limited significantly. Therefore, the integrity of the tight alveolar epithelium should be a key factor to be considered for the usage of ENaC agonists. The anticipated restore of alveolar fluid clearance may be seen at the earlier stage of ALI and lung edema mainly caused by injured pulmonary vasculature or post regeneration of alveolar epithelium by stem cells/progenitors.
Conclusions and Perspectives
ENaC is involved in edema formation and kidney disorders. cGMP, either by serving as a ligand to regulate cyclic nucleotide-gated ion channels or by acting upstream of the PKG signaling systems, has a role in the elimination of ENaC self-inhibition, suggesting a novel mechanism for CPT-cGMP to stimulate salt reabsorption. Compounds such as specific PKGII activators and/or self-inhibition inhibitors could be the basis for novel pharmaceutical interventions for combating diseases associated with impaired ENaC function. Future directions for study include optimizing cGMP structure to potentiate its efficacy in activating ENaC, evaluating its pharmaceutical relevance in preclinical and clinical models with impaired ENaC activity, and confirming the binding sites of cGMP in ENaC.
Acknowledgements
The authors are grateful for helpful discussions with Dr. Run-Zhen Zhao (University of Texas Health Science Center at Tyler), and Dr. Zai-Xing Chen (China Medical University). This work was partially supported by NIH grants HL87017 and HL095435, the American Heart Association (AHA 14GRNT20130034 to HLJ), and the National Science Foundation of China (grants 81270098 to HGN, 31270967 and 31571407 to QL).
Abbreviations
cAMP: cyclic adenosine monophosphate; cGMP: cyclic guanosine monophosphate; EC50: concentration for 50% of maximal effect; ENaC: epithelial sodium channel; PKA: protein kinase A; PKG: protein kinase G; NO: nitric oxide
Competing Interests
The authors declare no conflicts of interest.
References
1. Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S. et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375-414
2. Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769-824
3. Gudi T, Huvar I, Meinecke M, Lohmann SM, Boss GR, Pilz RB. Regulation of gene expression by cGMP-dependent protein kinase. Transactivation of the c-fos promoter. J Biol Chem. 1996;271:4597-600
4. Edwards AS, Scott JD. A-kinase anchoring proteins: protein kinase A and beyond. Curr Opin Cell Biol. 2000;12:217-21
5. Kim E, Park JM. Identification of novel target proteins of cyclic GMP signaling pathways using chemical proteomics. J Biochem Mol Biol. 2003;36:299-304
6. Horisberger JD, Chraibi A. Epithelial sodium channel: a ligand-gated channel? Nephron Physiol. 2004;96:37-41
7. Ronzaud C, Staub O. Ubiquitylation and control of renal Na+ balance and blood pressure. Physiology (Bethesda). 2014;29:16-26
8. Svenningsen P, Andersen H, Nielsen LH, Jensen BL. Urinary serine proteases and activation of ENaC in kidney-implications for physiological renal salt handling and hypertensive disorders with albuminuria. Pflugers Arch. 2015;467:531-42
9. Zennaro MC, Hubert EL, Fernandes-Rosa FL. Aldosterone resistance: structural and functional considerations and new perspectives. Mol Cell Endocrinol. 2012;350:206-15
10. Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR. et al. Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem. 2006;281:8233-41
11. Zhao RZ, Nie HG, Su XF, Han DY, Lee A, Huang Y. et al. Characterization of a novel splice variant of delta ENaC subunit in human lungs. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1262-72
12. Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467-70
