Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(6):639-652. doi:10.7150/ijbs.14025 This issue Cite
Research Paper
Dimethyloxaloylglycine Promotes the Angiogenic Activity of Mesenchymal Stem Cells Derived from iPSCs via Activation of the PI3K/Akt Pathway for Bone Regeneration
Jieyuan Zhang 1,*, Junjie Guan 1,*, Xin Qi 1,*, Hao Ding 1, Hong Yuan 2, Zongping Xie1, Chunyuan Chen 3, Xiaolin Li1 ![]() , Changqing Zhang 1
, Changqing Zhang 1 ![]() , Yigang Huang 1
, Yigang Huang 1 ![]()
1. Department of Orthopedics, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China;
2. Department of Dermatology, Zhongshan Hospital, Fudan University, Shanghai 200032, China;
3. Graduate School of Nanchang University, Nanchang, Jiangxi 330031, China.
* These authors contributed equally to this work.
Received 2015-10-1; Accepted 2016-2-27; Published 2016-4-8
Abstract

The vascularization of tissue-engineered bone is a prerequisite step for the successful repair of bone defects. Hypoxia inducible factor-1α (HIF-1α) plays an essential role in angiogenesis-osteogenesis coupling during bone regeneration and can activate the expression of angiogenic factors in mesenchymal stem cells (MSCs). Dimethyloxaloylglycine (DMOG) is an angiogenic small molecule that can inhibit prolyl hydroxylase (PHD) enzymes and thus regulate the stability of HIF-1α in cells at normal oxygen tension. Human induced pluripotent stem cell-derived MSCs (hiPSC-MSCs) are promising alternatives for stem cell therapy. In this study, we evaluated the effect of DMOG on promoting hiPSC-MSCs angiogenesis in tissue-engineered bone and simultaneously explored the underlying mechanisms in vitro. The effectiveness of DMOG in improving the expression of HIF-1α and its downstream angiogenic genes in hiPSC-MSCs demonstrated that DMOG significantly enhanced the gene and protein expression profiles of angiogenic-related factors in hiPSC-MSCs by sustaining the expression of HIF-1α. Further analysis showed that DMOG-stimulated hiPSC-MSCs angiogenesis was associated with the phosphorylation of protein kinase B (Akt) and with an increase in VEGF production. The effects could be blocked by the addition of the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002. In a critical-sized calvarial defect model in rats, DMOG-treated hiPSC-MSCs showed markedly improved angiogenic capacity in the tissue-engineered bone, leading to bone regeneration. Collectively, the results indicate that DMOG, via activation of the PI3K/Akt pathway, promotes the angiogenesis of hiPSC-MSCs in tissue-engineered bone for bone defect repair and that DMOG-treated hiPSC-MSCs can be exploited as a potential therapeutic tool in bone regeneration.
Keywords: Dimethyloxaloylglycine, Angiogenic activity, Induced pluripotent stem cells, PI3K/Akt, Bone regeneration.
Introduction
Critical-sized bone defects, which are caused by such factors as major trauma, tumor resection, and congenital defects, cannot be repaired without outside intervention [1]. Autologous or allogeneic bone grafting is the most commonly used surgical procedure to promote bone healing, but the application of this technique is hampered by their limited supply, risk of rejection and high cost [2]. Bone tissue engineering has emerged as a therapeutic strategy for repairing bone defects. Angiogenesis is a prerequisite step in bone healing, and the restoration of blood flow to bone defects provides a source of growth factors and nutrients [3]. Therefore, the promotion of angiogenesis in tissue-engineered bone is an interesting focus of research. Hypoxia inducible factor-1α (HIF-1α) is the master regulator of oxygen homeostasis in metazoans, and is expressed in almost all cell types [4, 5]. HIF-1α can activate the transcription of hundreds of downstream genes, including vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), stromal cell-derived factor-1 (SDF-1), basic fibroblast growth factor (bFGF), angiopoietin-1 (Ang-1), platelet-derived growth factor-BB (PDGF-BB), placental growth factor (PLGF), and stem cell factor (SCF) [6, 7], factors that are involved in neoangiogenesis. Among these factors, VEGF plays an important role in neovascularization and angiogenesis during the development of most tissues including bone [8, 9]. We then recognized that HIF-1α has more advantages for promoting angiogenesis compared with its downstream single gene. However, the most challenge is that, prolyl hydroxylase (PHD) is activated in normoxic states, resulting in the von Hippel-Lindau (VHL) protein binding to HIF-1α and thereby making HIF-1α subject to degradation.
Dimethyloxalylglycine (DMOG) is a cell penetrant oxoglutarate analogue that can inhibit PHD enzymes and thereby regulate the stability of HIF-1α in cells at normal oxygen tension [10] and induces downstream gene expression. Hence, DMOG is expected to be an alternative source for enhancing HIF-1α expression in cells for the treatment of ischemic diseases. Previous studies have demonstrated that DMOG can boost the expression of VEGF in MSCs and attenuate ischemic disease injury [11-14]. Our Recent studies have also showed that DMOG can increase the angiogenic activity of bone marrow stromal cells (BMSCs) and adipose derived stem cells (ADSCs) via promoting the expression of HIF-1α and its downstream genes in cells to improve the angiogenesis of tissue-engineered bone [15, 16]. In stem cell transplantation therapy, MSCs and embryonic stem cells (ESCs) have been the most commonly studied [17, 18]. Nevertheless, the realistic use of MSCs and ESCs is hampered by their invasive harvesting procedures and ethical and safety hurdles, respectively. Induced pluripotent stem cells (iPSCs) are similar to ESCs in terms of morphology, self-renewal, and differentiation capacity [19-21], and can be generated from any tissue type in the body. iPSCs show an unlimited growth capacity and are not associated with any ethical concerns, making them superior to traditional MSCs and ESCs; thus, these cells can serve as an inexhaustible source for bone tissue engineering. But iPSCs are potentially tumorigenicity [22], to overcome the obstacle, iPSC-MSCs have been explored [23-25], they incorporate the advantages of both MSCs and iPSCs. Specifically, a high yield of MSCs can be generated from iPSCs, and these iPSC-MSCs can proliferate for more than 120 population doublings, sustaining their self-renewal capacity and MSC characteristics [26], but these are also no longer tumorigenic [23, 27]. Patient-specific iPSC-MSCs can be used for autologous transplantation without immunological rejection. iPSC-MSCs have been previously used in tissue repair [26, 28], including bone regeneration [29, 30]. However, to date, there are no reports regarding the application of DMOG treated human iPSC-MSCs (DMOG-hiPSC-MSCs) in bone regeneration.
Porous β-tricalcium phosphate (β-TCP) is a synthetic and biodegradable ceramic material that has been widely used in bone defect repair [31]; the β-TCP scaffolds were used as cells carriers in our studies. In a critical-sized rat calvarial defect model, our findings provide the first demonstration that DMOG-hiPSC-MSCs can clearly promote angiogenesis and bone regeneration in vivo. The underlying mechanisms were analyzed in vitro. VEGF, a lead pro-angiogenesis factor, acts directly on endothelial cells to induce proliferation, migration, survival, and finally angiogenesis [32], and its expression is mediated by the PI3K/Akt signaling pathway [33-35]. Previous studies have also demonstrated that activation of PI3K/Akt signaling mediates VEGF protein expression at the transcriptional level through HIF-1α expression [36]. As indicated by our previous studies, DMOG enhances the angiogenic activity of MSCs through activating the expression of HIF-1α [15, 16]. DMOG can also promote Akt phosphorylation in MSCs [37]. We thus hypothesized that DMOG may exert a positive effect to stimulate angiogenic activity of hiPSC-MSCs through activation of the PI3K/Akt pathway.
The purpose of this study was to investigate the angiogenic activity of DMOG-hiPSC-MSCs in bone regeneration, as well as the underlying mechanisms. In conclusion, our results reveal that DMOG, via activation of the PI3K/Akt pathway, promotes the angiogenic activity of hiPSC-MSCs. DMOG-hiPSC-MSCs thus can be exploited as a potential therapeutic tool in bone regeneration.
