Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(10):1155-1167. doi:10.7150/ijbs.14654 This issue Cite
Research Paper
Class A Scavenger Receptor Exacerbates Osteoclastogenesis by an Interleukin-6-Mediated Mechanism through ERK and JNK Signaling Pathways
Shuyu Guo1,*, Yuanyuan Ni1,*, Jingjing Ben2,*, Yang Xia1, Tingting Zhou1, Dongyue Wang1, Jieli Ni1, Hui Bai2, Lin Wang1, Junqing Ma1 ![]() , Qi Chen2
, Qi Chen2 ![]()
1. Jiangsu Key Laboratory of Oral Diseases, Nanjing Medical University, Nanjing, China.
2. Atherosclerosis Research Center, Key Laboratory of Cardiovascular Disease and Molecular Intervention, Nanjing Medical University, Nanjing, China.
* SG, YN and JB contributed equally to this work.
Received 2015-12-11; Accepted 2016-5-27; Published 2016-8-26
Abstract

Osteoclasts originate from bone marrow monocyte/macrophage lineage cells, which are important for bone health. Class A scavenger receptor (SR-A) is a multifunctional molecule that functions during differentiation of monocyte into macrophages and osteoclasts. To further characterize the role of SR-A in osteoclasts, we used the murine tooth movement model (TM) and the murine anterior cruciate ligament transection model of osteoarthritis (ACLT OA). In these two models the bones involved are of different origin and have different properties. Bone resorption was decreased in SR-A-/- mice compared to SR-A+/+ mice. Further evaluation showed that the number of multinucleated osteoclasts in SR-A-/- mice, compared to SR-A+/+ mice, was significantly decreased both in vivo and in vitro. The levels of interleukin-6 (IL-6) produced by osteoclasts were reduced in SR-A-/- mice compared to SR-A+/+ mice. In the in vitro marrow-derived osteoclast formation assay and in both mouse models, osteoclastogenesis was restored to normal in SR-A-/- mice by administration of recombinant murine IL-6. Moreover, neutralization of IL-6 reduced the number of osteoclasts formed in SR-A+/+ mice of TM model. Both extracellular signal-regulated kinase (ERK) and c-Jun N-terminal protein kinase (JNK), but not p38, signaling pathways were downregulated in receptor activator of nuclear factor-κB ligand (RANKL)-stimulated SR-A-/- osteoclasts. Importantly, when treated with either ERK or JNK inhibitor, the numbers of osteoclasts generated from RANKL-induced bone marrow derived-macrophages of SR-A+/+ mice, and their IL-6 production, were significantly decreased. This suggests that SR-A activates the ERK and JNK signaling pathways, and promotes production of IL-6 by osteoclasts to further stimulate osteoclast formation.
Keywords: Osteoclasts, SR-A, IL-6, ERK, JNK.
Introduction
Bone homeostasis, which exists throughout a vertebrate's whole life, is accomplished by the balance of bone deposition by osteoblasts and bone resorption by osteoclasts [1, 2]. Bone homeostasis is associated with many activities and pathologies of the skeletal system, such as the onset of osteoarthritis and bone remodeling during orthodontic tooth movement. Osteoclasts are the primary bone resorbing cells derived from monocyte/macrophage lineage precursors [3]. Differentiation of murine bone marrow cells into osteoclasts requires two cytokines: macrophage-colony stimulating factor (M-CSF) produced by bone marrow cells, and receptor activator of nuclear factor-κB ligand (RANKL) produced by osteoblasts [4, 5]. M-CSF is essential for differentiation of macrophages, while RANKL supports cellular fusion to generate mature multinucleated osteoclasts. RANKL binds to RANK on osteoclast precursors, activating downstream signaling pathways, such as the mitogen activating protein kinase (MAPK) signaling pathway including c-Jun N-terminal protein kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 [6].
Class A scavenger receptor (SR-A) is one of the earliest characterized scavenger receptors. SR-A is a trimeric membrane glycoprotein, which is a multifunctional macrophage receptor that functions during monocyte differentiation into macrophages [7]. A range of substances has been reported as ligands for SR-A, which can influence a variety of functions in macrophages. Because osteoclasts are derived from the monocyte/macrophage lineage, the function of SR-A in osteoclast development is of potential interest. It has been reported that SR-A regulates osteoclastogenesis-related molecules such as RANK, nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1), and microphthalmia-associated transcription factor (MITF) [8]. SR-A is involved in some important pathophysiological processes, including inflammation, cell apoptosis, migration and adhesion [9-12]. SR-A is expressed in most macrophages and mononuclear osteoclast precursor cells. Moreover, osteoclastogenesis is reduced in SR-A-/- mice [8, 13]. Our research was designed to evaluate the functions and underlying molecular mechanisms of SR-A action on osteoclastogenesis using two murine experimental models: the tooth movement model (TM) and the anterior cruciate ligament transection model of osteoarthritis (ACLT OA).
In the present study, we have elucidated that SR-A on progenitor cells promotes osteoclast differentiation by activating the ERK and JNK signaling pathways, thereby stimulating the production of interleukin-6 (IL-6), which further promotes osteoclast differentiation.