13. Ji HL, Song W, Gao Z, Su XF, Nie HG, Jiang Y. et al. SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am J Physiol Lung Cell Mol Physiol. 2009;296:L372-83
14. Molina R, Han DY, Su XF, Zhao RZ, Zhao M, Sharp GM. et al. Cpt-cAMP activates human epithelial sodium channels via relieving self-inhibition. Biochim Biophys Acta. 2011;1808:1818-26
15. Nie HG, Chen L, Han DY, Li J, Song WF, Wei SP. et al. Regulation of epithelial sodium channels by cGMP/PKGII. J Physiol. 2009;587:2663-76
16. Nie HG, Zhang W, Han DY, Li QN, Li J, Zhao RZ. et al. 8-pCPT-cGMP stimulates alphabetagamma-ENaC activity in oocytes as an external ligand requiring specific nucleotide moieties. Am J Physiol Renal Physiol. 2010;298:F323-34
17. Broderick KE, Zhang T, Rangaswami H, Zeng Y, Zhao X, Boss GR. et al. Guanosine 3',5'-cyclic monophosphate (cGMP)/cGMP-dependent protein kinase induce interleukin-6 transcription in osteoblasts. Mol Endocrinol. 2007;21:1148-62
18. Schlossmann J, Feil R, Hofmann F. Signaling through NO and cGMP-dependent protein kinases. Ann Med. 2003;35:21-7
19. Reger AS, Yang MP, Koide-Yoshida S, Guo E, Mehta S, Yuasa K. et al. Crystal structure of the cGMP-dependent protein kinase II leucine zipper and Rab11b protein complex reveals molecular details of G-kinase-specific interactions. J Biol Chem. 2014;289:25393-403
20. Nie HG, Zhang W, Han DY, Li QN, Li J, Zhao RZ. et al. 8-pCPT-cGMP stimulates alphabetagamma-ENaC activity in oocytes as an external ligand requiring specific nucleotide moieties. Am J Physiol Renal Physiol. 2010;298:F323-34
21. Renauld S, Allache R, Chraibi C. Ile481 from the guinea pig alpha-subunit plays a major role in the activation of ENaC by cpt-cAMP. Cell Physiol Biochem. 2008;22:101-8
22. Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, Matalon S. delta ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1013-26
23. Edelheit O, Ben-Shahar R, Dascal N, Hanukoglu A, Hanukoglu I. Conserved charged residues at the surface and interface of epithelial sodium channel subunits-roles in cell surface expression and the sodium self-inhibition response. FEBS J. 2014;281:2097-111
24. Han DY, Nie HG, Su XF, Shi XM, Bhattarai D, Zhao M. et al. 8-(4-chlorophenylthio)-guanosine-3',5'-cyclic monophosphate-Na stimulates human alveolar fluid clearance by releasing external Na+ self-inhibition of epithelial Na+ channels. Am J Respir Cell Mol Biol. 2011;45:1007-14
25. Sheng S, Bruns JB, Kleyman TR. Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J Biol Chem. 2004;279:9743-9
26. Collier DM, Snyder PM. Extracellular chloride regulates the epithelial sodium channel. J Biol Chem. 2009;284:29320-5
27. Chraibi A, Horisberger JD. Na self inhibition of human epithelial Na channel: temperature dependence and effect of extracellular proteases. J Gen Physiol. 2002;120:133-45
28. Chraibi A, Horisberger JD. Dual effect of temperature on the human epithelial Na+ channel. Pflugers Arch. 2003;447:316-20
29. Roch A, Shlyonsky V, Goolaerts A, Mies F, Sariban-Sohraby S. Halothane directly modifies Na+ and K+ channel activities in cultured human alveolar epithelial cells. Mol Pharmacol. 2006;69:1755-62
30. Luger A, Turnheim K. Modification of cation permeability of rabbit descending colon by sulphydryl reagents. J Physiol. 1981;317:49-66
31. Friis S, Nielsen R. Effect of the putative Ca2+-receptor agonist Gd3+ on the active transepithelial Na+ transport in frog skin. J Membr Biol. 2001;184:291-7
32. Li JH, Lindemann B. Chemical stimulation of Na transport through amiloride-blockable channels of frog skin epithelium. J Membr Biol. 1983;75:179-92
33. Zeiske W, Lindemann B. Chemical stimulation of Na+ current through the outer surface of frog skin epithelium. Biochim Biophys Acta. 1974;352:323-6
