Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(6):701-709. doi:10.7150/ijbs.13635 This issue Cite
Research Paper
Ribose Accelerates Gut Motility and Suppresses Mouse Body Weight Gaining
Yan Liu1,2, Tong-Ruei R Li2, Cong Xu2, Tian Xu1,3 ![]()
1. State Key Laboratory of Genetic Engineering and Institute of Developmental Biology and Molecular Medicine, National Center for International Research, Fudan-Yale Center for Biomedical Research, School of Life Sciences, Fudan University, Shanghai 200433, China
2. Shanghai Yao Yuan Biotechnology (Drug Farm) Limited, Co. Room 701, 43 Handan Rd, Shanghai, 200437, China
3. Howard Hughes Medical Institute, Department of Genetics, Yale University School of Medicine, Boyer Center for Molecular Medicine, 295 Congress Avenue, New Haven, Connecticut 06536, USA.
Received 2015-8-23; Accepted 2016-2-24; Published 2016-4-28
Abstract

The increasing prevalence of obesity is closely related to excessive energy consumption. Clinical intervention of energy intake is an attractive strategy to fight obesity. However, the current FDA-approved weight-loss drugs all have significant side effects. Here we show that ribose upregulates gut motility and suppresses mice body weight gain. Ribokinase, which is encoded by Rbks gene, is the first enzyme for ribose metabolism in vivo. Rbks mutation resulted in ribose accumulation in the small intestine, which accelerated gut movement. Ribose oral treatment in wild type mice also enhanced bowel motility and rendered mice resistance to high fat diets. The suppressed weight gain was resulted from enhanced ingested food excretion. In addition, the effective dose of ribose didn't cause any known side effects (i.e. diarrhea and hypoglycemia). Overall, our results show that ribose can regulate gut motility and energy homeostasis in mice, and suggest that administration of ribose and its analogs could regulate gastrointestinal motility, providing a novel therapeutic approach for gastrointestinal dysfunction and weight control.
Keywords: Ribose, ribokinase, gut motility, body weight control.
Introduction
Obesity and overweight have become a global health concern in the past few decades. Obesity-related morbidities, such as diabetes, hypertension and cardiovascular diseases, dramatically decrease life expectancy [1-4]. The increasing prevalence of obesity in recent years is closely related to the changes of eating behavior, expanded energy intake and disturbed energy homeostasis. Interference of energy intake becomes an attractive approach for fighting obesity. There are currently three FDA-approved long-term-use weight loss drugs, targeting either the hypothalamus or the digestive tract [5, 6]. Qsymia and Lorcaserin targets neurotransmitter receptors in the central nervous system (CNS) to reduce appetite, but also leads to non-specific effects in several organ systems, e.g. blurred vision, dizziness, cognitive impairments and depression [7-9]. Comparing with the CNS which affects various organs, the digestive tract may be a better drug target. Nevertheless, orlistat, which inhibits fat absorption in the gut, may result in gastrointestinal disorder (e.g. steatorrhea and irregular bowel movements) and serious hepatic adverse events [10, 11]. It may also interfere with many other drugs and affect their bioavailabilities [12-14]. Thus, there are demands for new therapeutic strategies targeting energy homeostasis in the digestive tract.
Nucleic acid, carbohydrates, fat and protein are the major nutrition components derived from food. Although the metabolism of carbohydrates, fat and protein in energy homeostasis has been studied for decades [15-17], the roles of nucleic acid and its digestive products in regulating energy balance are poorly understood. D-ribose, a naturally occurring pentose monosaccharide, is a product of dietary nucleic acid digestion [18]. Previous studies indicated that ribose is a promising supplement in improving heart and skeletal muscle performances [19-23]. However, as one of the most abundant digestive products of nucleic acid, the function of ribose in the digestive tract and energy homeostasis is poorly understood.
Ribokinase is known as the first enzyme responsible for ribose metabolism in mammals. It belongs to the PfkB family of carbohydrate kinase, and can phosphorylate ribose into ribose-5-phosphate. Ribokinase is highly evolutionally conserved from bacteria, insects to mammals [24]. It is encoded by the Rbks gene, which has only one homolog in mice and human. Although ribokinase activity was firstly identified in in vitro assay using calf liver extracts more than 50 years ago [25], little work had been carried out on its function in mammals.
To better understand the role of ribose and ribokinase in mammals, we generated Rbks gene knock-out mice. Our data showed that after Rbks gene depletion, the motility of the small intestine enhanced. The accelerated gut movement was induced by disrupted ribose homeostasis, which was confirmed in wild type mice using ribose oral administration. In addition, when wild type mice were challenged by a high fat diet, the enhanced gut motility induced by ribose oral treatment lead to increased energy excretion and, in turn, affected body weight. Our study revealed, for the first time, the function of ribose in the digestive tract and energy homeostasis. Administration of ribose and its analogs could regulate gastrointestinal motility, providing a novel therapeutic approach for gastrointestinal dysfunction and weight control.