Materials and Methods
Animals
Mice deficient in type I and II SR-A were generated by disrupting exon 4 of SR-A, which is essential for trimeric receptor function, as described previously [14, 15]. The animals were maintained in the Nanjing Medical University animal facility with free access to mouse chow and water. All mice were handled in accordance with protocols approved by the Experimental Animal Care and Use Committee of Nanjing Medical University (2012-01-16).
Experimental protocol
Induction of tooth movement was based on previous work [16]. In short, an orthodontic appliance consisting of a Ni-Ti 0.25 x 0.76 mm coil spring (Grikin, Beijing, China) was bonded by dental adhesive resin (Transbond, Unitek/3M, CA, USA) between the upper right first molar (M1) and the upper incisors (UI), exerting a force of 0.35 N in the mesial direction. The left side was used as an internal control. The mice (10 week old) were euthanized after 7 and 14 days of force application. At least 5 mice were used for each time-point for every set of experiments.
OA was surgically induced by transection of the anterior cruciate ligament of the right knee joint of 10 week old, male mice [17]. All surgical procedures were performed using a stereoscopic microscope (SMZ1000, Nikon, Tokyo, Japan). Five ACLT OA and five sham controls (10 week old) were sacrificed at 14, 28 and 56 days after surgery.
Micro-CT analysis
To analyze the degree of bone resorption, we dissected the tibia and the maxillary bone free of soft tissue, fixed them overnight in 70% ethanol and scanned at high resolution (18 μm) using a micro-CT system (Skyscan 1176, Kontich, Belgium) at 50 kV and 456 μA. Images were reconstructed and analyzed by NRecon v1.6 and CTAn v1.13.8.1 software. We defined the region of interest (ROI) as the area of alveolar bone between the roots of the first maxillary molar in the TM model and the whole subchondral bone compartment of the medial tibial plateau in the ACLT OA model. We used a total of five consecutive images from the ROI for three-dimensional reconstruction and analysis. The parameters analyzed included: trabecular bone volume per total volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), and trabecular thickness (Tb.Th). The scan analysis operator was blinded to the treatments associated with each sample.
Histological analysis
Hind limbs or maxillary halves from both the operated side and the contralateral side of each animal were resected and fixed in 10% buffered formalin for 24 hours, decalcified in 10% EDTA (pH 7.4) for 21 days, embedded in paraffin and cut into sections of 6 μm thickness. The sections were stained with tartrate resistant acid phosphatase (TRAP), Goldner's trichrome, and Safranin O staining. The protocol was based on a published protocol [18]. TRAP-positive multinucleated cells containing more than 3 nuclei were counted (n = 5). Two examiners blinded to group status analyzed the slides.
The TM measurements were based on previous work [19]. We located the sagittal image plane that revealed the most root structure, and then magnified the images to precisely draw the lines. The line was drawn between the points of closest proximity of the two convex molar crown surfaces. We selected three random sagittal images of each animal for measurement, and then took the average of the results as the final measurement. Osteoclast and osteoblast counts were performed for positive cells attached to the alveolar bone surrounding the distal root in the TM model or within the area of subchondral bone of the tibia in the ACLT OA model [20, 21]. In Safranin O stained sections, the histologic scoring system for severity of cartilage destruction in osteoarthritis was applied according to the 0 ~ 6 score of Chambers et al. [22]. The severity was determined as the maximal structure score by calculating the average scores of five slides.
Mouse osteoclast culture protocol
Four-week-old SR-A+/+ and SR-A-/- mice were euthanized, and bone marrow cells were extracted from tibias and femurs as described previously [23]. Bone marrow cells flushed from the long bones were cultured in α-minimum essential medium (α-MEM) for 3 hours to remove adherent cells, and non-adherent cells were seeded into new plates and cultured in α-MEM with M-CSF (20 ng/ml, Sigma, MO, USA) for 3 days, resulting in bone marrow-derived macrophages (BMMs). BMMs were used for osteoclastogenesis by culturing them with M-CSF (20 ng/ml) and RANKL (20 ng/ml, Sigma, MO, USA). For immunoblot analysis, cells were then stimulated with 20 ng/ml M-CSF and RANKL for 0, 15, or 30 minutes at a density of 1 x 106/well in a 6-well plate. For TRAP-activity staining, cells were cultured with M-CSF (20 ng/ml) and RANKL (20 ng/ml) for 6 days at a density of 2 x 105 cells/well in a 24 well plate. For analysis of the function of IL-1 and IL-6 in SR-A-induced osteoclastogenesis, BMMs were cultured with M-CSF and RANKL in the presence of recombinant murine IL-1 (20 ng/ml, Peprotech, USA) or recombinant murine IL-6 (20 ng/ml, Peprotech, USA) for 6 days, and then stained for TRAP. For testing the function of MAPK signaling pathways, BMMs were cultured with added M-CSF (20 ng/ml) and RANKL (20 ng/ml) for 6 days in the absence or presence of ERK inhibitor (U0126, 5 μM, Cell Signaling Technology, MA, USA), JNK inhibitor (SP600125, 10 μM, Cell Signaling Technology), and then stained for TRAP. The protocol for in vitro TRAP staining was described in detail previously [24]. TRAP-positive multinucleated cells containing more than 3 nuclei were counted in 5 visual fields at 20× magnification (n = 3).
