Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(6):631-638. doi:10.7150/ijbs.14878 This issue Cite
Research Paper
In Vitro Antileukemia Activity of ZSTK474 on K562 and Multidrug Resistant K562/A02 Cells
Qianxiang Zhou1,2, Yali Chen1,2, Xi Chen1,2, Wennan Zhao1,2, Yuxu Zhong3, Ran Wang1, Meihua Jin1, Yuling Qiu1, ![]() , Dexin Kong1,2
, Dexin Kong1,2 ![]()
1. Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China;
2. Research Center of Basic Medical Sciences, Tianjin Medical University, Tianjin 300070, China;
3. State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, China.
Received 2016-1-4; Accepted 2016-2-29; Published 2016-4-8
Abstract

Chronic myelogenous leukemia (CML) is a malignant hematological disorder mainly caused by the Bcr-Abl tyrosine kinase. While Bcr-Abl inhibitors including Imatinib showed antitumor efficacy on many CML patients, resistance was frequently reported in recent years. Therefore, novel drugs for CML are still expected. ZSTK474 is a specific phosphatidylinositol 3-kinase (PI3K) inhibitor that we identified. In the present study, the efficacy of ZSTK474, alone or in combination with Imatinib, on K562 CML cells as well as on its multidrug resistance counterpart K562/A02 cells, was investigated. ZSTK474 inhibited the cell proliferation with an IC50 of 4.69 μM for K562 and 7.57 μM for K562/A02 cells, respectively. Treatment by ZSTK474 resulted in cell cycle arrest in G1 phase, which might be associated with upregulation of p27, and downregulation of cyclin D1. ZSTK474 also inhibited phosphorylation of Akt and GSK-3β, which might be involved in the effect on the above cell cycle-related proteins. Moreover, combination of ZSTK474 and Imatinib indicated synergistic effect on both cell lines. In conclusion, ZSTK474 exhibited antileukemia activity alone, and showed synergistic effect when combined with Imatinib, on CML K562 cells as well as the multidrug resistant ones, providing a potential therapeutic approach for CML patients.
Keywords: ZSTK474, antileukemia, G1 arrest, Imatinib, combination, PI3K.
Introduction
Chronic myelogenous leukemia (CML), a myeloproliferative disease, is mainly molecularly diagnosed by the Philadelphia (Ph) chromosome which originates from a reciprocal translocation of breakpoint cluster region (Bcr) gene on chromosome 22 with Abelson (Abl) gene on chromosome 9 and leads to a chimeric oncogene called Bcr-Abl [1-3]. This fusion gene encodes p210 Bcr-Abl protein showing intrinsic tyrosine kinase activity [4, 5]. As p210 Bcr-Abl protein is crucial for CML progression, drug development for CML therapy mainly focused on tyrosine kinase inhibitors targeting Bcr-Abl in the past 15 years. Imatinib, a Bcr-Abl inhibitor as the first generation molecular-targeted agent against CML, inhibits p210 Bcr-Abl protein by binding with Abl at ATP-binding site and has been demonstrated to be therapeutically efficient [6-8]. Nevertheless, reports about resistance to Imatinib in clinic have been increasing [9, 10]. Therefore, discovery of novel effective agents with low resistance is urgent for CML treatment.
Phosphatidylinositol 3-kinases (PI3Ks) are lipid kinases that preferentially phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3) which activates the downstream Akt, and therefore play important roles in controlling signal pathways involved in cell proliferation, etc [11]. Bcr-Abl was reported to promote carcinogenesis and CML cell proliferation through PI3K/Akt pathway [12, 13]. Therefore, PI3K might be a potential target for CML therapy. ZSTK474, a specific PI3K inhibitor we identified by use of JFCR39 drug discovery system [14, 15], was reported to show potent antitumor efficacy on various solid tumors [16, 17]. However, the antileukemia effect of ZSTK474 has not yet been reported.
Recently, we investigated the in vitro antileukemia activity of ZSTK474 on CML cell K562 as well as the multidrug resistant counterpart K562/A02. Moreover, the synergistic effect of combinational use of ZSTK474 and Imatinib was analyzed.