Materials and methods
Isolation and identification of hiPSC-MSCs
Human iPSC line (iPS-S-01) was provided by the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences [38]. The use of human iPSCs was approved by the Ethics Committee of Shanghai Sixth People's Hospital. After five days of culture, mTESR1 (StemCell Technologies, Canada) was replaced with MSC medium, which was prepared by supplementing Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA) with 10% FBS (Gibco, USA), 1% penicillin/streptomycin (Gibco, USA), 2 mM L-glutamine (Gibco, USA), and 0.1 mM non-essential amino acids (Gibco, USA), and the medium was changed every two days [25]. After two weeks of culture in MSC medium, the cells were harvested and expanded in 0.1% gelatin-coated dishes with MSC medium. The cells were continually passaged every five to seven days until they developed a homogeneous fibroblastic morphology. The cells were then evaluated in terms of MSC phenotype characteristics and differentiation potentials.
Surface antigens of hiPSC-MSCs were analyzed by flow cytometry. Cells at Passage 4 were blocked with 3% bovine serum albumin (BSA, Gibco, USA) for 30 min and incubated with the following primary antibodies according to the manufacturer's instructions: CD34, CD45, CD73, CD90, CD105, HLA-DR. Surface antigens were analyzed using a Guava easyCyteTM flow cytometer.
The functional differentiation of hiPSC-MSCs into osteogenic, chondrogenic, and adipogenic lineages was tested in osteogenic, chondrogenic, and adipogenic media, respectively. After incubating in specific culture media for three, four, and two weeks; hiPSC-MSCs were fixed and then detected by Alizarin Red, Toluidine Blue, and Oil Red O staining respectively.
The gene expression levels of markers associated tri-lineage differentiation were confirmed by qRT-PCR. hiPSC-MSCs were cultured in specific culture media for 7day, then he total RNA was isolated using Trizol (Invitrogen, USA), and 1μg of RNA in a final reaction volume of 20 μl was then reversed-transcribed into complementary DNA (cDNA) using the PrimeScript 1st Strand cDNA Synthesis kit (TaKaRa, Japan) according to the manufacturer's instructions. qRT-PCR was performed using SYBR Premix Ex Taq (Takara, Japan) in combination with an ABI 7500 Real-Time PCR System (Applied Biosystems, USA).Threshold cycles of primer probes were normalized to the housekeeping gene GAPDH and translated to relative values. The primers are synthesized as follows: OCN: forward, 5'-CCCCCTCTAGCCTAGGACC-3', and reverse, 5'-ACCAGGTAATGCCAGTTTGC-3'; ALP: forward, 5'-ACCACCACGAGAGTGAACCA-3', and reverse, 5'-CGTTGTCTGAGTACCAGTCCC-3'; Sox 9: forward, 5'-AGCGCCCCCACTTTTGCTCT-3', and reverse, 5'-GCTCGCCCTTGGGGAACGTG-3'; AGC: forward, 5'-TGCGGGTCAACAGTGCCTATC-3', and reverse, 5'-CACGATGCCTTTCACCACGAC-3'; LPL: forward, 5'-TGGAGGTACTTTTCAGCCAGGAT-3', and reverse, 5'-CGTGGGAGCACTTCACTAGCT-3'; PPARγ: forward, 5'-CACAAGAACAGATCCAGTGGTTGCAG-3', and reverse, 5'-AATAATAAGGTGGAGATGCAGG CTCC-3'; GAPDH: forward, 5'-ATCCCATCACCATCTTCC-3', and reverse, 5'-GAGTCCTTCCACGATACCA-3'. Each qRT-PCR was performed in triplicate for yield validation.
Effects of DMOG on hiPSC-MSCs proliferation and survival
To investigate the influence of DMOG on hiPSC-MSCs proliferation, hiPSC-MSCs were seeded on plates and then treated with or without DMOG (1000 μM; Sigma, USA) for 24, 48, and 72 h. The DMOG concentration was selected based on our previous studies [15, 16]. hiPSC-MSCs proliferation was measured using Cell Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology, China) and a Microplate Reader (450 nm; Bio-Rad, CA). Live/Dead cell staining was performed to assess the hiPSC-MSCs death ratio, a possible indication of the toxicity of DMOG. Briefly, Live/Dead cell staining solution (ScienCell, USA) was added to the medium, then incubation for thirty minutes. Images in six different randomly chosen fields per well were taken under the microscope. hiPSC-MSCs death was calculated by the ratio of dead cells to total cells. The influence of DMOG on hiPSC-MSCs survival in serum deprivation conditions was also evaluated using Live/Dead cell staining. hiPSC-MSCs were cultured in regular medium for 12 h. Subsequently, cells were washed with phosphatebuffered saline (PBS) and cultured in serum-free medium with 1000 μM DMOG for another 24, 48, and 72 h, before Live/Dead cell staining. Specifically, human bone marrow mesenchymal stem cells (hBMSCs) were used as the control to compare with hiPSC-MSCs in proliferation and survival evaluation. The use of hBMSCs was approved by the Ethics Committee of Shanghai Sixth People's Hospital. hBMSCs were harvested after obtaining informed consent from donors. hBMSCs were cultured in DMEM containing 10% FBS at 37°C in a humidified 5% CO2 incubator. The culture medium was replaced every 3 days with non-adherent cells removed. When 90% confluence was reached, cells were released from the culture substratum using 0.25% trypsin (Invitrogen, USA), and were then passaged at a 1:3 split. Cells of four to six passages were used for the following experiments. All of these experiments were performed in triplicate and repeated at least three times.
Effects of DMOG on angiogenic activity of hiPSC-MSCs
The qRT-PCR was carried out to detect the expression of angiogenic-related genes in hiPSC-MSCs under normal oxygen conditions. Briefly, after DMOG treated or untreated hiPSC-MSCs were cultured for 3 and 7 days, the total RNA was isolated using Trizol (Invitrogen, USA), and 1μg of RNA in a final reaction volume of 20 μl was then reversed-transcribed into complementary DNA (cDNA) using PrimeScript 1st Strand cDNA Synthesis kit (TaKaRa, Japan) according to the manufacturer's instructions. qRT-PCR was performed using SYBR Premix Ex Taq (Takara, Japan) in combination with ABI 7500 Real-Time PCR System (Applied Biosystems, USA).Threshold cycles of primer probes were normalized to the housekeeping gene GAPDH and translated to relative values. The primers are synthesized as follows: HIF-1α: forward, 5'-CCATGTGACCATGAGGAAAT-3', and reverse, 5'- CGGCTAGTTAGGGTACACTT-3'; VEGF: forward, 5'-CGCTCGGTGCTGGAATTTGA-3', and reverse, 5'-AGTGGGGAATGGCAAGCAAA -3'; SDF1: forward, 5'-ATGAACGCCAAGGTCGTGGTC-3', and reverse, 5'-CTTGTTTAAAGCTTTCTCCAGGTACT-3'; bFGF: forward, 5'-CAATTCCCATGTGCTGTGAC-3', and reverse, 5'-ACCTTGACCTCTCAGCCTCA-3'; PLGF: forward, 5'-CAGAGGTGGAAGTGGTACCCTTCC-3', and reverse, 5'-CGGATCTTTAGGAGCTGCATGGTGAC-3'; GAPDH: forward, 5'-ATCCCATCACCATCTTCC -3', and reverse, 5'-GAGTCCTTCCACGATACCA-3'. Each qRT-PCR was performed in triplicate for yield validation.