Quantitative reverse transcription PCR for mRNA analysis
Total RNA was isolated from osteoclasts using the TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer's protocol. The mRNA was converted to cDNA using a PrimeScript RT reagent kit. The gene expression level was analyzed by quantitative reverse transcription PCR (qRT-PCR) using the ABI-7300 Real-Time PCR System (Applied Biosystems, CA, USA). The primers used are listed below: GAPDH, 5'-GAAGGTGAAGGTCGGAGTC-3' and 5'-GAGATGGTGATGGGATTTC-3'; IL-1, 5'-GAAATGCCACCTTTTGACAGTG-3' and 5'-CTGGATGCTCTCATCAGGACA-3'; IL-6, 5'-GAGGATACCACTCCCAACAGACC-3', and 5'-AAGTGCATCATCGTTGTTCATACA-3'; TRAIL, 5'-ATGGTGATTTGCATAGTGCTCC-3' and 5'-GCAAGCAGGGTCTGTTCAAGA-3'; VEGF-C, 5'-CAGTGCATGAACACCAGCACA-3' and 5'-TAGACATGCACCGGCAGGAA-3'; LIF, 5'-TTGATCCCGACTCAAGCAACC-3' and 5'-CTGAAGCCGCTACCATGCAA-3'; IGF-1, 5'-TCACTGCCCAATTGAAATACGA-3' and 5'-TTAGGCCCAGACAGTTTAAACAAAG-3'; TSG-6, 5'-GGGATTCAAGAACGGGATCTTT-3' and 5'-TCAAATTCACATACGGCCTTGG-3'; TNF-α, 5'-CTGTGAAGGGAATGGGTGTT-3' and 5'-GGTCACTGTCCCAGCATCTT-3'; ECF-L, 5'-CAGGTCTGGCAATTCTTCTGAA-3' and 5'-GTCTTGCTCATGTGTGTAAGTGA-3'.
Enzyme-linked immunosorbent assay (ELISA)
BMMs were cultured with M-CSF (20 ng/ml) and RANKL (20 ng/ml) for 24 hours in the absence or presence of the ERK inhibitor (U0126, 5 μM, Cell Signaling Technology, MA, USA), the JNK inhibitor (SP600125, 10 μM, Cell Signaling Technology), or the p38 inhibitor (SB203580, 1 μM, Cell Signaling Technology). Then the supernatants of cells were collected, and the levels of IL-1 and IL-6 were determined using quantikine mouse interleukin-1 (IL-1) and interleukin-6 (IL-6) ELISA kits (Multisciences, Zhejiang, China) according to the manufacturer's instructions.
IL-6 injection
Two days after induction of tooth movement and experimental ACLT OA, SR-A+/+ and SR-A-/- mice were injected with 10 μl recombinant murine IL-6 (1 μg/ml, Peprotech, USA) dissolved in phosphate buffered saline (PBS), or 10 μl PBS only under the buccal periosteum of the first molar in the TM model at day 2, 4 and 6, or into the articular cavity of the knee-joint in the ACLT OA model at day 2, 4, 6, 8, 10, and 12.
IL-6 blocking
After induction of tooth movement, SR-A+/+ and SR-A-/- mice were randomized into two groups and received an intraperitoneal injection of monoclonal rat anti-mouse IL-6 antibody (1mg/kg, R&D Systems, MAB406) at day 2, 4 and 6. In the control group, mice received an injection of nonspecific rat IgG (R&D Systems, 6-001-A) at the same dose.
Immunoblot analysis
MAPK signaling pathway was analyzed in protein samples extracted from in vitro cultures of osteoclasts. Proteins were prepared in protein lysis buffer, resolved on SDS/PAGE, and electro-transferred onto polyvinylidene difluoride membranes. The details of the Western blot protocol were described previously [25]. Antibodies against p-p38, p38, p-JNK, JNK, p-ERK, ERK (Cell Signaling Technology), and β-actin (Santa Cruz, CA, USA) were used as primary antibodies. Semi-quantitative measurements were carried out by Image J software.
Statistical analysis
All values were expressed as means ± SD. Experiments were conducted separately at least 3 times. Statistical analysis was performed using Student's t-test (SPSS 19.0), and P values of less than 0.05 were considered statistically significant.