Materials and Methods
Reagents
ZSTK474 and Adriamycin (ADR) were purchased from Selleck (London, ON, Canada). MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) reagent was from Amresco (Solon, OH, USA). Antibodies against Akt, phospho-Akt (Ser473), phospho-GSK-3β (Ser9), β-actin; anti-mouse and anti-rabbit HRP-conjugated secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against pRb (pS780), cyclin D1 and p27 were from BD Biosciences Pharmingen (San Jose, CA, USA). Antibody against Lamin B was from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture
The human chronic myelogenous leukemia K562 cell line was purchased from Cell Resource Center, Peking Union Medical College (Beijing, China). K562/A02, an ADR-selected multi drug resistance (MDR) cell sub-line, was a gift from the Institute of Hematology, Chinese Academy of Medical Sciences. Cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, 1% kanamycin (100 μg/ml), and 1% glutamine (0.44 μg/ml) in a 5% CO2 incubator at 37℃. In the case of K562/A02 cell culture, ADR (1 μg/ml, final concentration) was added in the medium to maintain the MDR phenotype. The cells were further cultured in ADR-free medium for 2 weeks before experiment.
Cell proliferation assay
Cell proliferation assay was performed using MTT assay as described in our previous reports [18, 19]. Two hundred microliters of cell suspension (2×104 cells/ml) were seeded in 96-well plate, and incubated together with various concentrations of ZSTK474 or Imatinib for 48 h at 37℃. After addition of MTT, the cells were further incubated for 4 h and dissolved in DMSO. The resulting absorbance at 490 nm was measured by microplate reader iMark (BIO-RAD, Hercules, CA, USA).
Flow cytometry analysis
Cell cycle distribution was carried out by flow cytometry analysis as described by us previously [20]. Briefly, 2 ml of cell suspension (5×105 cells/ml) was seeded in 6-well plate. After treated with ZSTK474, cells were harvested, washed with ice-cold PBS and fixed with 70% ethanol overnight. The cell suspension was centrifuged, and the resulting cell pellet was resuspended in 25 μg/ml of PI solution containing 0.5% Triton X-100 and 2% RNase A. The treated cells were incubated for 30 minutes in the dark at 4 °C to be available for cell cycle analysis with flow cytometer BD Accuri C6 (BD Biosciences, San Jose, CA, USA). Data were analyzed by using Modfit software.
Western blot analysis
Western blot analysis was carried out as we reported previously [21, 22]. Total protein and nuclear protein from K562 and K562/A02 cells were prepared using RIPA lysis buffer (Roche Diagnostics, Basel, Switzerland) and NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA), respectively. Protein concentrations were determined with BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of protein were subjected to 10%-12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto PVDF membrane (Millipore, Billerica, MA, USA). The membrane was blocked in 3% non-fat dried milk for 1 h, probed with the specified primary antibodies overnight at 4℃ followed by exposure to the respective anti-mouse or anti-rabbit secondary antibody. The blots were visualized using ECL (enhanced chemiluminescence) reagents and digitalized by scanning.
Quantitative real time-polymerase chain reaction (qRT-PCR)
Total RNA of the cells was extracted using TriZol reagent (Life Technologies, Carlsbad, CA, USA) and quantified by a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative RT-PCR was performed as described by us previously [23]. Briefly, 500 ng of RNA was reverse-transcribed to cDNA by using PrimeScriptTM RT Master Mix Kit (Takara, Tokyo, Japan) in accordance with the manufacturer's protocol. The reaction was conducted in a system of 20 µl volume containing 100 ng cDNA, 200 nM of former primer and reverse primer, and 10 µl of 2× Power SYBR® Premix Ex TaqTM (Takara, Tokyo, Japan), using CFX96TM Real-Time PCR Detection System (BIO-RAD, Hercules, CA, USA). GAPDH was used as a housekeeping gene to normalize RNA expression. Sequences of the PCR primers are: GAPDH: sense primer 5'-GCACCGTCAAGGCTGAGAAC-3', antisense primer 5'-GGTGAAGACGCCAGTGGA-3'; p27: sense primer 5'-TGCAACCGACGATTCTTCTACTCAA-3', antisense primer 5'-CAAGCAGTGATGTATCTGATAAAC-3'. The relative gene expression levels were quantified by using the comparative Ct (ΔΔCt) method.