34. Ferreira KT, Guerreiro MM, Svensson WM. The mechanism of action of Cu2+ on the frog skin. Biochim Biophys Acta. 1979;552:341-5
35. Sheng S, Carattino MD, Bruns JB, Hughey RP, Kleyman TR. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am J Physiol Renal Physiol. 2006;290:F1488-96
36. Molina R, Han DY, Su XF, Zhao RZ, Zhao M, Sharp GM. et al. Cpt-cAMP activates human epithelial sodium channels via relieving self-inhibition. Biochim Biophys Acta. 2011;1808:1818-26
37. Garty H, Benos DJ. Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiol Rev. 1988;68:309-73
38. Collier DM, Snyder PM. Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J Biol Chem. 2009;284:792-8
39. Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens. 2007;16:444-50
40. Mukherjee A, Mueller GM, Kinlough CL, Sheng N, Wang Z, Mustafa SA. et al. Cysteine palmitoylation of the gamma subunit has a dominant role in modulating activity of the epithelial sodium channel. J Biol Chem. 2014;289:14351-9
41. Chen Z, Zhao R, Zhao M, Liang X, Bhattarai D, Dhiman R. et al. Regulation of epithelial sodium channels in urokinase plasminogen activator deficiency. Am J Physiol Lung Cell Mol Physiol. 2014;307:L609-17
42. Chen J, Kleyman TR, Sheng S. Gain-of-function variant of the human epithelial sodium channel. Am J Physiol Renal Physiol. 2013;304:F207-13
43. Wang YB, Leroy V, Maunsbach AB, Doucet A, Hasler U, Dizin E. et al. Sodium transport is modulated by p38 kinase-dependent cross-talk between ENaC and Na,K-ATPase in collecting duct principal cells. J Am Soc Nephrol. 2014;25:250-9
44. Graffe CC, Bech JN, Lauridsen TG, Pedersen EB. Urinary excretion of AQP2 and ENaC in autosomal dominant polycystic kidney disease during basal conditions and after a hypertonic saline infusion. Am J Physiol Renal Physiol. 2014;302:F917-27
45. Dirlewanger M, Huser D, Zennaro MC, Girardin E, Schild L, Schwitzgebel VM. A homozygous missense mutation in SCNN1A is responsible for a transient neonatal form of pseudohypoaldosteronism type 1. Am J Physiol Endocrinol Metab. 2011;301:E467-73
46. Bubien JK, Ismailov II, Berdiev BK, Cornwell T, Lifton RP, Fuller CM. et al. Liddle's disease: abnormal regulation of amiloride-sensitive Na+ channels by beta-subunit mutation. Am J Physiol. 1996;270:C208-13
47. Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569-600
48. Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350-9
49. Matalon S, Lazrak A, Jain L, Eaton DC. Invited review: biophysical properties of sodium channels in lung alveolar epithelial cells. J Appl Physiol. 2002;93:1852-9
50. Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403-23
51. Matalon S, Bartoszewski R, Collawn JF. Role of Epithelial Sodium Channels (ENaC) In the Regulation of Lung Fluid Homeostasis. Am J Physiol Lung Cell Mol Physiol. 2015 ajplung 00319 02015
52. Czikora I, Alli A, Bao HF, Kaftan D, Sridhar S, Apell HJ. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am J Respir Crit Care Med. 2014;190:522-32
53. Giraldez T, Dominguez J, Alvarez de la Rosa D. ENaC in the brain-future perspectives and pharmacological implications. Curr Mol Pharmacol. 2013;6:44-9
54. Arias-Diaz J, Vara E, Torres-Melero J, Garcia C, Baki W, Ramirez-Armengol JA, Role of Epithelial Sodium Channels (ENaC) In the Regulation of Lung Fluid Homeostasis. Nitrite/nitrate and cytokine levels in bronchoalveolar lavage fluid of lung cancer patients. Cancer. 1994;74:1546-51
55. Sandera P, Hillinger S, Stammberger U, Schoedon G, Zalunardo M, Weder W, Role of Epithelial Sodium Channels (ENaC) In the Regulation of Lung Fluid Homeostasis. 8-Br-cyclic GMP given during reperfusion improves post-transplant lung edema and free radical injury. J Heart Lung Transplant. 2000;19:173-8