Results
Lack of SR-A is associated with reduced bone loss
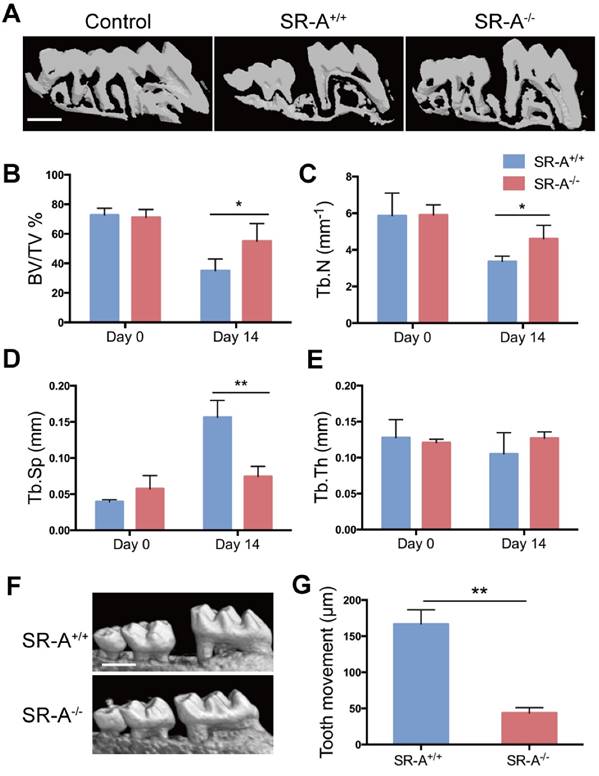
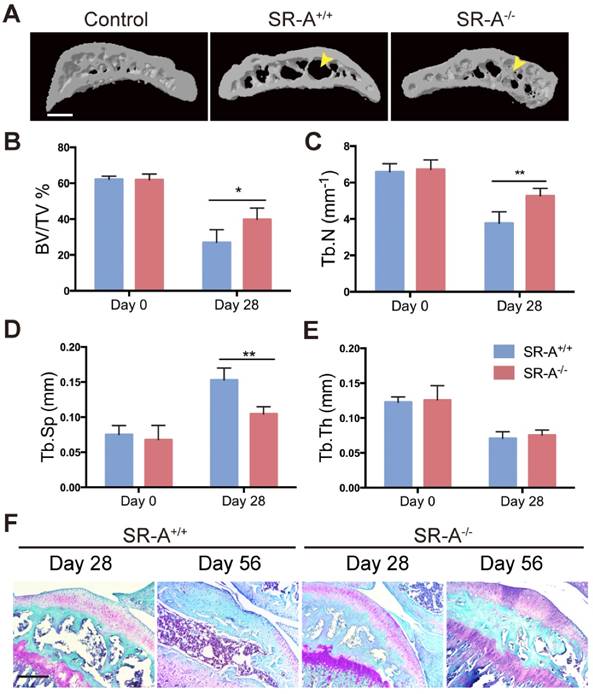
To examine the function of SR-A on bone homeostasis, two animal models TM and ACLT OA were chosen, and both wild-type and SR-A-/- mice were used. Micro-computed tomography (micro-CT) was performed to observe the bone architecture and quantitatively analyze the relevant parameters of alveolar and tibial subchondral bone. In the TM model, SR-A+/+ mice exhibited a marked decrease of alveolar bone within the roots of the first molar at day 14, while there were smaller effects on alveolar bone in SR-A-/- mice (Figure 1A). SR-A-/- mice showed an increased BV/TV value (bone volume over total volume, P < 0.05, Figure 1B), Tb.N value (trabecular number, P < 0.05, Figure 1C), and reduced Tb.Sp value (trabecular separation, P < 0.01, Figure 1D). The tooth movement in SR-A-/- mice was much smaller than in SR-A+/+ mice (P < 0.01, Figure 1F, G). A similar phenotype appeared in the ACLT OA model. At 28 days, SR-A-/- mice displayed a smaller subchondral bone resorption area (Figure 2A), the BV/TV (P < 0.05) and Tb.N values (P < 0.01) were increased, and the Tb.Sp (P < 0.01) value was decreased in SR-A-/- mice compared to wild-type mice (Figure 2B, C, D). However, there was no significant difference between the Tb.th (trabecular thickness) values in SR-A-/- and in SR-A+/+ mice in both experimental models (Figure 1E, 2E). Loss of proteoglycan in cartilage manifested at 28 days and was aggravated at 56 days in SR-A+/+ mice, while there was no significant change in SR-A-/- mice. There was much less cartilage destruction in SR-A-/- mice than in SR-A+/+ mice (Figure 2F). Collectively, these data point to a positive role of SR-A on bone resorption.
Mice with SR-A deletion have reduced bone resorption and distance of tooth movement in the TM model. (A) Three-dimensional micro-CT image of alveolar bone (sagittal view) at 14 days after the operation inducing tooth movement. The maxilla without operation was used as control. Scale bars equal 500 μm. (B-E) Quantitative analyze of bone volume over total volume (BV/TV), and trabecular number (Tb.N), separation (Tb.Sp), and thickness (Tb.Th), of alveolar bone by micro-CT. The blue bar represents the SR-A+/+ mice, while the red bar represents the SR-A-/- mice. (F) Micro-CT image of alveolar bone at 14 days after TM. Scale bars equal 500 μm. (G) Quantitative analysis of the distance of the first molar mesial movement in SR-A-/- and SR-A+/+ mice. *P < 0.05, **P < 0.01 compared to the SR-A+/+ group at the corresponding time points. All data are shown as the mean ± SD.