Synergism assay
Synergism assay was conducted to analyze whether combination of ZSTK474 with Imatinib can achieve an improved therapeutic effect. Cell growth inhibition activities of each single drug and the combination at a fixed ratio (IC50 ZSTK474׃IC50 Imatinib) were tested, respectively. For drug combination experiments, the combination index (CI) was calculated using CalcuSyn software according to the method of Chou and Talalay [24]. CI > 1 indicates antagonism, CI = 1 indicates additivity, CI < 1 indicates synergism. All experiments were carried out in triplicate.
Statistical analysis
Data were presented as means ± standard deviation (SD), representative of at least three independent experiments. Student's t-test was carried out for statistical significance analysis with GraphPad Prism 5 (GraphPad, San Diego, CA, USA). Value of p < 0.05 was regarded as statistically significant.
Results
Anti-proliferative activity of ZSTK474 on K562 and K562/A02 cells
K562 is a chemosensitive cell line, while K562/A02 cell, an ADR-selected MDR cell sub-line, was reported to have MDR phenotype due to the reduced intracellular drug accumulation and wide cross-resistance [25]. In order to confirm this, we exposed K562 and K562/A02 cells to various concentrations of ADR for 48 h, then determined the inhibitory activities of ADR on both cell lines by using MTT assay. As shown in Fig. 1A, ADR showed different potency in inhibition against proliferation of K562 and K562/A02 cells, with the IC50 to be 0.17 μM and 9.88 μM, respectively. K562/A02 exhibited about 50 fold resistance to ADR compared with K562 cell line, suggesting the MDR characteristic of K562/A02.
Anti-proliferative activity of ZSTK474 on K562 and the resistant K562/A02 cells. (A) K562/A02 cells showed resistance to ADR. Cell viability was determined by MTT assay after treatment with various concentration of ADR for 48 h. (B) ZSTK474 inhibited the proliferation of both K562 and K562/A02 cells in a dose dependent manner. The cells were treated with various concentrations of ZSTK474 for 48 h. MTT assay was carried out to measure the cell viability. Data are mean ± SD, representative of three independent experiments.
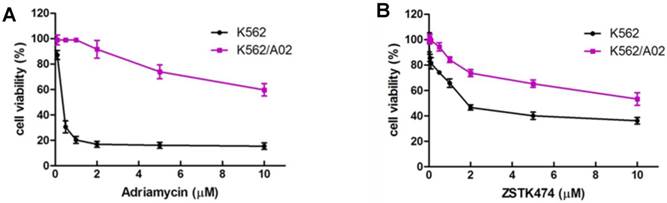
We then investigated the anti-proliferative activity of ZSTK474 on K562 and K562/A02 cells. The cells of both cell lines were treated with 0.01, 0.05, 0.1, 0.5, 1, 2, 5 and 10 μM concentrations of ZSTK474 for 48 h, and the cell viability was analyzed with MTT assay. As shown in Fig. 1B, ZSTK474 reduced cell viability of both cell lines in a dose-dependent manner, with the IC50 to be 4.69 μM for K562 and 7.57 μM for K562/A02. Compared with ADR, ZSTK474 showed more potent inhibition against K562/A02 cell proliferation.