Western blot analysis
hiPSC-MSCs cultured with or without 1000 μM DMOG under normal oxygen conditions were analyzed by western blotting. The total cellular proteins were first extracted. Briefly, the cells were lysed in ice-cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 0.1% SDS) containing 1 mM phenylmethylsulphonyl fluoride (PMSF) (Sigma, USA) and phosphatase inhibitor cocktail (Sigma, USA). The cell lysates were cleared by centrifugation at 4°C and 12,000 rpm for 15 min. The protein concentrations of the lysates were quantified using a BCA assay Kit. The cell proteins were separated by standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After incubation in 5% BSA blocking solution for 1 h, the membranes were incubated overnight at 4°C with anti-HIF-1α, anti-VEGF, anti-p-Akt, anti-Akt, and anti-β-actin antibodies (all from Abcam, UK). The signal inhibitor used for PI3K/Akt was LY294002 (20 μM; Sigma, USA). The membranes were then washed three times with PBS-Tween-20 and incubated with horseradish peroxidase (HPR)-conjugated secondary antibodies (Sigma, USA) at 37°C. The immunoreactive bands were visualized using the ECL chemiluminescence reagent (Millipore, USA).
Effects of DMOG on angiogenesis of endothelial cell
hiPSC-MSCs were cultured in DMEM with or without 1000 μM DMOG for three days, the medium was removed and added fresh DMEM without DMOG cultured for another three days, then the medium was collected as conditioned medium (CM). The receptors (VEGFR2, CXCR4, bFGFR, VEGFR1) expressions of VEGF, SDF1, bFGF, and PLGF in endothelial cells after treatment with hiPSC-MSCs CM or DMOG-hiPSC-MSCs CM for 24 h were also detected by qRT-PCR as mentioned above. The primers are synthesized as follows: VEGFR2: forward, 5'-GTGATCGGAAATGACACTGGAG-3', and reverse, 5'-CATGTTGGTCACTAACAGAAGCA-3'; CXCR4: forward, 5'-GGTGGTCTATGTTGGCGTCT-3', and reverse, 5'-TGGAGTGTGACAGCTTGGAG-3'; bFGFR: forward, 5'-GACGGCTCCTACCTCAA-3', and reverse, 5'-GCTGTAGCCCATGGTGTTG-3'; VEGFR1: forward, 5'- TTTAAAAGGCACCCAGCACAT -3', and reverse, 5'-TTACTCACCATTTCAGGCAAAGAC-3'; GAPDH: forward, 5'-ATCCCATCACCATCTTCC -3', and reverse, 5'-GAGTCCTTCCACGATACCA-3'. Each qRT-PCR was performed in triplicate for yield validation.
The use of human umbilical vein endothelial cells (HUVECs) was approved by the Ethics Committee of Shanghai Sixth People's Hospital. HUVECs were harvested after obtaining informed consent from donors. Culture plates (24-well) were pre-coated with Matrigel, and HUVECs (5×104 cells per well) were seeded in hiPSC-MSCs CM or DMOG-hiPSC-MSCs CM and cultured for 6 h. Separate wells of control cells were grown in DMEM alone. To confirm the role of the PI3K/Akt signaling pathway in angiogenesis, hiPSC-MSCs were cultured with inhibitor LY294002 for 24 h prior to DMOG and LY294002 addition, then the CM was collected after 3 days of culture in fresh medium and used for tube formation assay. Total branching points and total tubule lengths in 5 randomly chosen fields were quantified.
Effects of DMOG on hiPSC-MSCs attachment
To investigate the influence of DMOG on hiPSC-MSCs attachment on β-TCP scaffolds, each scaffold was placed in a well of a 24-well plate and immersed in normal medium for 1 day before cell seeding. Then 1.0 × 105 hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs were seeded on scaffolds. After 12 and 24 h, the scaffolds were washed with PBS to remove the non-adhesion cells, then the cells in medium were calculated using countess automated cell counter (Invitrogen, USA). The morphological characteristics of the attached cells were observed by scanning electron microscope (SEM; FEI Quanta 450).
Animal experiments
All of the animal procedures were approved by the Animal Research Committee of Sixth People's Hospital, Shanghai Jiao Tong University. Classical porous β-TCP scaffolds (5 mm in diameter and 2 mm in depth) with an average pore size of 500 μm and 75% porosity were used as cell carriers in in vivo studies. hiPSC-MSCs were treated with 1000 μM DMOG (Gibco, USA) for 72 h. The cells were then seeded at a density of 1.0 × 105 cells/ml onto each β-TCP under sterile conditions and incubated for at least 4 h to ensure total absorption. To avoid the potential immune rejection of xenogenic hiPSC-MSCs, cyclosporin A (10mg/kg of weight; Novartis, Switzerland) was intraperitoneally injected into adult male Sprague Dawley (SD) rats three days before the transplantation, which was continued until the end of the study. No signs of infections were identified during the course of the experiment. Adult male SD rats weighing 250-300 g were anesthetized. Two 5-mm-diameter critical-sized calvarial defects were created on each side of the cranium using dental trephine. The scaffolds were then implanted into the defects. Thirty-six rats were randomly divided into four groups to receive the following implants: (1) β-TCP (n=9), (2) β-TCP with hiPSC-MSCs (n=9), (3) β-TCP with DMOG-hiPSC-MSCs (n=9), and (4) β-TCP with DMOG-hiPSC-MSCs+LY294002 (n=9). Eight weeks after surgery, six rats per group were sacrificed and their craniums were harvested. The craniums were first analyzed by micro-CT, and six craniums per groups were divided into two parts: one for non-decalcified histological analysis and the other for decalcified immunohistochemical analysis. To compare blood vessel formation in vivo, three eight-week rats per groups were perfused with Microfil (Flow Tech, USA). In brief, all of the rats were anesthetized for rib cage opening. The descending aorta was clamped, and an angiocatheter was used to penetrate the left ventricle. After the inferior vena cava was incised, heparinized saline was perfused until the venous effluent was free of blood. Then 20 mL of Microfil was perfused at 2 mL/min. Finally, the rats were maintained overnight at 4°C to ensure polymerization of the contrast agent.
Sequential fluorescent labeling
Two, four and six weeks after the operation, the rats were intraperitoneally injected with tetracycline (25 mg/kg of body weight; Sigma, USA), alizarin red (30 mg/kg of body weight; Sigma, USA) and calcein (20 mg/kg of body weight, Sigma, USA). The trichromatic sequential fluorescent labeling method was then used to observe the mineralized tissue.
Micro-CT analysis
Eight weeks after the operation, six rats per group were euthanized. The craniums were fixed in 10% neutral buffered formalin solution for at least 48 h. The undecalcified samples and decalcified samples perfused with Microfil of the craniums were scanned using micro-CT (Skyscan, Belgium) at 18-μm resolution and 9-μm resolution, respectively. The threshold of 800 was defined as bone tissue. Three-dimensional grayscale images were generated using auxiliary software. Percentages of new bone volume relative to tissue volume (BV/TV) in the bone defects were calculated.
Histological and immunohistochemical analysis
One part of the fixed craniums was dehydrated through a graded alcohol series and then embedded in polymethylmethacrylate (PMMA). After hardening, the sagittal sections of the central segment were cut into 150- to 200-μm-thick slices using a microtome (Leica, Germany), then glued onto a plastic support and polished to a final thickness of approximately 40 μm. First, a confocal laser scanning microscope (CLSM) (Leica, Germany) was used to observe the fluorescent labeling of the sections using excitation/emission wavelengths for chelating fluorochromes of 405/580 nm (tetracycline, yellow), 543/617 nm (alizarin red, red), and 488/517 nm (calcein, green), respectively. The sections were then stained with van Gieson's picrofuchsin to assess new bone formation (the red and black colors indicated new bone and β-TCP, respectively). The other part of the craniums was decalcified for two to three weeks in 10% EDTA solution, dehydrated with gradient alcohols, embedded in paraffin, and sectioned into 4-mm-thick sections. The CD31 (ab64543, 1:200; Abcam, UK), VEGF (ab46154, 1:100; Abcam, UK) and HIF-1α (ab463, 1:200; Abcam, UK) were used for the detection of newly formed vessels in new bone formation.