Mice with SR-A deletion have changes in subchondral bone architecture in the ACLT OA model. (A) Three-dimensional micro-CT image of tibial subchondral bone medial compartment (sagittal view) at 28 days after sham operation or ACLT surgery. Yellow arrows indicate altered bone resorption of the SBP (subchondral bone plate). Scale bars equal 500 μm. (B-E) Quantitative analysis of the parameters of subchondral bone by micro-CT. Blue bars designate SR-A+/+ mice, while red bars designate SR-A-/- mice. n = 5 in each group. (F) Safranin O staining of sagittal sections of the medial compartment of the tibia at 28 and 56 days. Red stain indicates proteoglycan, and blue stain indicates bone. Scale bars equal 500 μm. *P < 0.05, **P < 0.01 compared to the SR-A+/+ group at the corresponding time points. All data are reported as the mean ± SD.
SR-A deletion suppressed osteoclast differentiation
To further explore our hypothesis, we examined histological differences between the two experimental models to determine whether SR-A deletion may directly influence bone cell activities. The number of osteoclasts was examined in SR-A+/+ and SR-A-/- mice in both the TM (Figure 3A, B) and ACLT OA model (Figure 3C, D). In both cases, the number of tartrate-resistant acid phosphatase (TRAP)-positive cells was significantly higher in SR-A+/+ mice (P < 0.01, Figure 3A, B, C, D). Moreover, the size of TRAP-positive osteoclasts was much smaller in SR-A-/- mice. Furthermore, we found no significant differences in osteoblast numbers between SR-A+/+ and SR-A-/- mice (Figure 3A, B, C, D).
SR-A deletion affects osteoclastogenesis in vivo and in vitro. (A) TRAP (top) and Goldner's trichrome staining (bottom) of osteoclasts and osteoblasts in alveolar bone of mice 7 days after surgery inducing tooth movement. Scale bars equal 100 μm. (B) Quantitative analysis of the number of TRAP+ cells and osteoblasts along the mesial area of the distal root of the first molar. Oc N/BPm (/mm), osteoclast number per bone perimeter; Ob N/BPm (/mm), osteoblast number per bone perimeter; Oc.S/BS, osteoclast surface per bone surface; Ob.S/BS, osteoblast surface per bone surface. (C) TRAP (top) and Goldner's trichrome staining (bottom) of osteoclasts and osteoblasts in subchondral bone of mice 14 days after ACLT surgery. Scale bars equal 100 μm. (D) Quantification of the number of TRAP+ cells and osteoblasts per section within subchondral bone is shown on the right. (E) TRAP activity staining of differentiated osteoclasts in vitro. Scale bars equal 50 μm. (F) Quantification of TRAP-positive multinucleated cells in vitro is shown on the right. OC per field, osteoclast number per field. Blue bars designate SR-A+/+ mice, while red bars designate SR-A-/- mice. n = 5 in each group. **P < 0.01 compared to the SR-A+/+ group. All data are reported as mean ± SD.
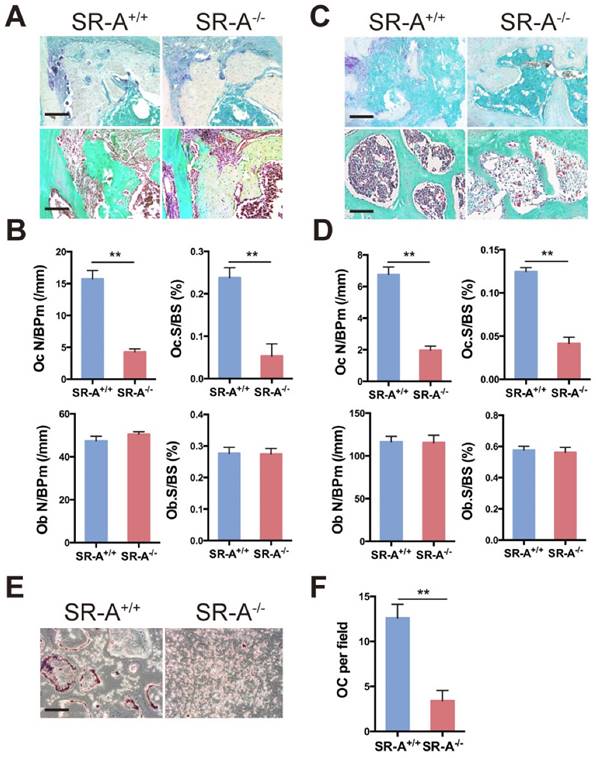
To analyze the role of SR-A in osteoclast differentiation in vitro, we cultured bone marrow cells from SR-A+/+ and SR-A-/- mice in the presence of M-CSF (20 ng/ml) for 3 days, and then continued to treat the cells with M-CSF and RANKL (20 ng/ml) for another 6 days. Compared with SR-A+/+ cells, most SR-A-/- bone marrow-derived osteoclasts were immature and small, and there were only a few multinucleated mature osteoclasts (Figure 3E). We then quantified the number of multinucleated osteoclasts and found that the number of multinucleated osteoclasts derived from the bone marrow of SR-A-/- mice was significantly lower than the number of multinucleated osteoclasts derived from the bone marrow of SR-A+/+ mice (Figure 3F).