Cell cycle arrest induced by ZSTK474 in K562 and K562/A02 cells
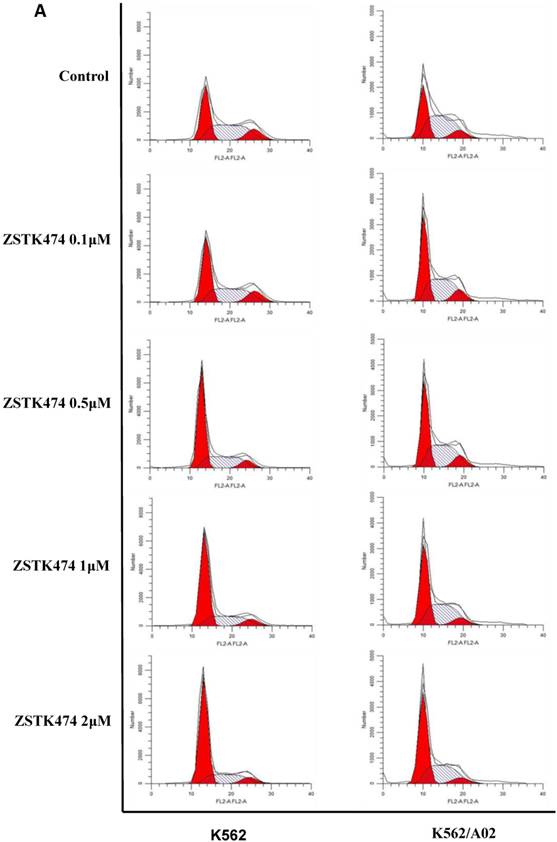
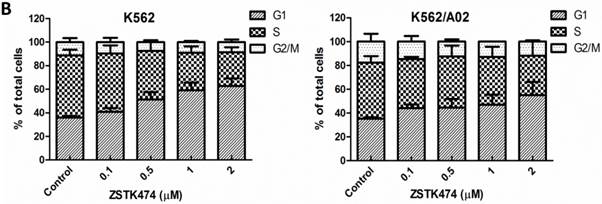
Cell cycle progression is necessary for cell proliferation. To investigate the effect of ZSTK474 on cell cycle, we analyzed the cell cycle distribution in both K562 and K562/A02 cells by flow cytometry. The cells were treated with 0, 0.1, 0.5, 1, 2 μM of ZSTK474 for 48 h, stained with PI, and analyzed by flow cytometer. As a result, ZSTK474 induced G1 arrest in both K562 and K562/A02 cells dose-dependently (Fig. 2A and Fig. 2B).
ZSTK474 induced cell cycle arrest at G1 phase in K562 and K562/A02 cells. (A) Cell cycle distribution analysis by flow cytometer. K562 and K562/A02 cells were incubated with indicated concentrations of ZSTK474 for 48 h. The cells were harvested, stained with PI and analyzed by flow cytometer. Cell cycle distribution was further analyzed by Modfit software. (B) The percentage of total cells at G1, S, and G2/M phases. Data are mean ± SD, representative of three independent experiments.
ZSTK474 affected the cell cycle-related proteins in K562 and K562/A02 cells
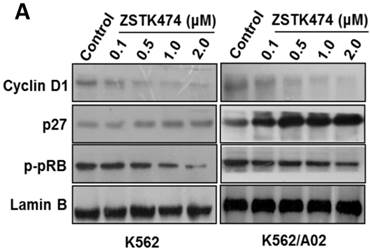
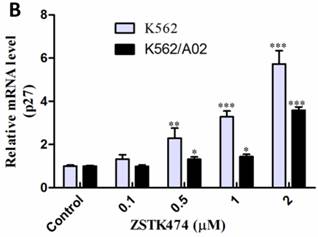
Cell cycle progression is regulated positively by CDK (cyclin-dependent kinases)-cyclins, and negatively by CDK inhibitors including p27. To investigate the mechanism involved in ZSTK474-induced G1 arrest, we examined the effect on cyclin D1, p27, as well as the downstream pRb by Western blot. As shown in Fig. 3A, after treatment with ZSTK474 for 48 h, the expression of p27 increased, while the level of cyclin D1 and phosphorylated pRb decreased, in the nucleus of both K562 and K562/A02 cells, in a dose-dependent manner. The effect of ZSTK474 on p27 expression at mRNA level was also examined by use of qRT-PCR. Fig. 3B showed that the RNA expression levels of p27 in K562 and K562/A02 cells were enhanced obviously after ZSTK474 treatment (p < 0.05, compared with vehicle group). It could be concluded that upregulation of p27, and downregulation of cyclin D1 might be involved in G1 arrest induced by ZSTK474 in K562 and K562/A02 cells.
ZSTK474 affected the cell cycle-related proteins in K562 and K562/A02 cells. (A) Western blot analysis of the cell cycle-related proteins. K562 and K562/A02 cells were treated with indicated concentrations of ZSTK474 for 48 h. The levels of cyclin D1, p27, and p-pRb in nucleus were determined by western blot. (B) qRT-PCR analysis of p27 expression at mRNA level. Total RNA of the cells was extracted using TriZol reagent and quantified by a Nanodrop spectrophotometer. Five hundred ng of RNA was reverse-transcribed to cDNA. The PCR reaction was conducted in a 20 µl system containing 100 ng cDNA, 200 nM of former primer and reverse primer, using CFX96TM Real-Time PCR Detection System. GAPDH was used as a housekeeping gene to normalize RNA expression. The relative gene expression levels were quantified by using the comparative Ct (ΔΔCt) method. Results represent mean ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, compared with control.