Results
Generation of Rbks knock-out mice
The murine ribokinase protein contains 323 amino acids (a.a.), with the substrate binding sites (26-308 a.a.), the dimer interface sites (29-181 a.a.), and the ATP binding sites (200-303 a.a.). The Rbks gene knock-out mice were generated by removing exon2, which results in deleting 31-76 a.a. encoded by exon2 and frame shift after 76 a.a. from splicing exon1 to exon3 (Fig. S1; Materials and Methods). Rbks gene targeted embryonic stem cells purchased from European Conditional Mouse Mutagenesis Program were injected into C57BL/6 blastocysts. The targeting construct contained an E. coli β-galactosidase reporter and a neomycin expression cassette (galeo cassette) flanked by FRT sites. Immediate after the galeo cassette, the Rbks exon2 was flanked by loxP sites (Fig. S1A). The chimeric Rbksgaleo/+ male mice were mated with C57BL/6 females to generate stable colonies for LacZ gene expression assay and for further Rbks knock-out mice breeding (Fig. S1B). The galeo reporter cassette was removed by mating with ubiquitously active CAG-flpe mice to produce RbksF/+ mutants. RbksF/+ mice were mated with Actin-cre mice to delete exon2 by cre-loxP recombination (Fig. S1A; Materials and Methods). Homozygous Rbks knock-out (Rbks∆2/∆2) mice were generated by mating of heterozygous animals. The deletion of exon2 was verified by genotyping-PCR and RT-PCR with primers locating on exon2 (Fig. S1C, D). The Rbks knock-out mice are viable, fertile, and have no obvious morphological defects.
High expression level of the Rbks gene in the gut
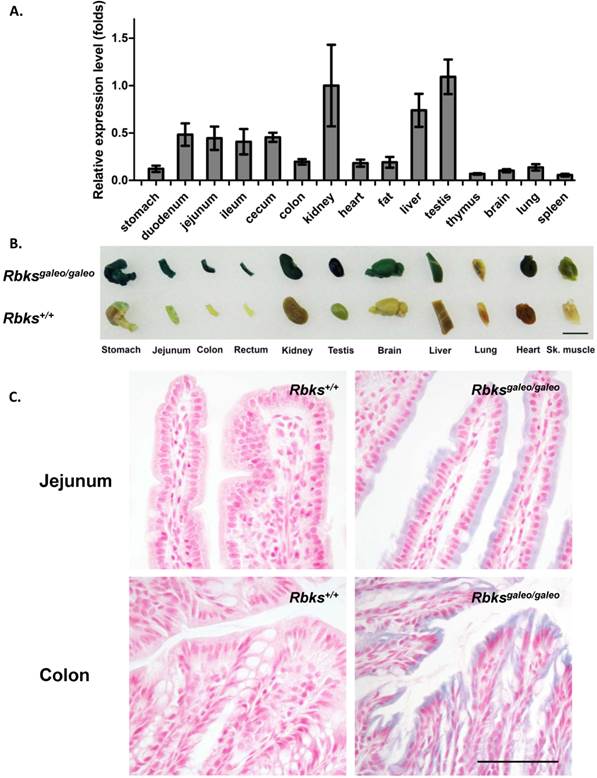
Using quantitative real-time PCR, Rbks expression level was measured in multiple mouse tissues (Fig. 1A). A high level of Rbks RNA was detected in the kidney, liver, testis, small intestine (duodenum, jejunum and ileum) and cecum. The Rbks expression in these tissues was confirmed by whole mount X-gal staining with Rbksgaleo/ galeo mice tissues (Fig. 1B), in which the β-galactosidase reporter is driven by the endogenous Rbks promoter. The β-galactosidase activity was also detected in the brain, heart, skeletal muscle and lung (Fig. 1B). To determine the cellular expression of Rbks in the gut, the whole mount X-gal stained tissues were fixed and prepared for paraffin sections. Rbks promoter-driven β-galactosidase signals were localized to the intestine villi (Fig. 1C).
Expression pattern of Rbks gene in mice. (A) Quantitative real-time (qRT) PCR revealed a high RNA level of Rbks gene in kidney, liver, testis, small intestine (duodenum, jejunum and ileum) and cecum of wild type mice. Primers for qRT PCR located on exon5 and exon6. (B) The expression pattern of Rbks gene was also revealed by whole mount X-gal staining with Rbksgaleo/galeo mice tissues. The wild type litter mates served as controls. Black bar: 1cm. (C) The whole mount staining intestines were further processed for paraffin sections, and the cellular β-galactosidase signals were identified in intestinal brush boarders. Black bar: 100μm.