Statistical analysis
All of the data are shown as the mean ± standard deviation (SD). Differences between groups were assessed by one-way analysis of variance (ANOVA) using the GraphPad Prism software; p values < 0.05 were considered statistically significant.
Results
Characterization of hiPSC-MSCs
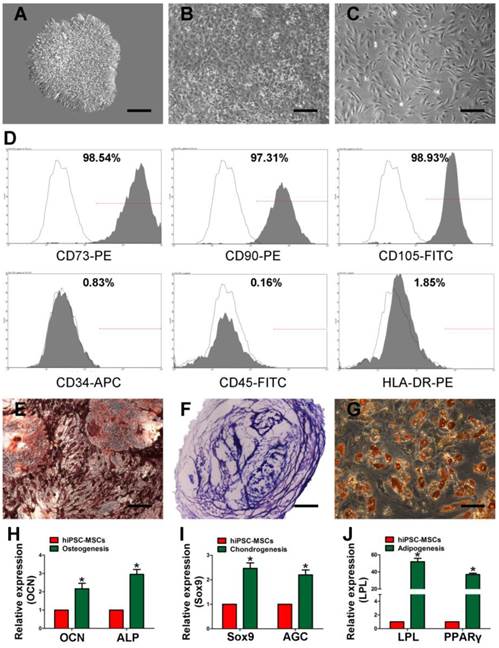
Using a modified one-step induction protocol [25], almost 100% human iPS cells successfully differentiated into hiPSC-MSCs. Under the induction conditions, hiPSCs showed a tendency to form packed clones with decreased nuclear-to-cytoplasmic volume ratios and formed a monolayer with a larger spindle-shaped morphology at the border of the colonies after culture in MSC medium for a few days. After culturing for 14 days, the cells were continually passaged until homogeneous fibroblastic morphologies were observed (Figure 1A-C). The differentiation of hiPSCs into MSCs was evaluated by flow cytometry. MSCs were identified as cells positive for CD73, CD90, and CD105 and negative for CD34, CD45, and HLA-DR (Figure 1D). Tri-lineage MSC differentiation experiments were performed to assess the multipotency of the derived cells. The cells showed the potential of osteogenic, chondrogenic, and adipogenic (Figure 1E-G). The osteo-, chondro-, and adipogenic differentiation-related genes analysis demonstrated that the gene expression of OCN and ALP (Figure 1H), Sox9 and AGC (Figure 1I), LPL and PPARγ (Figure 1J) were upregulated in induced iPSC-MSCs, respectively. These results suggest that the derived hiPSC-MSCs possessed MSC properties and multipotency.
Characterization of human induced pluripotent stem cell-derived mesenchymal stem cells (hiPSC-MSCs). Light microscopy images demonstrating that morphological changes that occur during hiPSCs differentiation into fibroblast-like cells. (A) Representative cell morphology of hiPSCs before differentiation. (B) Intermediate phase of the differentiating the hiPSCs into MSCs. (C) Typical fibroblast-like morphology of the cells. (D) Flow cytometric analysis of the surface markers in hiPSC-MSCs. Assessment of the tri-lineage differentiation capacity of iPSC-MSC-like cells. (E) Alizarin Red staining for bone matrix after 3 weeks in culture with osteogenic medium. (F) Toluidine blue staining for proteoglycans after 4 weeks in culture with chondrogenic medium. (G) Oil Red O staining for adipocytes after 2 weeks in culture with adipogenic medium. The qRT-PCR results for OCN and ALP (H), Sox9 and AGC (I), LPL and PPARγ (J) after 7 days in culture with osteo-, chondro-, and adipogenic mediun. Scale bar: 500 μm.
DMOG suppresses hiPSC-MSCs proliferation and enhances hiPSC-MSCs survival
The influence of DMOG on hiPSC-MSCs proliferation was measured with the CCK-8. It showed that hiPSC-MSCs had higher proliferative ability than hBMSCs at 24, 48, and 72 h. Meanwhile, hiPSC-MSCs proliferation was significantly suppressed after 48 and 72 h of incubation with DMOG (Figure 2A). Cell death was detected using Live/Dead Cell Staining. There were no significant differences in the death ratio of hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs (Figure 2B), which indicated 1000 μM DMOG had no obvious toxicity in hiPSC-MSCs. The effects of DMOG on serum-deprivation-induced cell death was also determined. DMOG can reduce hiPSC-MSCs death in serum deprivation conditions, which indicated that DMOG enhanced cell survival during cell stress (Figure 2C).
Effects of DMOG on the proliferation, survival and angiogenic-related gene and protein expression of hiPSC-MSCs. (A) Effects of DMOG on the proliferation of hiPSC-MSCs was determined using CCK-8 after 24, 48, and 72 h. Effects of DMPG on the death ratio of hiPSC-MSCs in normal condition (B) or in serum deprivation condition (C) was determined using Live/Dead cell staining after 24, 48, and 72 h. (*p < 0.05 compared with the hiPSC-MSCs group, #p < 0.05 compared with the hBMSCs group). (D) Effects of DMOG on the angiogenic-related genes (HIF-1α, VEGF, SDF1, bFGF, and PLGF) or angiogenic-unrelated genes (Sox9) expression in hiPSC-MSCs after 3 and 7 days were detected using qRT-PCR (*p < 0.05). (E) Effects of DMOG on the protein expression of HIF-1α and VEGF in hiPSC-MSCs after 12, 24, and 72 h were detected using western blotting.
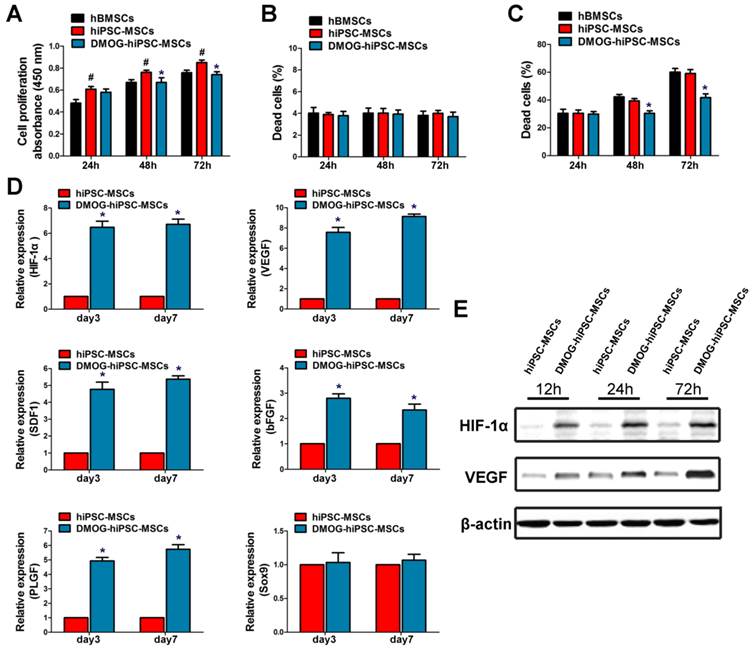
DMOG enhances mRNA expression of angiogenic factors in hiPSC-MSCs
The mRNA levels of angiogenic-related genes in DMOG-hiPSC-MSCs were detected in vitro by qRT-PCR. The expression of HIF-1α, VEGF, SDF1, bFGF and PLGF were all markedly increased in the DMOG-hiPSC-MSCs group compared with that in hiPSC-MSCs group (Figure 2D). The mRNA expression of these genes was then maintained at a high level from day 3 to day 7. Notably, expression of angiogenic-unrelated gene in hiPSC-MSCs, such as Sox9 did not change with treatment, which indicated that DMOG may specifically enhanced hiPSC-MSCs angiogenesis at normal oxygen tension.