SR-A deficiency reduced IL-1 and IL-6 production in osteoclasts
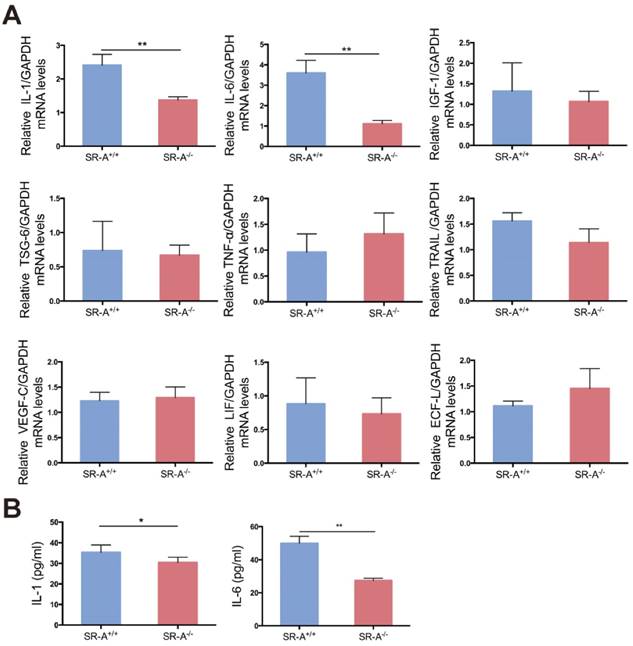
Some factors play a vital role in osteoclastogenesis [26-30], and it has been demonstrated that the function of SR-A is closely related to some of these factors in other biological processes [31-34]. In order to analyze the effect of SR-A on osteoclastogenesis, we performed real-time quantitative PCR analysis on nine factors of interest including interleukin-1 (IL-1), interleukin-6 (IL-6), insulin-like growth factor-1 (IGF-1), tumor necrosis factor-alpha-stimulated gene-6 (TSG-6), tumor necrosis factor-α (TNF-α), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), vascular endothelial growth factor-C (VEGF-C), leukemia inhibitory factor (LIF) and eosinophil chemotactic factor-L (ECF-L). As demonstrated in Figure 4A, IL-1 and IL-6 mRNA levels were significantly lower in osteoclasts of SR-A-/- mice compared to osteoclasts from SR-A+/+ mice. Consistent with our quantitative RT-PCR findings, we observed that the secretion of IL-1 and IL-6 protein from osteoclasts of SR-A-/- mice was significantly lower than from osteoclasts of SR-A+/+ mice as determined by enzyme-linked immunosorbent assay (ELISA) assay (Figure 4B).
IL-1 and IL-6 production is decreased in SR-A -/- osteoclasts. (A) The level of IL-1, IL-6, IGF-1, TSG-6, TNF-α, TRAIL, VEGF-C, LIF and ECF-L mRNA expression in osteoclasts was examined by real-time PCR at 24 hours of culture with M-CSF and RANKL after treatment with M-CSF for 3 days. (B) Concentrations of IL-1 and IL-6 protein in supernatant of osteoclasts determined by ELISA at 24 hours. Blue bars designate SR-A+/+ mice, while red bars designate SR-A-/- mice. n = 5 in each group. **P < 0.01 compared to the SR-A+/+ group. All data are reported as the mean ± SD.