Enhanced gut motility in Rbks targeting mice
Ribose is one of the major digestive products of dietary nucleic acids [18]. The high expression level of Rbks gene implied potential roles of ribose and its metabolites in the digestive tract. However, no overt changes in small intestine length and gut morphology were observed after Rbks gene depletion (Fig S2).
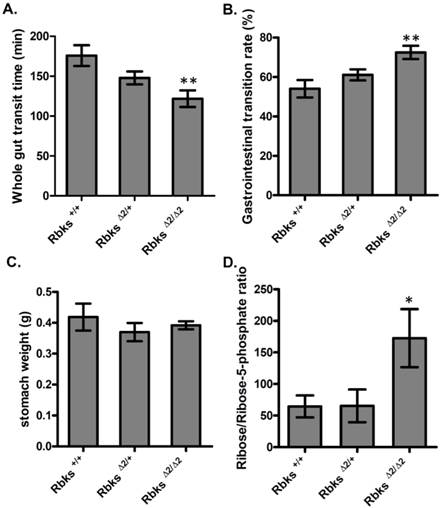
We further examined whether gut function was affected after Rbks depletion. Firstly, the whole gut motility was assessed by the whole gut transition time (WGTT) test. We found WGTT was significantly decreased by 32% in Rbks knock-out mice when compared to wild type controls (n=8, P<0.05) (Fig. 2A), which indicated that whole gut motility was enhanced. To further locate which part of the digestive tract caused the increased motility, gastrointestinal (GI) transition rate was measured at 30 minutes after charcoal gavage. The GI transition rate refers to the upper gut (gastric-small intestine) movements, and the rate was dramatically increased in Rbks knock-out mice by 33% when compared to wild type controls (n=4, P<0.05) (Fig. 2B). In addition, 30 minutes after charcoal gavage, there was not significant differences in gastric weight, which indicated during 30 minutes, the gastric empty was not affected (Fig. 2C).
Gut motility enhanced in Rbks gene targeting mice. After Rbks exon2 depletion, the whole gut transition time was significantly decreased (A), while the gastrointestinal transition rate increased (B) in Rbks∆2/∆2 mice when compared with their litter mates. (C) There was not overt changes in gastric weight after charcoal gavage between knock-out mice and controls. (D) The ribose/ribose-5-phosphate ratio was elevated in the small intestines of Rbks∆2/∆2 mice. *=P<0.05, **=P<0.01. Significances indicated are based on Student's t-test. Data represent the average ± s.e.m.
Accelerated gut movement in wild type mice with ribose administration
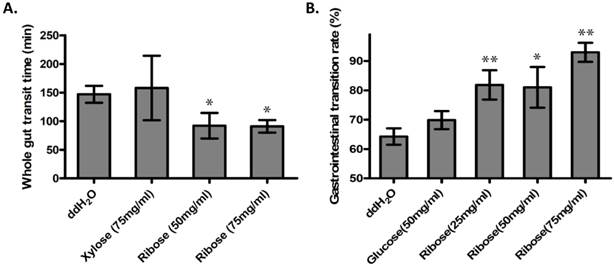
Ribokinase phosphorylates ribose to ribose-5-phosphate (R-5-P), which is the first step of ribose metabolism in vivo. Hence, the Rbks knock-out would result in ribose accumulation. Consistent with this notion, the ribose/ribose-5-phosphate ratio was dramatically increased in the small intestines after Rbks mutation (Fig. 2D) (n=10, P<0.05). We therefore assumed the increased ribose proportion may lead to the enhanced bowel motility in the mutants. The assumption was demonstrated using ribose oral administration in wild type mice. The ribose oral gavage decreased WGTT and up-regulated GI transition rate, while the other sugar controls (xylose or glucose) with the same concentrations did not affect gut motility (Fig. 3A, 3B).
Ribose oral administration increased gut motility in wild type mice. (A) Ribose oral administration (50 mg/ml and 75 mg/ml) dramatically reduced whole gut transition time when compared with ddH2O controls, while the same concentration of xylose control didn't affect the transition time. (B) Ribose oral gavage (25 mg/ml, 50 mg/ml and 75 mg/ml) significantly accelerate gastrointesintal transition rate, while there's no obvious change in the glucose group. *=P<0.05, **=P<0.01. Significances indicated are based on Student's t-test. Data represent the average ± s.e.m.