DMOG promotes expression of HIF-1α and VEGF in hiPSC-MSCs
After the treatment with 1000 μM DMOG for 12, 24 and 72 h, the expression of HIF-1a and VEGF in hiPSC-MSCs was detected by western blotting. The results showed that the level of HIF-1α protein was enhanced in response to DMOG treatment. The expression of HIF-1α in hiPSC-MSCs increased to stable levels after treatment with DMOG for 24 h, and the expression levels of HIF-1α exhibited no significant difference after 24 h and 72 h (Figure 2E). The level of VEGF protein expression was also enhanced after DMOG treatment with increasing treatment duration (Figure 2E).
PI3K/Akt signaling pathway are involved in angiogenic activity of hiPSC-MSCs by DMOG
The qRT-PCR and western blot results demonstrated that DMOG can promote angiogenic activity of hiPSC-MSCs, we are interested in whether the PI3K/Akt signaling pathway is involved in the process. As shown in Figure 3A, the protein levels of p-AKT, HIF-1α, and VEGF were significantly increased after incubation with 1000 μM DMOG for 12 h. To confirm whether the activation of the PI3K/Akt signaling pathway is required for enhanced angiogenic activity, inhibitor LY294002 was used to inhibit the PI3K/Akt signaling pathway in hiPSC-MSCs prior to treatment with DMOG. Then the protein levels of p-AKT, HIF-1α, and VEGF were markedly suppressed.
Activation of the PI3k/Akt pathway in the angiogenic activity of the DMOG-hiPSC-MSCs group and the effects of conditioned medium (CM) on tube formation by HUVECs. (A) Expression profile of key proteins in the PI3k/Akt pathway in the hiPSC-MSCs and DMOG-treated hiPSC-MSCs groups at 12 h; the hiPSC-MSCs and DMOG-treated hiPSC-MSCs groups were cultured with or without LY294002. (B) Effects of DMOG on the receptors (VEGFR2, CXCR4, bFGFR, VEGFR1) expressions in HUVECs after 24 h were detected using qRT-PCR. (C) HUVECs tube formation was studied by growing cells in Matrigel in DMEM, hiPSC-MSCs CM, or DMOG-treated hiPSC-MSCs CM for 6 h, and quantitative analysis by (D) total branching points and (E) tube lengths. Scale bar: 250μm. (*p < 0.05)
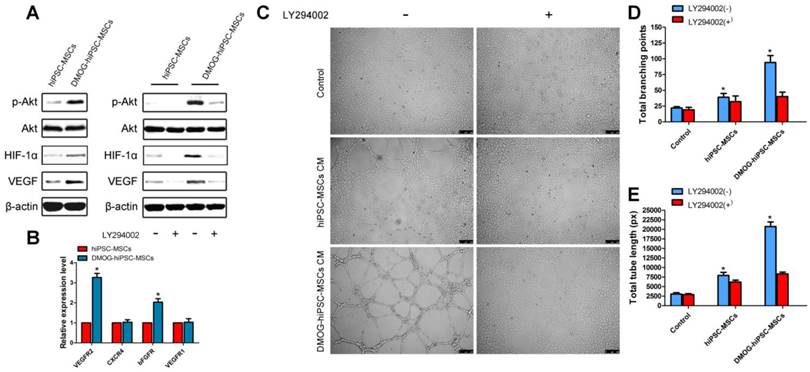
The receptors expressions in endothelial cells were also detected in this study, and we found that the expression of VEGFR2 and bFGFR were obvious upregulated treatment with DMOG-hiPSC-MSCs CM. However, the expressions of CXCR4 and VEGFR1 have no significant change (Figure 3B). After 6 h of culture on Matrigel, DMOG-hiPSC-MSCs CM cultured HUVECs formed many tubes with obvious structures. The effect could be blocked by the addition of a PI3k/Akt inhibitor, indicating that the PI3K/Akt signaling pathway is required for DMOG promoted angiogenic activity of hiPSC-MSCs (Figure 3C-E).
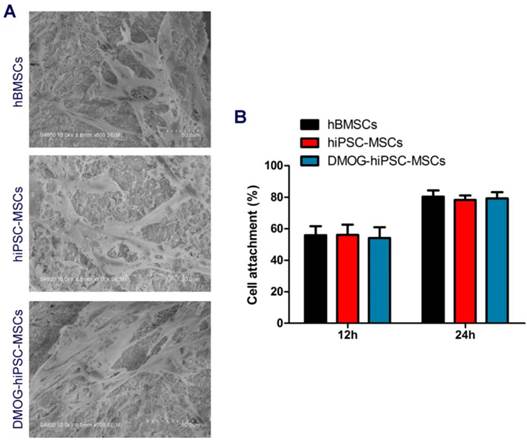
DMOG did not affect hiPSC-MSCs attachment
The hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs all presented well-spread morphology on the scaffolds (Figure 4A). As shown in Figure 4B, no statistically significant difference was found in the cell adhesion rates of hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs on β-TCP.
Effects of DMOG on the hiPSC-MSCs attachment on β-TCP. (A) SEM images of the attachment of hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs on β-TCP after culturing for 7 days. (B) The cell adhesion rates in hBMSCs, hiPSC-MSCs, and DMOG-hiPSC-MSCs on β-TCP after culturing for 12 and 24 h.
DMOG-hiPSC-MSCs improve bone regeneration of critical-sized rat calvarial defects in vivo
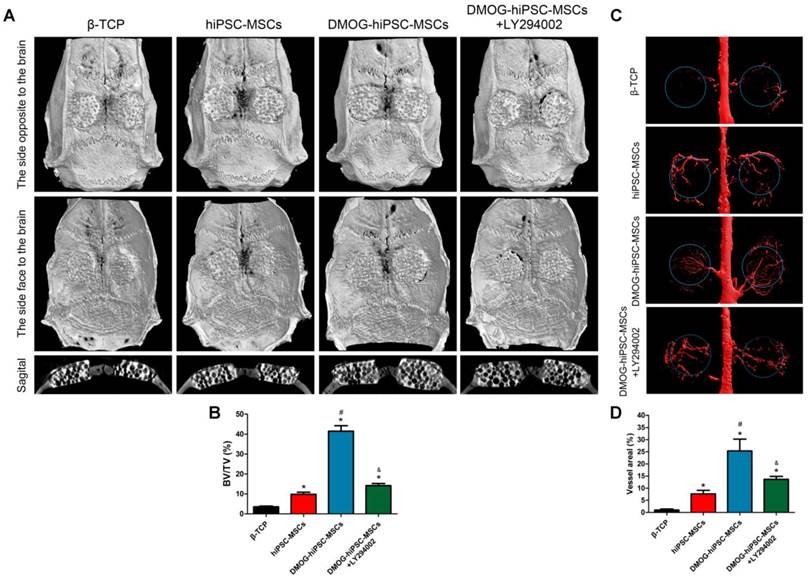
The morphology of the newly formed bone in the defect area was reconstructed with micro-CT. From the sagittal view, significantly more newly formed bone filling the pores of β-TCP was observed in the DMOG-hiPSC-MSCs group compared with that found in the hiPSC-MSCs and β-TCP groups. Moreover, the LY294002 treatment decreased the new bone growth into β-TCP (Figure 5A). A quantitative analysis of the newly formed bone was performed using the micro-CT analysis system. The BV/TV ratio in the DMOG-hiPSC-MSCs group was markedly higher than that in other three groups, we also found that BV/TV of hiPSC-MSCs group was lower than that of DMOG-hiPSC-MSCs+LY294002 group (Figure 5B). The results indicated that the presence of DMOG can improve the bone healing capacity of hiPSC-MSCs in vivo.
Micro-CT evaluation of the repaired craniums and blood vessels formation at 8 weeks post-implantation. (A) Three-dimensional reconstruction and sagittal images showed the different reparative effects of the β-TCP, hiPSC-MSCs, DMOG-hiPSC-MSCs, and DMOG-hiPSC-MSCs+LY294002. (B) The bone volume-to-total volume ratio (BV/TV) varied between the groups. (C) The new blood vessels in the calvarial defects are presented by Three-dimensional reconstruction images. (D) Morphometric analysis was performed to determine the local blood vessel area in the bone defects (*p < 0.05 compared with the β-TCP group, #p < 0.05 compared with hiPSC-MSCs and DMOG-hiPSC-MSCs+LY294002groups, &p < 0.05 compared with hiPSC-MSCs group).