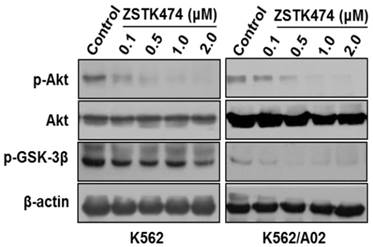
Since ZSTK474 is a specific PI3K inhibitor, and PI3K/Akt pathway is known to play an important role in cell cycle progression, in order to further investigate the mechanism involved in ZSTK474-induced G1 arrest, we examined the effect on the signal proteins downstream of PI3K/Akt pathway. As shown in Fig. 4, ZSTK474 inhibited the phosphorylation of Akt dose-dependently, while total Akt amount was not affected, in both K562 and K562/A02 cells. Furthermore, phosphorylation of GSK-3β was also reduced by ZSTK474 in a dose-dependent manner (Fig. 4). GSK-3β is known as an upstream activator of cyclin D1, and a downstream effector of Akt [26, 27]. P27 was also known to be regulated by Akt [27]. Therefore, our results suggested that PI3K/Akt/GSK-3β/cyclin D1 as well as PI3K/Akt/p27 pathways might be involved in the cell proliferation inhibition and G1 arrest by ZSTK474 in K562 and K562/A02 cells.
ZSTK474 inhibited phosphorylation of Akt and GSK-3β in K562 and K562/A02 cells. K562 and K562/A02 cells were cultured in the presence of various concentrations of ZSTK474 for 48 h. The cells were collected, and the cell lysates were prepared to be available for western blot analysis for p-Akt, Akt, p-GSK-3β, as well as β-actin levels.
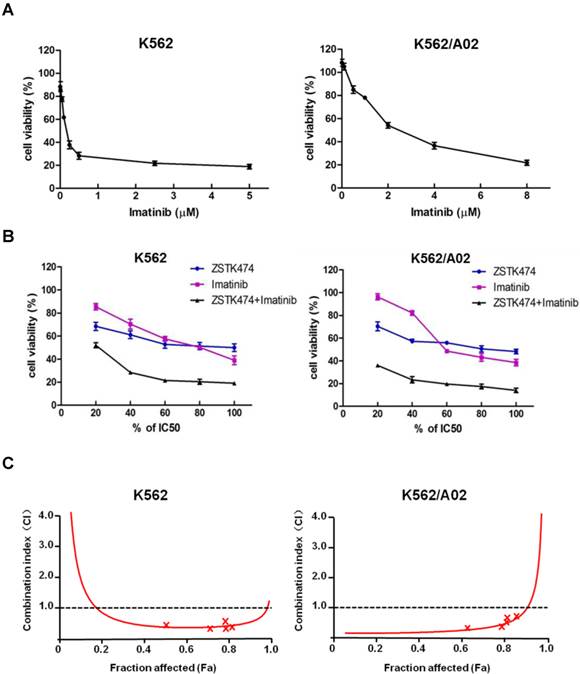
Synergistic effect of ZSTK474 and Imatinib in K562 and K562/A02 cells
While Imatinib is widely used for CML therapy, resistance has been frequently reported [28]. To investigate whether combination of ZSTK474 with Imatinib is able to enhance the antitumor efficacy or not, we determined the synergistic effect of ZSTK474 and Imatinib on K562 and K562/A02 cells. The two cell lines were cultured in the presence of ZSTK474, Imatinib, or both compounds at 37℃ for 48 h. The different effects on cell viability were determined by MTT assay. As shown in Fig. 5A, Imatinib inhibited K562 and K562/A02 cells dose-dependently, with the IC50 values to be 0.20 and 2.87 μM, respectively (Table 1). Then, we chose a fixed ratio of IC50ZSTK474 to IC50Imatinib (23.45׃1), and used 20%, 40%, 60%, 80%, 100% of IC50 value of each drug to investigate the combination effect. As shown in Fig. 5B, combination of ZSTK474 and Imatinib led to a higher inhibition against cell proliferation of both K562 and K562/A02 cells, compared with each drug alone. To analyze the synergistic effect, Chou and Talalay method was used in which drug interactions were depicted as combination index (CI) [24]. Fig. 5C showed that the CI values were less than 1 at all the indicated Fa (Fraction affected) points, suggesting their combination was synergistic. Particularly, the CI values at growth inhibition of 50% (ED50), 75% (ED75) and 90% (ED90) were calculated to be 0.37, 0.39, 0.54 for K562 cells, and 0.08, 0.16, 0.39 for K562/A02 cells, respectively (Table 1). The linear correlation coefficients (r value) of all the curves were >0.95 (Table 1).