Suppressed weight gain with ribose 5 weeks treatment
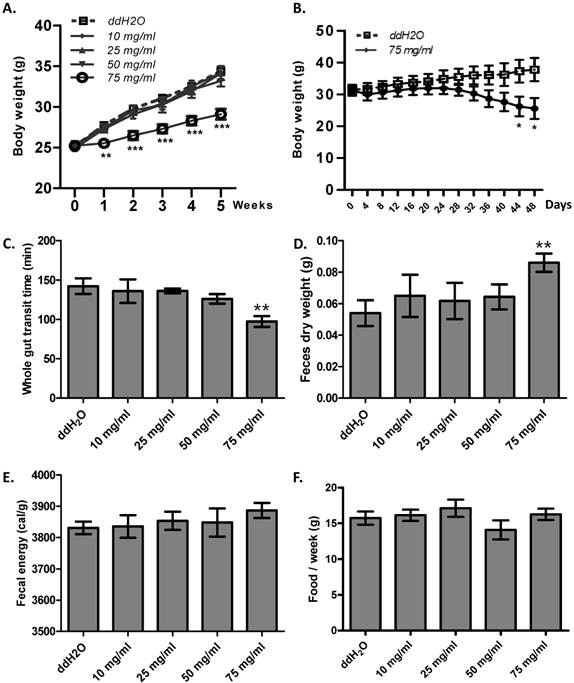
Enhanced gut motility may result in malabsorption and accelerated defecation, and, in turn, disturbed energy homeostasis [26]. Thus, we hypothesized that ribose long-term administration in wild type mice may influence body weight. To test this, we gave ribose to wild type mice via drinking water for 5 weeks. We found that ribose administration (75 mg/ml) dramatically depressed body weight gain in wild type mice when fed with high fat diets, while lower concentrations (10 mg/ml, 25 mg/ml, 50 mg/ml) did not affect body weight (Fig. 4A). To investigate the cause of the suppressed weight gain, we checked the gut motility and the defecation rate. 75 mg/ml ribose treatment led to shorter whole gut transition time and more fecal outputs when compared to the distilled water controls (Fig. 4C, 4D). In addition, the fecal energy was also slightly enhanced (3886 ± 24 cal/g in 75 mg/ml group, 3830 ± 20 cal/g in ddH2O group), although the difference was not statistically significance (n=12) (Fig. 4E). The increased defecation and fecal energy resulted in more energy lost in excreta. Since food consumption is not changed between ribose treated and wild type mice (Fig. 4F), more energy excretion leads to less energy storage and in turn suppressed body weight. Previous reports in human revealed that high dose of ribose may result in diarrhea or hypoglycemia [21, 27, 28]. To exclude the possibility that the body weight change was caused by these adverse effects, the blood glucose level and fecal water content was measured. After 5 weeks of ribose drinking, the fecal water content was not obviously changed in 75 mg/ml ribose-treated mice when compared to the distilled water controls (Fig. S3A). Also, the blood glucose level was not obviously affected (Fig. S3B).
Ribose disturbed metabolism balance and suppressed mouse body weight gain. (A) Ribose (75 mg/ml) 5 weeks administration in drinking water rendered wild type mice resistance to high-fat diets. (B) Ribose (75 mg/ml) administration in drinking water for 48 days resulted in weight loss in obese mice (6 week old Lepr mutant male mice). (C and D) The whole gut transit time and the dry weight of fecal outputs within 5 hours were dramatically changed after 5 weeks 75 mg/ml ribose treatments. (E) Fecal energy was slightly increased with 75 mg/ml ribose treatment, while no statistic significances were detected. (F) Food consumption per week (the 5th week) was not altered with ribose administration. **=P<0.01, ***=P<0.001. Significances indicated are based on Student's t-test. Data represent the average ± s.e.m.
Discussion
Ribose is a naturally occurring pentose that is provided through various dietary sources. As a good supplement for ATP synthesis, ribose has been used in human for improving the cardiac functions and enhancing the skeletal muscle performances [19-23]. While oral administration of ribose is generally safe, these studies also revealed that high dose ribose may induce diarrhea [21, 22], suggesting a potential role of ribose in the gut. However, little is known about the function of ribose in the digestive tract.
Ribokinase (Rbks) is the first enzyme responsible for ribose metabolism in mammalians. It converts ribose into ribose-5-phosphate, which can be further incorporated into pentose phosphate pathway or nucleotides biosynthesis [29]. However, ribose-5-phosphate could also be generated from glucose through the de novo nucleotide synthesis pathway. Indeed, we established the Rbks gene knock-out mice and found that the mutant animals are viable and fertile. However, accelerated motility of the intestine was observed in the mutants. Both quantitative real-time PCR and X-gal staining experiments revealed that Rbks gene has a high expression level in the murine digestive tract. Whole gut motility has been increased about 32% in the mutants in comparison to the wild-type litter mate controls. Furthermore, gastrointestinal transition assay also revealed an increase in transition rate by 33%. Together, these data showed accelerated bowl movements after Rbks gene depletion.
Rbks knock-out mice exhibited an enhanced ribose/ribose-5-phosphate ratio in the small intestine. To investigate whether the ribose accumulation was responsible for the increased bowel motility, we orally administrated C57BL/6 wild type mice with ribose. By measuring the whole gut transition time and gastrointestinal transition rate, we found that ribose upregulates gut movements. This finding provides an explanation for diarrhea in humans with high dose ribose oral administration.