Neovascularization at site of injury
New vessel formation in the defect area was visualized by Microfil perfusion. Three-dimensional micro-CT images showed that neovascularization in the DMOG-hiPSC-MSCs, DMOG-hiPSC-MSCs+LY294002 and hiPSC-MSCs groups were markedly greater than that in the β-TCP group, and the DMOG-hiPSC-MSCs group showed higher neovascularization than the DMOG-hiPSC-MSCs+LY294002 and hiPSC-MSCs group (Figure 5C). The quantification of the blood vessel area confirmed the 3D results (Figure 5E). In particular, the vessel area in the LY294002 treatment group was higher than that in normal hiPSC-MSCs group. This may be due to DMOG contained in cells or β-TCP that affected surrounding autologous BMSCs or EC neovascularization.
Fluorochrome labeling histomorphometrical analysis
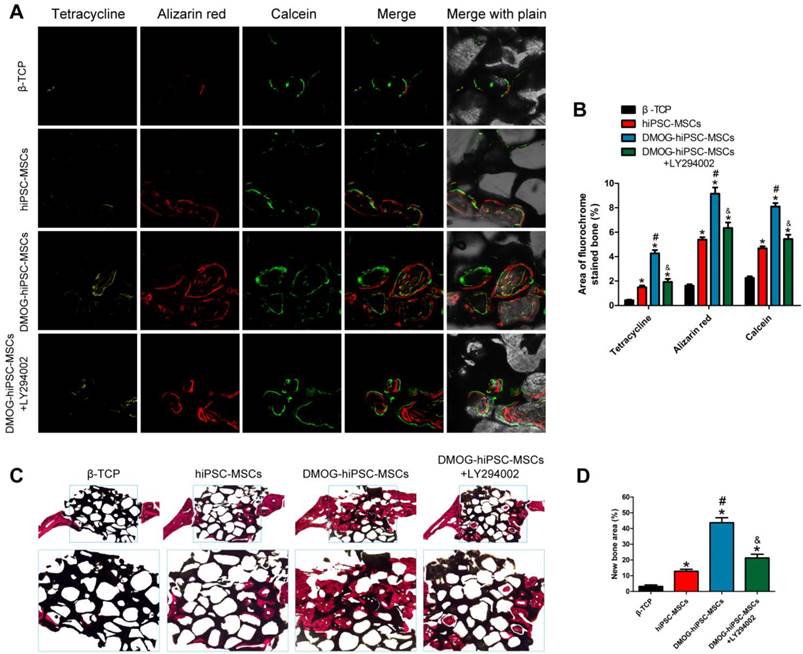
New bone formation and mineralization were quantitatively analyzed at weeks 2, 4 and 6 post-operation through fluorescence labling (Figure 6A and B). After two weeks, the percentage of tetracycline labeling (yellow, Figure 6A, column 1) in the DMOG-hiPSC-MSCs group was greater than that in other three groups, whereas there was also significant difference between the DMOG-hiPSC-MSCs+LY294002 and hiPSC-MSCs groups. After four weeks, the highest percentage of alizarin red labeling (red, Figure 6A, column 2) was observed in the DMOG-hiPSC-MSCs group compare with other three groups, and there was also a significant difference between the DMOG-hiPSC-MSCs+LY294002 and hiPSC-MSCs groups. After six weeks, the percentage of calcein labeling (green, Figure 6A, column 3) in the DMOG-hiPSC-MSCs group was significantly higher than that in other three groups, and there was also a significant difference between the DMOG-hiPSC-MSCs+LY294002 and hiPSC-MSCs groups. These results also suggest that the presence of DMOG can improve the bone healing capacity of hiPSC-MSCs in vivo.
New bone formation and mineralization was determined histomorphometrically through fluorochrome-labeling analysis and histological analysis of newly formed bone. (A) Column 1 (yellow), 2 (red) and 3 (green) represent staining with tetracycline after 2 weeks, alizarin red after 4 weeks, and calcein after 6 weeks, respectively. Column 4 represents the merged images of the three fluorochromes for the same group, and column 5 represents merged images from a plain microscope. (B) The graph shows the percentage of each fluorochrome area for the different groups. (C) The undecalcified craniums were sliced, and sections were stained with van Gieson's picrofuchsin. The new bone and β-TCP are shown in red and black, respectively. (D) There were significant differences in the new bone area between the β-TCP, hiPSC-MSCs, DMOG-treated hiPSC-MSCs and DMOG-hiPSC-MSCs+LY294002 groups (*p < 0.05 compared with the β-TCP group, #p < 0.05 compared with hiPSC-MSCs and DMOG-hiPSC-MSCs+LY294002groups, &p < 0.05 compared with hiPSC-MSCs group).
Histological and immunohistochemical analysis
The van Gieson's picrofuchsin staining of non-decalcified craniums apparently showed that bone regeneration was markedly increased by the addition of DMOG (Figure 6C). The percentage of newly formed bone area was markedly higher in the DMOG-hiPSC-MSCs group than in other three groups (Figure 6D).
The immunohistochemistry staining of decalcified craniums for the angiogenic markers CD31, VEGF, and HIF-1α showed that all markers were highly expressed in the DMOG-hiPSC-MSCs and DMOG-hiPSC-MSCs+LY294002 groups, low expression in hiPSC-MSCs group, but nearly no positive staining could be found in the β-TCP group. Moreover, the DMOG-hiPSC-MSCs group also showed higher expression levels of angiogenic markers than the hiPSC-MSCs-MSCs+LY294002 group (Figure 7).
Immunohistochemical analysis of CD31, VEGF and HIF-1α expression in each group 8 weeks post-operation. There were nearly no positive staining for CD31, VEGF and HIF-1α found in β-TCP Group, a few of positive staining observed in hiPSC-MSCs Group, and more positive staining in DMOG-hiPSC-MSCs and DMOG-hiPSC-MSCs-LY294002 Groups. Scale bar: 150 μm.

Discussion
In the present study, we investigated whether DMOG could promote the angiogenic activity of hiPSC-MSCs for bone regeneration and explored the underlying mechanisms involved. Our findings provide the first demonstration that DMOG can activate the PI3K/Akt pathway to stimulate the angiogenic activity of hiPSC-MSCs and thereby contribute to bone regeneration in a critical-sized rat calvarial defect model in vivo.
iPSCs are a type of newly defined stem cells that was first generated by Yamanaka and colleagues [39]. iPSCs show an unlimited growth capacity and are not associated with any ethical concerns, making them superior to traditional MSCs and ESCs. iPSCs have been transplanted to repair bone and cartilage defects, and regenerated bone and cartilage tissues are found at the wound sites [40-42]. However, iPSCs have the potential to be expanded indefinitely and are thus potentially tumorigenic [43]. iPSC-MSCs then emerged, and these cells offer the advantages of both MSCs and iPSCs and are not tumorigenic [23, 27]. Our study demonstrated that hiPSC-MSCs display MSC-like properties and multipotency. Specifically, iPSCs can infinitely expand in vitro and offer an unlimited source for MSCs generation, which is very important because large numbers of expanded cells are needed for stem cell transplantation therapy. iPSC-MSCs have been demonstrated to play beneficial effects in tissue repair therapy [26, 28] and have also been adopted to construct tissue-engineered bone for bone defect repair, but the repair effect of hiPSC-MSCs in vivo has not yet been verified [29, 30]. Our study first examined the effect of bone defect repair by hiPSC-MSCs transplantation in vivo. Moreover, in bone tissue engineering, limited numbers of functional cells could be seeded on bone substitutes; thus, modified iPSC-MSCs with enhanced angiogenic activity are necessary.