Exogenous IL-6 rescued decreased osteoclastogenesis in SR-A-/- mice, while IL-6 blockade attenuated osteoclastogenesis in SR-A+/+ mice
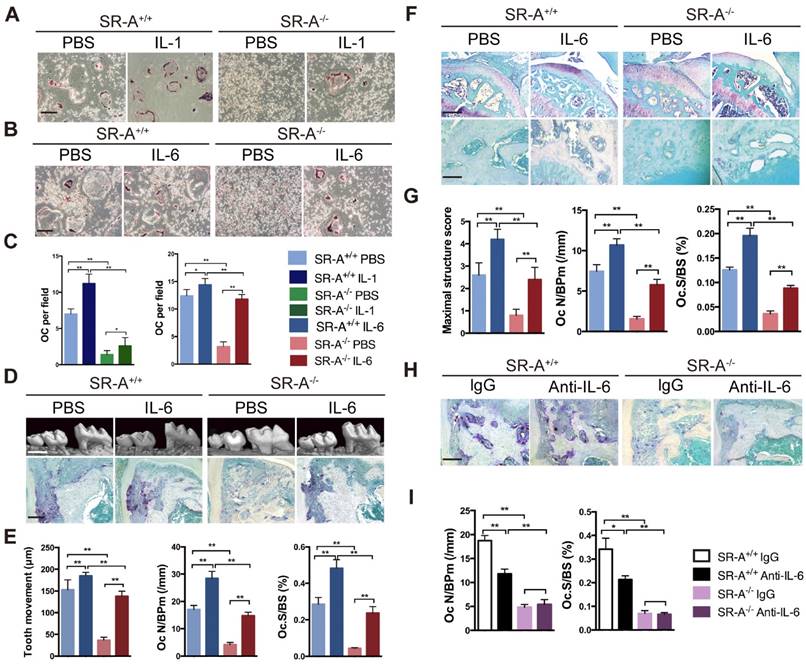
We evaluated whether the deficiency in osteoclastogenesis in SR-A-/- mice could be rescued by IL-1 or IL-6 restoration in vitro. We observed that the number of osteoclasts in the SR-A+/+ group increased after treatment with IL-1. In contrast, the number of osteoclasts in the SR-A-/- group treated with IL-1 increased not so obvious (Figure 5A, C). However, treatment with IL-6 significantly increased the number of osteoclast in SR-A+/+ group, and also rescued osteoclastogenesis in RANKL-induced SR-A-/- bone marrow cells (Figure 5B, C). IL-6 has been reported to regulate SR-A function in macrophages [31], so IL-6 was chosen for additional studies. We then performed IL-6 restoration experiments in vivo. In the TM model, the distance of the tooth movement increased after injection of recombinant murine IL-6, and exogenous IL-6 resulted in a remarkable increase in the number of osteoclasts, compared to phosphate buffered saline (PBS) injection in control mice (Figure 5D, E). In the ACLT OA model, severity of cartilage destruction and proteoglycan loss in SR-A-/- mice injected with IL-6 was much more pronounced than in SR-A-/- mice treated with PBS (Figure 5F, G). Moreover, the number of osteoclasts also increased in the IL-6 treatment group (Figure 5F, G). To obtain further prove for the role of IL-6 in osteoclast formation, we examined whether osteoclastogenesis was affected in SR-A+/+ and SR-A-/- mice subjected to the TM model after injection with either an anti-IL-6 antibody (1mg/kg) or control IgG (1mg/kg). As expected, neutralization of IL-6 reduced the number of osteoclasts formed in SR-A+/+ mice, but had no significantly effect in SR-A-/- mice (Figure 5H, I).
IL-6 rescues the phenotype induced by SR-A deletion, while IL-6 blockade inhibits osteoclastogenesis. (A) TRAP staining of bone marrow-derived osteoclasts in SR-A+/+ and SR-A-/- mice treated with PBS or IL-1. Scale bars equal 50 μm. (B) TRAP staining of bone marrow-derived osteoclasts in SR-A+/+ and SR-A-/- mice treated with PBS or IL-6. Scale bars equal 50 μm. (C) Quantitative analysis of bone marrow-derived osteoclasts in SR-A+/+ and SR-A-/- mice treated with IL-1 or IL-6 (PBS as control). n =5 per group. (D) Top, micro-CT image of alveolar bone of mice treated with PBS or IL-6 and analyzed 14 days after surgery inducing TM. Scale bars equal 500 μm. Bottom, TRAP staining of osteoclasts in alveolar bone in SR-A+/+ and SR-A-/- mice injected of IL-6 or PBS only and stained 7 days after surgery inducing TM. Scale bars equal 100 μm. (E) Quantitative analysis of the mesial movement of the first molar and the number of osteoclasts in SR-A+/+ and SR-A-/- mice treated with PBS or IL-6 and analyzed 7 or 14 days after surgery inducing TM. (F) Top, Safranin O staining of articular cartilage in sagittal sections of the tibial medial compartment of mice treated with PBS or IL-6 and analyzed 56 days after ACLT surgery. Scale bars equal 500 μm. Bottom, TRAP staining of osteoclasts in subchondral bone of mice treated with PBS or IL-6 and analyzed 14 days after ACLT surgery. Scale bars equal 100 μm. (G) Quantitative analysis of the maximal score for structural cartilage damage and the number of osteoclast in the tibial medial compartment of mice treated with PBS or IL-6 and analyzed 14 days or 56 days after ACLT surgery. (H) TRAP staining of osteoclasts in alveolar bone in SR-A+/+ and SR-A-/- mice injected with anti-IL-6 antibody or nonspecific rat IgG and stained 7 days after surgery inducing TM. Scale bars equal 100 μm. (I) Quantitative analysis of osteoclasts numbers in SR-A+/+ and SR-A-/- mice treated with anti-IL-6 antibody or nonspecific rat IgG and analyzed 7 days after surgery inducing TM. n = 5 per group. *P < 0.05, **P < 0.01 compared with SR-A-/- mice. All data are reported as the mean ± SD.