One possible interpretation for how ribose affects the intestinal movement is that concentrated sugars may disturb the osmotic balance inside the intestines [30, 31]. However, oral administration of other monosaccharide, i.e. xylose and glucose, at similar concentrations didn't influence gut motility. Thus, the effect of ribose oral administration on gut motility could not be simply attributed to a hyperosmotic pressure. It raises the possibility that ribose could act as a signal for regulating gut motility.
Efficiency of digestion is correlated with the length of food transition time in the gut. Increased gut motility, thus, could reduce digestion efficiency and therefore acting as a way for weight control. It has been reported that weight loss in Huntington patients could be attributed to deregulation of gut motility [26]. The toxicological studies of ribose oral administration in rats did not observe any irreversible adverse effects, however, the 13 weeks study did note a weight loss [32, 33]. Our finding that ribose oral administration can enhance gut motility suggests that it could be used as a weight control agent. Indeed, our data showed that when the wild type mice were challenged with high-fat diets, ribose treatment (75 mg/ml) for 5 weeks via drinking water suppressed body weight gain. Similar result was also observed with Lepr mutant mice (Fig. 4B). Furthermore, consistent with our hypothesis, the disruption of energy balance in these animals was the result of a significant increase in energy excretion. Finally, the effective ribose dose didn't lead to any known adverse effect, i.e. hypoglycemia and diarrhea [21, 22, 27, 28].
In addition, although Rbks gene revealed a high expression level in the liver, no overt liver impairment was observed after gene depletion (Fig. S4A, 4B). Also, ribose administration didn't alter the lipid metabolism in the liver (Fig. S4C - 4F). Similarly, no overt kidney impairments were observed after gene depletion (Fig. S5A, 5B).
Overall, our study has indicated a significant role of ribose in regulating intestinal motility and energy homeostasis and suggested ribose as a potential weight-loss drug targeting this new mechanism.
Materials and methods
Ethics Statement
All experiments were approved by and conducted in compliance with the Yale Animal Resources Center and the Institutional Animal Care and Use Committee under protocol number 11050 and 11250.
Generation of Rbks targeting mice
Rbks targeting embryonic stem (ES) cell clone was purchased from the European Mouse Mutant Cell Repository (EuMMCR, product ID:24874, clone number: EPD0116_3_A10) [34]. The Rbks mutated ES cell clones containing the Rbksgaleo allele were injected into C57BL/6 blastocysts to generate Rbksgaleo/+ chimeric mice. The chimeric Rbksgaleo/+ mice were identified by genotyping PCR using primer pairs Rbks-F (CAAAGGCGGCCATAGTCTGCAAGG) and Rbks-R (TGAACTGATGGCGAGCTCAGACC) to detect the loxP locus, and using 5'A (CCCAAACTTAGTACTTAGCGCTCC), 3'A (TATCAAGTGTGGGACTTGAAGAGG) and LAR3 (CAACGGGTTCTTCTGTTAGTCC) for the galeo cassette. Male Rbksgaleo/+ mice were bred with C57BL/6 female mice to produce stable Rbks targeting colonies. The galeo reporter cassette was depleted by mating with ubiquitously active CAG-flpe mice (Taconic, stock number: 7089) to generate RbksF/+ mice. In order to delete Rbks exon2 by cre-loxP recombination, RbksF/+ were bred with Actin-cre mice to produce Rbks∆2/+ heterozygous mutants. Homozygous Rbks gene targeting mice (Rbks ∆2/∆2) were obtained by heterozygous mating. The progenies were genotyped by PCR with 2F (TGAGTCTAAGTGCTGAGTGG) and 2R (GGTTCATAGAGGCAGAATGTAG) to detect the exon2-depleted allele, and with 5'A and 3'A to detect the exon2 wild type allele.
RT-PCR
Total RNAs were isolated from progenitors of Rbks∆2/+ mice heterozygous mating. The RNA samples were treated with TURBO DNA-free kit (Life tech, AM1907) to digest the genomic DNA contamination. cDNA templates were prepared using iScript cDNA synthesis kit (BIORAD, 170-8891) and amplified with the combination of specific primers for Rbks exon2 and exon7, or for 18s rRNA as an internal control. The primer pairs are: Rbks-RT-F (TCGCTTGCCAAAAACTGG) and Rbks-RT-R (TGGTGTCTACAGCCTTGACAG) for Rbks gene, and 18s-F (AGGGGAGAGCGGGTAAGAGA) and 18s-R (GGACAGGACTAGGCGGAACA) for 18s rRNA.