Combination of ZSTK474 and Imatinib led to a synergistic anti-proliferative effect on K562 and K562/A02 cells. (A) Imatinib inhibited the proliferation of K562 and K562/A02 cells. K562 and K562/A02 cells were exposed to indicated concentrations of Imatinib. Cell viability was determined by MTT assay. Data are mean ± SD representative of three independent experiments. (B) Combination with ZSTK474 enhanced the inhibitory activity of Imatinib. K562 and K562/A02 cells were incubated with various concentrations of ZSTK474 and Imatinib (20%, 40%, 60%, 80%, 100% IC50 of each drug), alone or in combination. Cell viability was determined by MTT assay. Data are mean ± SD representative of three independent experiments. (C) Analysis of the combinational effect by using CalcuSyn software. CI-Fa plots are shown for K562 cells (left) and K562/A02 (right). The line of CI=1 is shown. CI: combination index, Fa: Fraction affected.
Combination indexes (CI) for ZSTK474 and Imatinib.
| Cell line | Drug | IC50 (μM) | r | CI | ||
|---|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | ||||
| K562 | ZSTK474 | 4.69 | 0.969 | |||
| Imatinib | 0.20 | 0.998 | 0.37 | 0.39 | 0.54 | |
| ZSTK474+Imatinib | 0.78 | 0.971 | ||||
| K562/A02 | ZSTK474 | 7.57 | 0.981 | |||
| Imatinib | 2.87 | 0.969 | 0.08 | 0.16 | 0.39 | |
| ZSTK474+Imatinib | 0.24 | 0.962 | ||||
Discussion
Current therapeutic approaches for CML mainly include chemotherapy with Bcr-Abl targeted drug or cytotoxic agents, and stem cell transplant [29]. Although the efficacy of the first generation Bcr-Abl inhibitor Imatinib is undoubted for CML therapy, resistance remains to be a challenge [30]. Causes of the drug resistance include P-glycoprotein (P-gp) upregulation, Abl kinase domain mutations, and Bcr-Abl overexpression [31]. While new generations of inhibitors targeting Bcr-Abl have been developed and used in clinic [32], it has been reported that blocking Bcr-Abl kinase activity is not sufficient to eliminate leukemogenic events [2, 33]. Since PI3K/Akt pathway is closely related to cell survival and cell cycle progression, PI3K has become a promising target for cancer therapy [34, 35]. Currently, over a dozen of PI3K inhibitors are in clinical trials. However, to our knowledge, there is no report about the antitumor effect of PI3K inhibitors on CML until now.
In the present study, we investigated the antitumor effect of ZSTK474 on K562 CML cells. Besides, K562/A02 cells were also utilized to examine the effect of ZSTK474 on the multidrug resistant cells. K562/A02 cell line was established and maintained by treating with ADR, an agent with wide anti-cancer spectrum. Our result indicated that K562/A02 cells are resistant to Imatinib, compared with the sensitivity of the parent cell K562. In contrast, ZSTK474 showed similar efficacy on K562/A02 cells to that on K562 cells, suggesting that ZSTK474 might overcome the multidrug resistance caused by ADR. Sui H, et al reported that activation of PI3K/Akt/NF-κB pathway promotes P-gp expression, and inhibition of this pathway results in the reversing P-gp mediated multidrug resistance [36]. Therefore, the reversal effect of ZSTK474 on K562/A02 cells might be associated with the downregulation of P-gp.