56. Jain L, Chen XJ, Brown LA, Eaton DC. Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am J Physiol. 1998;274:L475-84
57. Chen L, Bosworth CA, Pico T, Collawn JF, Varga K, Gao Z. et al. DETANO and nitrated lipids increase chloride secretion across lung airway cells. Am J Respir Cell Mol Biol. 2008;39:150-62
58. Kemp PJ, Kim KJ, Borok Z, Crandall ED. Re-evaluating the Na+ conductance of adult rat alveolar type II pneumocytes: evidence for the involvement of cGMP-activated cation channels. J Physiol. 2001;536:693-701
59. Chen L, Song W, Davis IC, Shrestha K, Schwiebert E, Sullender WM. et al. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am J Respir Cell Mol Biol. 2009;40:588-600
60. Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, Eaton DC. et al. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol. 2004;30:720-8
61. Sakuma T, Zhao Y, Sugita M, Sagawa M, Toga H, Ishibashi T. et al. Malnutrition impairs alveolar fluid clearance in rat lungs. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1268-74
62. Guo LJ, Alli AA, Eaton DC, Bao HF. ENaC is regulated by natriuretic peptide receptor-dependent cGMP signaling. Am J Physiol Renal Physiol. 2013;304:F930-7
63. Guo Y, DuVall MD, Crow JP, Matalon S. Nitric oxide inhibits Na+ absorption across cultured alveolar type II monolayers. Am J Physiol. 1998;274:L369-77
64. Rafii B, Gillie DJ, Sulowski C, Hannam V, Cheung T, Otulakowski G. et al. Pulmonary oedema fluid induces non-alpha-ENaC-dependent Na(+) transport and fluid absorption in the distal lung. J Physiol. 2002;544:537-48
65. Schwiebert EM, Potter ED, Hwang TH, Woo JS, Ding C, Qiu W. et al. cGMP stimulates sodium and chloride currents in rat tracheal airway epithelia. Am J Physiol. 1997;272:C911-22
66. Kemp PJ, Kim KJ, Borok Z, Crandall ED. Re-evaluating the Na(+) conductance of adult rat alveolar type II pneumocytes: evidence for the involvement of cGMP-activated cation channels. J Physiol. 2001;536:693-701
67. Xu W, Leung S, Wright J, Guggino SE. Expression of cyclic nucleotide-gated cation channels in airway epithelial cells. J Membr Biol. 1999;171:117-26
68. Yamada T, Konno N, Matsuda K, Uchiyama M. Frog atrial natriuretic peptide and cGMP activate amiloride-sensitive Na(+) channels in urinary bladder cells of Japanese tree frog, Hyla japonica. J Comp Physiol B. 2007;177:503-8
69. Yamada T, Matsuda K, Uchiyama M. Frog ANP increases the amiloride-sensitive Na(+) channel activity in urinary bladder cells of Japanese tree frog, Hyla japonica. Gen Comp Endocrinol. 2007;152:286-8
70. Lazrak A, Nielsen VG, Matalon S. Mechanisms of increased Na(+) transport in ATII cells by cAMP: we agree to disagree and do more experiments. Am J Physiol Lung Cell Mol Physiol. 2000;278:L233-8
71. Bubien JK, Watson B, Khan MA, Langloh AL, Fuller CM, Berdiev B. et al. Expression and regulation of normal and polymorphic epithelial sodium channel by human lymphocytes. J Biol Chem. 2001;276:8557-66
72. Robins GG, MacLennan KA, Boot-Handford RP, Sandle GI. Rapid stimulation of human renal ENaC by cAMP in Xenopus laevis oocytes. J Physiol Biochem. 2013;69:419-27
73. del Monaco S, Assef Y, Kotsias BA. Epithelial sodium channel in a human trophoblast cell line (BeWo). J Membr Biol. 2008;223:127-39
Author contact
![]() Corresponding author: Hong-Long (James) Ji, 11937 US Highway 271, 604 BMR, Tyler, TX 75708-3154. E-mail: james.jiedu. Telephone: 903-877-7418. Fax: 903-877-5438.
Corresponding author: Hong-Long (James) Ji, 11937 US Highway 271, 604 BMR, Tyler, TX 75708-3154. E-mail: james.jiedu. Telephone: 903-877-7418. Fax: 903-877-5438.