Accumulating evidence demonstrates that angiogenesis plays an important role in bone regeneration and repair. The angiogenic activity of MSCs is regulated by various signaling pathways, such as TGF-β, fibroblast growth factor-2 (FGF-2), prostaglandins E1 and E2, insulin-like growth factor-1 (IGF-1), endothelin-1, and vitamin D3. A proximate control point in all of the pathways is associated with the HIF-1α pathway [44]. Some studies have transduced the HIF-1α gene into MSCs using lentivirus vectors and showed that the existence of HIF-1α enhances the angiogenesis of MSCs and subsequently produces satisfactory bone regeneration effects [45]. However, after HIF-1α is transduced into MSCs using lentivirus vectors, the risk of tumorigenesis remains, which confines the clinical application of HIF-1α to a certain extent. Growth factors are routinely considered in tissue engineering approaches to accelerate the process of angiogenesis. Local VEGF application has been successfully exploited to promote angiogenesis in bone regeneration [46, 47]. Nevertheless, the applications of growth factors are hampered by several complications, including high dose requirements, lower half-life, protein instability, higher costs and undesired side effects [48, 49]. Recently, a variety of small molecules have emerged as alternatives to growth factors for enhancing angiogenesis. Small molecules are simple biochemical components that elicit specific cellular responses through signaling cascades, which shows that small-molecule-based strategies can reduce several of the side effects associated with a high dose growth factors, result in maintained bioactivity in a biological environment, and minimize cost issues associated with complex biological growth factors [48, 49]. Additionally, the use of small molecules is more convenient than strategies requiring growth factors or gene therapy. The small molecule DMOG is a cell-permeable PHD inhibitor that can upregulate the expression of the HIF-1α protein post-transcriptionally under normoxic conditions. Because our previous studies indicated that DMOG-treated BMSCs or ADSCs can improve the angiogenic activity of tissue-engineered bone by activating HIF-1α, we evaluated whether DMOG-hiPSC-MSCs can be used for bone regeneration via the promoting of angiogenic activity. Experiments in vitro demonstrated that DMOG slightly inhibited hiPSC-MSCs proliferation but didn't cause the death of hiPSC-MSCs, even can inhibit hiPSC-MSCs death induced by serum deprivation conditions, which indicated that DMOG has no obvious cytotoxic effect on hiPSC-MSCs viability. The gene and protein expression profiles of the DMOG-hiPSC-MSCs showed that DMOG acts to stabilize the HIF-1α expression at normal oxygen tension in hiPSC-MSCs, leading to the subsequent angiogenesis. In the present study, we further explored the underlying mechanisms of DMOG-induced angiogenesis in vitro. The PI3K/Akt signal transduction pathway widely exists in cells and mediates the response to a wide range of stimuli. Previous studies have demonstrated that the PI3k/Akt pathway can exert a critical effect on angiogenesis in adult organisms while actively promoting endothelial repair and regeneration [50, 51]. Studies have also indicated that activation of PI3K/Akt signaling mediates VEGF protein expression at the transcriptional level through HIF-1α expression [36], and that DMOG can promote Akt phosphorylation in MSCs [37]. We thus hypothesized that DMOG may enhance the angiogenic activity of hiPSC-MSCs via activation of the PI3k/Akt pathway. Our results demonstrated that elevated protein levels of p-AKT and VEGF were found in the presence of DMOG and that the addition of a PI3k/Akt inhibitor blocked the expression of these proteins. These data suggested that DMOG promotes angiogenic activity of hiPSC-MSCs via activation of PI3k/Akt pathway.
The ability of DMOG-hiPSC-MSCs in bone regeneration in vivo was determined using a critical-sized rat calvarial defect model. Quantitative analysis by micro-CT based on the BV/TV ratio showed that there was only a slight newly formed bone in the β-TCP group. The presence of DMOG significantly enhanced new bone formation compared with that obtained in the normal hiPSC-MSC group. Histological analysis then demonstrated that only a slight amount of newly formed bone covered the defect areas in the β-TCP group, whereas the hiPSC-MSCs and DMOG-hiPSC-MSCs groups exhibited a significant promotion of bone formation over the defect areas, and DMOG treatment resulted in coverage of most of the defect areas, consistent with the micro-CT findings. Microfil perfusion was used to evaluate the neovascularization at the site of injury, and the 2D quantification of the percentage of new vessel area in the defect region demonstrated that the hiPSC-MSCs and DMOG-hiPSC-MSCs treatments lead to a significantly higher amount of newly formed vessels compared with that found in the β-TCP group, and the largest areas of newly formed vessels were found in the DMOG-hiPSC-MSCs group. All of the above data demonstrated that DMOG-hiPSC-MSCs can improve the bone regeneration of critical-sized rat calvarial defects in vivo by promoting angiogenesis. It is noteworthy that, the treatment of LY294002 could significantly suppressed the DMOG-induced neovascularization and bone regeneration, which implied that the in vivo DMOG-stimulated hiPSC-MSCs angiogenesis was associated with PI3K/Akt pathway. In particular, we also found that the DMOG-hiPSC-MSCs+LY294002 group showed better neovascularization and bone regeneration than hiPSC-MSCs group, which may be due to the DMOG stimulated surrounding autologous BMSCs or EC neovascularization. Although we showed that MSCs derived from iPS, bone marrow and adipose tissue can be treated with DMOG to enhance angiogenic activity for bone defect repair, which one exerts the best effect requires further studies.
Conclusion
In the present study, hiPSC-MSCs, which incorporate the advantages of both MSCs and iPSCs were used as seed cells for constructing tissue-engineered bone. Small molecular DMOG has no obvious cytotoxic effect on hiPSC-MSCs viability and can enhance the angiogenic activity of hiPSC-MSCs. Further in vitro and in vivo analyses demonstrated that the activation of the PI3K/Akt pathway contributes to the enhanced angiogenic activity of DMOG-hiPSC-MSCs. In a critical-sized rat calvarial defect model, DMOG-hiPSC-MSCs significantly promoted bone regeneration through the promotion of angiogenesis, which suggests that DMOG-hiPSC-MSCs is a potential reliable alternative source for applications in bone regeneration and bone defect repair.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81572120). “Priority Among Priorities” Clinical Medical Center Construction Project of the Shanghai Municipality. Doctoral Innovation Fund of Shanghai Jiao Tong University School of Medicine (BXJ201341). The authors wish to thank Ting Guo (University of Maryland-College Park), for the help on translating.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McKee MD. Management of segmental bony defects: the role of osteoconductive orthobiologics. J Am Acad Orthop Surg. 2006;14:S163-167
2. Oreffo RO, Triffitt JT. Future potentials for using osteogenic stem cells and biomaterials in orthopedics. Bone. 1999;25:5S-9S
3. Yu H, VandeVord PJ, Mao L, Matthew HW, Wooley PH, Yang SY. Improved tissue-engineered bone regeneration by endothelial cell mediated vascularization. Biomaterials. 2009;30:508-517
4. Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082-4090
5. Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621-627
6. Madeddu P. Therapeutic angiogenesis and vasculogenesis for tissue regeneration. Exp Physiol. 2005;90:315-326
7. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677-684
8. Ferrara N. Molecular and biological properties of vascular endothelial growth factor. J Mol Med (Berl). 1999;77:527-543
9. Riddle RC, Khatri R, Schipani E, Clemens TL. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J Mol Med (Berl). 2009;87:583-590
10. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-472
11. Song YR, You SJ, Lee YM, Chin HJ, Chae DW, Oh YK. et al. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant. 2010;25:77-85
12. Liu X, Wang JA, Ji XY, Yu SP, Wei L. Preconditioning of bone marrow mesenchymal stem cells by prolyl hydroxylase inhibition enhances cell survival and angiogenesis in vitro and after transplantation into the ischemic heart of rats. Stem Cell Res Ther. 2014;5:111
13. Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D. et al. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132-143
14. HoWangYin KY, Loinard C, Bakker W, Guerin CL, Vilar J, D'Audigier C. et al. HIF-prolyl hydroxylase 2 inhibition enhances the efficiency of mesenchymal stem cell-based therapies for the treatment of critical limb ischemia. Stem Cells. 2014;32:231-243
15. Ding H, Chen S, Song WQ, Gao YS, Guan JJ, Wang Y. et al. Dimethyloxaloylglycine improves angiogenic activity of bone marrow stromal cells in the tissue-engineered bone. Int J Biol Sci. 2014;10:746-756
16. Ding H, Gao YS, Wang Y, Hu C, Sun Y, Zhang C. Dimethyloxaloylglycine increases the bone healing capacity of adipose-derived stem cells by promoting osteogenic differentiation and angiogenic potential. Stem Cells Dev. 2014;23:990-1000
17. Kahle M, Wiesmann HP, Berr K, Depprich RA, Kubler NR, Naujoks C. et al. Embryonic stem cells induce ectopic bone formation in rats. Biomed Mater Eng. 2010;20:371-380
18. Marrelli M, Paduano F, Tatullo M. Cells isolated from human periapical cysts express mesenchymal stem cell-like properties. Int J Biol Sci. 2013;9:1070-1078
19. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA. et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141-146
20. Kang L, Wang J, Zhang Y, Kou Z, Gao S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135-138
21. Cao H, Yang P, Pu Y, Sun X, Yin H, Zhang Y. et al. Characterization of bovine induced pluripotent stem cells by lentiviral transduction of reprogramming factor fusion proteins. Int J Biol Sci. 2012;8:498-511
22. Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885-896
23. Villa-Diaz LG, Brown SE, Liu Y, Ross AM, Lahann J, Parent JM. et al. Derivation of mesenchymal stem cells from human induced pluripotent stem cells cultured on synthetic substrates. Stem Cells. 2012;30:1174-1181
24. Liu Y, Goldberg AJ, Dennis JE, Gronowicz GA, Kuhn LT. One-step derivation of mesenchymal stem cell (MSC)-like cells from human pluripotent stem cells on a fibrillar collagen coating. PLoS One. 2012;7:e33225
25. Zou L, Luo Y, Chen M, Wang G, Ding M, Petersen CC. et al. A simple method for deriving functional MSCs and applied for osteogenesis in 3D scaffolds. Sci Rep. 2013;3:2243
26. Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y. et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010;121:1113-1123
27. Yen ML, Hou CH, Peng KY, Tseng PC, Jiang SS, Shun CT. et al. Efficient derivation and concise gene expression profiling of human embryonic stem cell-derived mesenchymal progenitors (EMPs). Cell Transplant. 2011;20:1529-1545
28. Himeno T, Kamiya H, Naruse K, Cheng Z, Ito S, Kondo M. et al. Mesenchymal stem cell-like cells derived from mouse induced pluripotent stem cells ameliorate diabetic polyneuropathy in mice. Biomed Res Int. 2013;2013:259187
29. Liu J, Chen W, Zhao Z, Xu HH. Reprogramming of mesenchymal stem cells derived from iPSCs seeded on biofunctionalized calcium phosphate scaffold for bone engineering. Biomaterials. 2013;34:7862-7872
30. Liu J, Chen W, Zhao Z, Xu HH. Effect of NELL1 gene overexpression in iPSC-MSCs seeded on calcium phosphate cement. Acta Biomater. 2014;10:5128-5138
31. Walsh WR, Vizesi F, Michael D, Auld J, Langdown A, Oliver R. et al. Beta-TCP bone graft substitutes in a bilateral rabbit tibial defect model. Biomaterials. 2008;29:266-271
32. Liu GT, Chen HT, Tsou HK, Tan TW, Fong YC, Chen PC. et al. CCL5 promotes VEGF-dependent angiogenesis by down-regulating miR-200b through PI3K/Akt signaling pathway in human chondrosarcoma cells. Oncotarget. 2014;5:10718-31
33. Hou Y, Ryu CH, Jun JA, Kim SM, Jeong CH, Jeun SS. IL-8 enhances the angiogenic potential of human bone marrow mesenchymal stem cells by increasing vascular endothelial growth factor. Cell Biol Int. 2014;38:1050-1059
34. Jun EK, Zhang Q, Yoon BS, Moon JH, Lee G, Park G. et al. Hypoxic conditioned medium from human amniotic fluid-derived mesenchymal stem cells accelerates skin wound healing through TGF-beta/SMAD2 and PI3K/Akt pathways. Int J Mol Sci. 2014;15:605-628
35. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114-127
36. Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279:45643-45651
37. Liu XB, Wang JA, Ogle ME, Wei L. Prolyl hydroxylase inhibitor dimethyloxalylglycine enhances mesenchymal stem cell survival. J Cell Biochem. 2009;106:903-911
38. Liao J, Wu Z, Wang Y, Cheng L, Cui C, Gao Y. et al. Enhanced efficiency of generating induced pluripotent stem (iPS) cells from human somatic cells by a combination of six transcription factors. Cell Res. 2008;18:600-603
39. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676
40. Ko JY, Park S, Im GI. Osteogenesis from human induced pluripotent stem cells: an in vitro and in vivo comparison with mesenchymal stem cells. Stem Cells Dev. 2014;23:1788-1797
41. Ko JY, Kim KI, Park S, Im GI. In vitro chondrogenesis and in vivo repair of osteochondral defect with human induced pluripotent stem cells. Biomaterials. 2014;35:3571-3581
42. Tsumaki N, Okada M, Yamashita A. iPS cell technologies and cartilage regeneration. Bone. 2015;70:48-54
43. Jung Y, Bauer G, Nolta JA. Concise review. Induced pluripotent stem cell-derived mesenchymal stem cells: progress toward safe clinical products. Stem Cells. 2012;30:42-47
44. Wang Y, Wan C, Gilbert SR, Clemens TL. Oxygen sensing and osteogenesis. Ann N Y Acad Sci. 2007;1117:1-11
45. Zou D, Zhang Z, Ye D, Tang A, Deng L, Han W. et al. Repair of critical-sized rat calvarial defects using genetically engineered bone marrow-derived mesenchymal stem cells overexpressing hypoxia-inducible factor-1alpha. Stem Cells. 2011;29:1380-1390
46. Street J, Bao M, deGuzman L, Bunting S, Peale FV Jr, Ferrara N. et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci U S A. 2002;99:9656-9661
47. Peng H, Usas A, Olshanski A, Ho AM, Gearhart B, Cooper GM. et al. VEGF improves, whereas sFlt1 inhibits, BMP2-induced bone formation and bone healing through modulation of angiogenesis. J Bone Miner Res. 2005;20:2017-2027
48. Aravamudhan A, Ramos DM, Nip J, Subramanian A, James R, Harmon MD. et al. Osteoinductive small molecules: growth factor alternatives for bone tissue engineering. Curr Pharm Des. 2013;19:3420-3428
49. Segar CE, Ogle ME, Botchwey EA. Regulation of angiogenesis and bone regeneration with natural and synthetic small molecules. Curr Pharm Des. 2013;19:3403-3419
50. Namkoong S, Lee SJ, Kim CK, Kim YM, Chung HT, Lee H. et al. Prostaglandin E2 stimulates angiogenesis by activating the nitric oxide/cGMP pathway in human umbilical vein endothelial cells. Exp Mol Med. 2005;37:588-600
51. Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS. et al. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol. 2006;291:H2836-2846
Author contact
![]() Corresponding authors: Yigang Huang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. Tel.: +86-21-24056319; Fax: +86-21-64363802; E-mail: yuan.hongsh.cn Co-correspondence: Changqing Zhang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: zhangcqgkcom Xiaolin Li, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: li-xiaolincom.
Corresponding authors: Yigang Huang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. Tel.: +86-21-24056319; Fax: +86-21-64363802; E-mail: yuan.hongsh.cn Co-correspondence: Changqing Zhang, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: zhangcqgkcom Xiaolin Li, Department of Orthopedic Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China. E-mail: li-xiaolincom.