ZSTK474 induced G1 arrest in both K562 and K562/A02 cells. Meanwhile, treatment with similar concentrations of ZSTK474 led to upregulation of p27, downregulation of cyclin D1, and the decreased phosphorylation of pRb. Since cyclin D1 is a substrate of GSK-3β and p27 is reported to be upregulated by Akt [27], the induction of G1 arrest by ZSTK474 might be associated with the blockade of PI3K/Akt pathway.
Accumulating reports showed that proper drug combination often leads to incredible efficacy compared with the single use of a drug or a drug having dual targets [37, 38]. Our result showed that combination of ZSTK474 and Imatinib was highly synergistic. Interestingly, synergism of these two drugs on the resistant K562/A02 cells seem to be more effective than that on K562 cells. Such effects might be associated with the following mechanisms: 1. ZSTK474 reverses the resistance of K562/A02 cells to ADR and Imatinib by downregulating P-gp expression; 2. ZSTK474 and Imatinib act on different targets for CML therapy: PI3K and Bcr-Abl. The synergic effect of ZSTK474 combination with Imatinib implies the promising application in therapy of CML patients with multidrug resistance.
In summary, our study showed that ZSTK474 exhibited anti-proliferative and G1 arrest effect on both K562 and K562/A02 cells via blocking PI3K/Akt pathway. Combination of ZSTK474 with Imatinib indicated synergistic effect. In addition, previous in vivo experiments [15, 16] and preliminary clinical result [unpublished data] have shown that ZSTK474 is generally safe and quite tolerable. Our report suggests a promising therapeutic approach for CML therapy in the future.
Acknowledgements
K562/A02 cell line is a gift of Professor Dongsheng Xiong, Institute of Hematology, Chinese Academy of Medical Sciences, China. This work was supported by grants from National Natural Science Foundation of China (81373441, 81202542, 81402901), the Natural Science Foundation of Tianjin (12JCZDJC25800, 13JCYBJC24800), China Postdoctoral Science Foundation Funded Project (2014M551035, 2014M551037), and the Tianjin Medical University Research Fund (2013ky07).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holyoake TL, Helgason GV. Do we need more drugs for chronic myeloid leukemia? Immunol Rev. 2015;263:106-23
2. Rangatia J, Bonnet D. Transient or long-term silencing of BCR-ABL alone induces cell cycle and proliferation arrest, apoptosis and differentiation. Leukemia. 2006;20:68-76
3. Maru Y. Molecular biology of chronic myeloid leukemia. Cancer Sci. 2012;103:1601-10
4. Yang K, Fu LW. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit Rev Oncol Hematol. 2015;93:277-92
5. Chereda B, Melo JV. Natural course and biology of CML. Ann of Hematol. 2015;94(Suppl 2):S107-21
6. Mathisen MS, Kantarjian HM, Cortes J, Jabbour EJ. Practical issues surrounding the explosion of tyrosine kinase inhibitors for the management of chronic myeloid leukemia. Blood Rev. 2014;28:179-87
7. O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F. et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. New Engl J Med. 2003;348:994-1004
8. Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C. et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. New Engl J Med. 2002;346:645-52
9. Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A. et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66:11314-22
10. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN. et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science (New York, NY). 2001;293:876-80
11. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7-24
12. Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK. et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151-61
13. Andreu EJ, Lledo E, Poch E, Ivorra C, Albero MP, Martinez-Climent JA. et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005;65:3264-72
14. Kong D, Yamori T. JFCR39, a panel of 39 human cancer cell lines, and its application in the discovery and development of anticancer drugs. Bioorgan Med Chem. 2012;20:1947-51
15. Kong DX, Yamori T. ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor identified using the JFCR39 drug discovery system. Acta Pharmacol Sin. 2010;31:1189-97
16. Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H. et al. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer I. 2006;98:545-56
17. Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99:1734-40
18. Chen X, Tang SA, Lee E, Qiu Y, Wang R, Duan HQ. et al. IVSE, isolated from Inula japonica,suppresses LPS-induced NO production via NF-kappaB and MAPK inactivation in RAW264.7 cells. Life Sci. 2015;124:8-15
19. Wang X, Tang SA, Wang R, Qiu Y, Jin M, Kong D. Inhibitory Effects of JEUD-38, a New Sesquiterpene Lactone from Inula japonica Thunb, on LPS-Induced iNOS Expression in RAW264.7 Cells. Inflammation. 2015;38:941-8
20. Tang SA, Zhou Q, Guo WZ, Qiu Y, Wang R, Jin M. et al. In vitro antitumor activity of stellettin B, a triterpene from marine sponge Jaspis stellifera, on human glioblastoma cancer SF295 cells. Mar drugs. 2014;12:4200-13
21. Kong D, Yamori T, Yamazaki K, Dan S. In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA. Invest New Drug. 2014;32:1134-43
22. Zhao W, Guo W, Zhou Q, Ma SN, Wang R, Qiu Y. et al. In vitro antimetastatic effect of phosphatidylinositol 3-kinase inhibitor ZSTK474 on prostate cancer PC3 cells. Int J Mol Sci. 2013;14:13577-91
23. Aoki S, Kong D, Suna H, Sowa Y, Sakai T, Setiawan A. et al. Aaptamine, a spongean alkaloid, activates p21 promoter in a p53-independent manner. Biochem Bioph Res Co. 2006;342:101-6
24. Tentori L, Ricci-Vitiani L, Muzi A, Ciccarone F, Pelacchi F, Calabrese R. et al. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC cancer. 2014;14:151
25. Peng H, Yuan X, Shi R, Wei X, Ren S, Yan C. et al. PHII-7 inhibits cell growth and induces apoptosis in leukemia cell line K562 as well as its MDR- counterpart K562/A02 through producing reactive oxygen species. Eur J Pharmacol. 2013;718:459-68
26. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7
27. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL. et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590-603
28. Alvarado Y, Kantarjian H, O'Brien S, Faderl S, Borthakur G, Burger J. et al. Significance of suboptimal response to imatinib, as defined by the European LeukemiaNet, in the long-term outcome of patients with early chronic myeloid leukemia in chronic phase. Cancer. 2009;115:3709-18
29. Kimura S, Ando T, Kojima K. Ever-advancing chronic myeloid leukemia treatment. Int J Clin Oncol. 2014;19:3-9
30. Hochhaus A, O'Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L. et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054-61
31. Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM. et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070-9
32. Goldman JM, Marin D. Is imatinib still an acceptable first-line treatment for CML in chronic phase? Oncology. 2012;26:901-7
33. Bueno-da-Silva AE, Brumatti G, Russo FO, Green DR, Amarante-Mendes GP. Bcr-Abl-mediated resistance to apoptosis is independent of constant tyrosine-kinase activity. Cell Death Differ. 2003;10:592-8
34. Park SJ, Jin ML, An HK, Kim KS, Ko MJ, Kim CM. et al. Emodin induces neurite outgrowth through PI3K/Akt/GSK-3beta-mediated signaling pathways in Neuro2a cells. Neurosci Lett. 2015;588:101-7
35. Son YO, Wang L, Poyil P, Budhraja A, Hitron JA, Zhang Z. et al. Cadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3beta/beta-catenin signaling. Toxicol Appl Pharm. 2012;264:153-60
36. Sui H, Pan SF, Feng Y, Jin BH, Liu X, Zhou LH. et al. Zuo Jin Wan reverses P-gp-mediated drug-resistance by inhibiting activation of the PI3K/Akt/NF-kB pathway. BMC Complement Altern Med. 2014;14:279
37. Prahallad A, Bernards R. Opportunities and challenges provided by crosstalk between signalling pathways in cancer. Oncogene. 2015 published online first: Doi: 10.1038/onc.2015.151
38. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205-14
Author contact
![]() Corresponding authors: Dexin Kong, Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China, e-mail: kongdexinedu.cn; Tel: 86-22-83336659; Yuling Qiu, Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China, e-mail: qiuyulingedu.cn; Tel: 86-22-83336673.
Corresponding authors: Dexin Kong, Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China, e-mail: kongdexinedu.cn; Tel: 86-22-83336659; Yuling Qiu, Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China, e-mail: qiuyulingedu.cn; Tel: 86-22-83336673.