Quantitative real-time PCR
To detect the expression pattern of the Rbks gene, total RNAs were isolated from C57BL/6 mice (stock no. 000664, Jackson lab). The candidate tissues were stomach, duodenum, jejunum, ileum, cecum, colon, kidney, heart, fat, liver, testis, thymus, brain, lung and spleen. cDNAs were prepared using iScript cDNA synthesis kit (BioRad) to serve as templates for the quantitative real-time (qRT) PCR. The qRT PCR was performed on step one real-time PCR system (Applied biosystem) with iTaq universal SYBR green supermix (BioRad). Primers to detect Rbks gene located on exon5 (GCCGAGCCAAAGTGATGATATG) and exon6 (TGGAGAGGGTATAGAACTGGGG). 18s rRNA was used as a baseline standard for qRT PCR to normalize the samples. The expression level of Rbks in each tissue was normalized by the folds of gene expression level in kidney. For the measurements of the lipid metabolism gene expression level, total RNAs were isolated from the liver. Primer sequences for the lipid metabolism genes are as followed: CD36-L1 (ATGGGCTGTGATCGGAACTG); CD36-R1 (TTTGCCACGTCATCTGGGTTT); Cpt1a-L1 (CTCCGCCTGAGCCATGAAG); Cpt1a-R1 (CACCAGTGATGATGCCATTCT); LPL-L1 (GGGAGTTTGGCTCCAGAGTTT); LPL-R1 (TGTGTCTTCAGGGGTCCTTAG).
Whole mount X-gal staining
The tissue whole mount X-gal staining was followed by the previous protocol with a little modification [35]. Rbksgaleo/galeo mice and the wild type control tissues were isolated immediately after euthanization. After being briefly rinsed in the ice cold PBS, tissues were fixed by 4% PFA for 1 hour on ice, and washed with the wash buffer (2mM MgCl2, 0.02% NP40, 0.05% deoxycholate, PBS, pH7.4) for three times, 10 minutes each on ice. Tissues were immersed in the X-gal staining buffer (20 mg/ml X-gal (in DMSO), 5mM potassium ferrocyanide and 5mM potassium ferricyanide in wash buffer) in 37 ºC until blue signals were observed in Rbksgaleo/galeo mice tissues. The control wild type tissues were incubated in the staining buffer for the same time as the Rbksgaleo/galeo tissues (e.g. wild type kidney was treated for the same time with the Rbksgaleo/galeo kidney). For paraffin sections preparation, the X-gal stained tissues were briefly rinsed in ice cold PBS, and then post-fixed in 4% PFA in 4 ̊C overnight. The paraffin sections were cut at 10μm. Nuclear fast red was used for counterstaining.
Measurement of small intestine length
Male Rbks∆2/∆2 mice (9 weeks old) were euthanized by cervical dislocation. After recording body weights, the small intestine was isolated. The mesentery was carefully removed without stretching the small intestine. The total length of the small intestine was measured from the pyloric junction to the caecal end.
Measurement of ribose/ribose-5-phosphate ratio
Male mice (8~10 weeks) small intestine specimens were briefly washed with ice cold PBS and fully diced in a 2 ml Eppendorf tube by scissors. After the addition of ice cold 75% methanol (500 μl for 100 mg tissues), the tissues were homogenized at 30 Hz for 4 minutes using 5 mm stainless steel balls on a tissuelyser (Tissuelyser LT, Qiagen), followed by centrifugation at 12500 rpm for 10 minutes in 4 °C. The clear supernatants were diluted in 75% methanol by 50 times for measurements. The ribose and ribose-5-phosphate ratio was detected by the mass spectrometer (4000 QTRAP LC/MS/MS, AB SCIEX), using ribose (Sigma-Aldrich) and ribose-5-phoshpate (Sigma-Aldrich) dilutions to set up the standard curve.
Whole gut transition time
The whole gut transit time was measured following the previous report with a little modification [36]. Male mice (8 weeks) were fasted for 18 hours before the experiment with free access to water. The carmine dye was employed as a marker, and 0.3 ml carmine solution (6% carmine in 0.5% methylcellulose) was orally administered to each mouse. After oral gavage, mice were returned to the individually housed mesh bottom cages, which were placed on a white sheet. Whole gut transit time is the time taken from carmine meal gavage to the appearance of the first red fecal pellets. For wild type mice experiments, C57BL/6 mice (The Jackson laboratory, stock number 000664) were orally treated with ribose (50 mg/ml, 75 mg/ml), xylose (75 mg/ml) or ddH2O mixed with the carmine meal.
Gastrointestinal transition rate
The gastrointestinal transition rate was measured using the charcoal gavage as described before [36], with some modifications. Male mice (8 weeks) were fasted individually for 18 hours with free access to water, and then received an orally force feeding with 0.15 ml of a suspension of charcoal meal (10% charcoal in 5% gum arabic). Thirty minutes after the gavage, mice were euthanized by cervical dislocation. The stomach weights were recorded. The small intestine was removed from the pyloric junction to the caecal end. Gastrointestinal transition rate was the leading edge of charcoal transition length divided by the total length of small intestine. For wild type mice experiments, C57BL/6 mice were orally treated with ribose (25 mg/ml, 50 mg/ml, 75 mg/ml), glucose (50 mg/ml) or ddH2O mixed with the charcoal meal.
Ribose long-term feeding trail
C57BL/6 male mice (8 weeks) were housed individually and were divided into 4 groups, receiving drinking water with ribose (10 mg/ml, 25 mg/ml, 50 mg/ml, 75 mg/ml) or ddH2O for 5 weeks. Mice were fed with high fat diets. The body weights and the amount of food consumption were monitored every week. After 3 weeks treatments, mice were transferred to individually-house clean cages for one day housing. The fecal pellets were collected and dried in 56 ºC for 24 hours. The dried fecal pellets were used for fecal energy test using C500 calorimeter (IKA). Mice were returned to the ribose treating trial for another 2 weeks. After 5 weeks ribose drinking in total, mice were euthanized by cervical dislocation. The blood glucose levels were recorded and the 3.5 cm distal rectums were isolated. The rectum fecal contents were collected to measure the fecal number and fecal wet weight. After being dried in 56 ºC for 24 hours, the rectum fecal content dry weights were recorded. Fecal water content (%) = (wet weight-dry weight)/wet weight×100. After 5 weeks ribose drinking, the gut motility and fecal outputs were measured. Gut motility was indicated by measuring the whole gut transit time using the method described above. The fecal pellets within 5 hours were collected and dried in 56 ºC for 24 hours to measure the dry weights. When testing ribose in obese mice, Lepr mutants were used as the obese model. Lepr mutant male mice were generated by Piggybac mutagenesis system in FVB background. 6 weeks old male mutants received 75 mg/ml ribose via drinking water for 48 days and the body weights were monitored every four days. Body composition (fat mass) was measured before and after the 48 days drinking trial.
The liver triglyceride level measurements
The liver triglyceride level was measured followed the manufacture's protocol (Meilun, China). The triglyceride level (mmol) was normalized by the tissue weight (g).
Statistics analyses
All data, presented as average ± s.e.m. were analyzed with GraphPad Prism 6. Unpaired Student's t test was used to determine differences between groups in each panel. A value of * (p<0.05) or ** (p<0.01) denoted statistical significance.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Yale Core Center and animal genomics services for Musculoskeletal Disorders for histology sections and transgenic injection, the Xu lab members for critical reading of the manuscript. Y.L. is a Yale exchange pre-doctoral fellow from Fudan University supported by China Scholarship Council. T.X. is a Howard Hughes Medical Institute investigator.
Competing interests
Yan Liu, Tong-Ruei R Li and Cong Xu are employees of Shanghai Yao Yuan Biotechnology (Drug Farm) Limited, Co.
References
1. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840-6
2. Wilson PW, D'Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Archives of internal medicine. 2002;162:1867-72
3. Han GM, Gonzalez S, DeVries D. Combined effect of hyperuricemia and overweight/obesity on the prevalence of hypertension among US adults: result from the National Health and Nutrition Examination Survey. Journal of human hypertension. 2014;28:579-86
4. Lavie CJ, McAuley PA, Church TS, Milani RV, Blair SN. Obesity and cardiovascular diseases: implications regarding fitness, fatness, and severity in the obesity paradox. Journal of the American College of Cardiology. 2014;63:1345-54
5. Manning S, Pucci A, Finer N. Pharmacotherapy for obesity: novel agents and paradigms. Therapeutic advances in chronic disease. 2014;5:135-48
6. Bray GA. Medical treatment of obesity: the past, the present and the future. Best practice & research Clinical gastroenterology. 2014;28:665-84
7. Nathan PJ, O'Neill BV, Napolitano A, Bullmore ET. Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS neuroscience & therapeutics. 2011;17:490-505
8. Fleming JW, McClendon KS, Riche DM. New obesity agents: lorcaserin and phentermine/topiramate. The Annals of pharmacotherapy. 2013;47:1007-16
9. Shyh G, Cheng-Lai A. New antiobesity agents: lorcaserin (Belviq) and phentermine/topiramate ER (Qsymia). Cardiology in review. 2014;22:43-50
10. Richelsen B, Tonstad S, Rossner S, Toubro S, Niskanen L, Madsbad S. et al. Effect of orlistat on weight regain and cardiovascular risk factors following a very-low-energy diet in abdominally obese patients: a 3-year randomized, placebo-controlled study. Diabetes care. 2007;30:27-32
11. Filippatos TD, Derdemezis CS, Gazi IF, Nakou ES, Mikhailidis DP, Elisaf MS. Orlistat-associated adverse effects and drug interactions: a critical review. Drug safety. 2008;31:53-65
12. Zhi J, Melia AT, Koss-Twardy SG, Arora S, Patel IH. The effect of orlistat, an inhibitor of dietary fat absorption, on the pharmacokinetics of beta-carotene in healthy volunteers. Journal of clinical pharmacology. 1996;36:152-9
13. Melia AT, Koss-Twardy SG, Zhi J. The effect of orlistat, an inhibitor of dietary fat absorption, on the absorption of vitamins A and E in healthy volunteers. Journal of clinical pharmacology. 1996;36:647-53
14. MacWalter RS, Fraser HW, Armstrong KM. Orlistat enhances warfarin effect. The Annals of pharmacotherapy. 2003;37:510-2
15. Guan X. The CNS glucagon-like peptide-2 receptor in the control of energy balance and glucose homeostasis. American journal of physiology Regulatory, integrative and comparative physiology. 2014;307:R585-R96
16. Hara T, Kashihara D, Ichimura A, Kimura I, Tsujimoto G, Hirasawa A. Role of free fatty acid receptors in the regulation of energy metabolism. Biochimica et biophysica acta. 2014;1841:1292-300
17. Duraffourd C, De Vadder F, Goncalves D, Delaere F, Penhoat A, Brusset B. et al. Mu-opioid receptors and dietary protein stimulate a gut-brain neural circuitry limiting food intake. Cell. 2012;150:377-88
18. Heaf DJ, Davies JI. The effect of RNA supplementation of rat diets on the composition of body fluids. The British journal of nutrition. 1976;36:381-402
19. Omran H, Illien S, MacCarter D, St Cyr J, Luderitz B. D-Ribose improves diastolic function and quality of life in congestive heart failure patients: a prospective feasibility study. European journal of heart failure. 2003;5:615-9
20. MacCarter D, Vijay N, Washam M, Shecterle L, Sierminski H, St Cyr JA. D-ribose aids advanced ischemic heart failure patients. International journal of cardiology. 2009;137:79-80
21. Gross M, Reiter S, Zollner N. Metabolism of D-ribose administered continuously to healthy persons and to patients with myoadenylate deaminase deficiency. Klinische Wochenschrift. 1989;67:1205-13
22. Zollner N, Reiter S, Gross M, Pongratz D, Reimers CD, Gerbitz K. et al. Myoadenylate deaminase deficiency: successful symptomatic therapy by high dose oral administration of ribose. Klinische Wochenschrift. 1986;64:1281-90
23. Dodd SL, Johnson CA, Fernholz K, St Cyr JA. The role of ribose in human skeletal muscle metabolism. Medical hypotheses. 2004;62:819-24
24. Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein science: a publication of the Protein Society. 1993;2:31-40
25. Agranoff BW, Brady RO. Purification and properties of calf liver ribokinase. The Journal of biological chemistry. 1956;219:221-9
26. van der Burg JM, Winqvist A, Aziz NA, Maat-Schieman ML, Roos RA, Bates GP. et al. Gastrointestinal dysfunction contributes to weight loss in Huntington's disease mice. Neurobiology of disease. 2011;44:1-8
27. Segal S, Foley J, Wyngaarden JB. Hypoglycemic effect of D-ribose in man. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine (New York, NY). 1957;95:551-5
28. Gross M, Zollner N. Serum levels of glucose, insulin, and C-peptide during long-term D-ribose administration in man. Klinische Wochenschrift. 1991;69:31-6
29. Segal S, Foley J. The metabolism of D-ribose in man. The Journal of clinical investigation. 1958;37:719-35
30. Lee SR, Britton WM. Magnesium-induced catharsis in chicks. The Journal of nutrition. 1987;117:1907-12
31. Hammer HF, Santa Ana CA, Schiller LR, Fordtran JS. Studies of osmotic diarrhea induced in normal subjects by ingestion of polyethylene glycol and lactulose. The Journal of clinical investigation. 1989;84:1056-62
32. Griffiths JC, Borzelleca JF, St Cyr J. Sub-chronic (13-week) oral toxicity study with D-ribose in Wistar rats. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2007;45:144-52
33. Griffiths JC, Borzelleca JF, St Cyr J. Lack of oral embryotoxicity/teratogenicity with D-ribose in Wistar rats. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2007;45:388-95
34. Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337-42
35. Constance Cepko ER, Donna M. Fekete, Suzanne Bruhn. Detection of ß-galactosidase and alkaline phosphatase activities in tissue. http://genepath.med.harvard.edu/~cepko/protocol/xgalplap-stain.htm
36. Nagakura Y, Naitoh Y, Kamato T, Yamano M, Miyata K. Compounds possessing 5-HT3 receptor antagonistic activity inhibit intestinal propulsion in mice. European journal of pharmacology. 1996;311:67-72
Author contact
![]() Corresponding author: tian.xuedu, +1-(203) 887-6295.
Corresponding author: tian.xuedu, +1-(203) 887-6295.