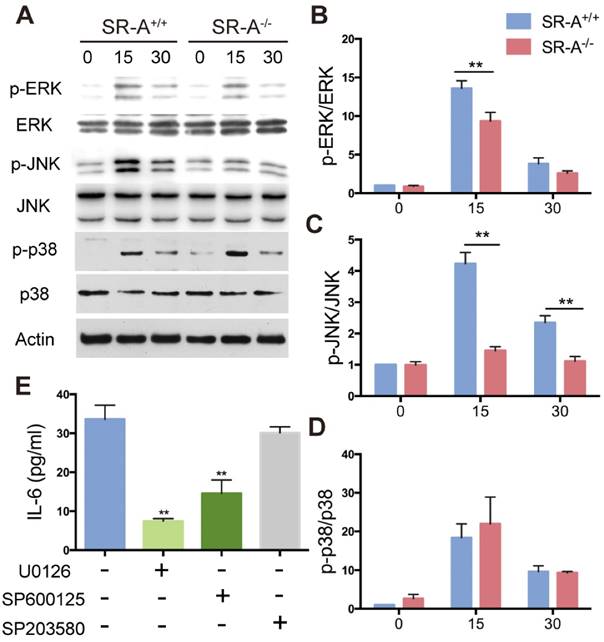
Alterations in RANKL-induced MAPK signaling in osteoclasts from SR-A-/- mice
Three subgroups of MAPKs (ERK, JNK and P38) are known to be involved in osteoclast development [5]. Western blotting was performed to investigate MAPKs signaling pathways and further clarify the molecular mechanisms through which SR-A gene disruption affected osteoclast differentiation. When stimulated with RANKL for 15 minutes, phosphorylation of ERK increased markedly in SR-A+/+ osteoclasts, which is in accordance with a previous report [8]. In contrast, phosphorylation of ERK in SR-A-/- osteoclasts was attenuated after 15 minutes of RANKL stimulation. We also found that phosphorylation of JNK increased markedly at 15 and 30 minutes after RANKL stimulation in SR-A+/+ osteoclasts. In contrast, phosphorylation of JNK in SR-A-/- osteoclasts was reduced after 15 or 30 minutes of RANKL stimulation. There was no significant difference in the phosphorylation of p38 in SR-A-/- and in SR-A+/+ osteoclasts after RANKL stimulation (Figure 6A). These observations were confirmed by quantitative analysis (Figure 6B, C, D). In good agreement with these observations, the level of IL-6 protein in supernatants of osteoclast cultures decreased after addition of ERK or JNK inhibitor, but not after addition of p38 inhibitor (Figure 6 E).
SR-A promotes production of IL-6 by RANKL-induced activation of ERK and JNK signaling pathways. (A) BMMs of SR-A+/+ and SR-A-/- mice were seeded at 1 x 106 cells per well in 6-well plates treated with M-CSF (20 ng·ml-1) for 3 days and then stimulated with RANKL (20 ng·ml-1) for 0, 15, or 30 minutes. Protein levels of p-ERK, ERK, p-JNK, JNK, p-p38, and p38 were determined by Western blotting, and β-actin was used as a control. (B, C, D) p-ERK, p-JNK, and p-p38 levels were determined by normalizing for total ERK, JNK, p38 expression. (E) Supernatants of osteoclast cultures treated with ERK (U0126, 5 μM), JNK (SP600125, 10 μM) or p38 inhibitor (SB203580, 1 μM) were collected and analyzed for IL-6 production by ELISA. n = 3 - 5 in each group. * P < 0.05, **P < 0.01. All data are reported as the mean ± SD.
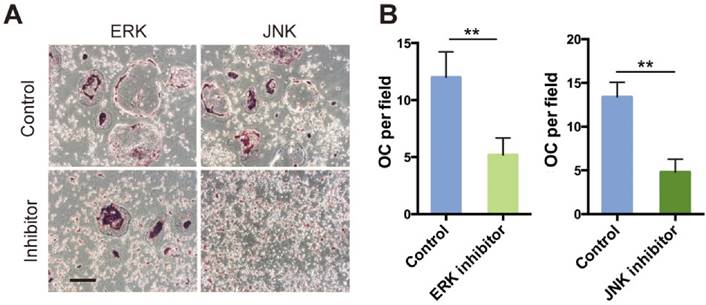
Inhibition of ERK and JNK signaling delayed osteoclast differentiation
We hypothesized that attenuated osteoclast formation in SR-A-/- bone marrow cultures occurs via inhibition of ERK and JNK signaling pathways. To test this hypothesis, we independently treated osteoclast progenitor cells from SR-A+/+ mice with ERK inhibitor (U0126, 5 μM) or JNK inhibitor (SP600125, 10 μM), and then examined the number of osteoclasts (Figure 7A). Osteoclast formation was markedly inhibited and the size of cells was reduced (Figure 7B). These results suggest that SR-A activates the ERK and JNK signaling pathways, inducing osteoclasts to produce IL-6 that promotes further osteoclast formation.
ERK and JNK inhibitors reduce the number of RANKL-induced osteoclasts. (A) RANKL-induced bone marrow cells of SR-A+/+ mice were incubated with inhibitors of JNK (SP600125, 10 μM) or ERK (U0126, 5 μM) for 6 days, respectively, and then stained for TRAP. Scale bars equal 50 μm. (B) Quantitative analysis of the number of TRAP-positive cells. n = 5 in each group. **P < 0.01. All data are reported as the mean ± SD.
Discussion
Osteoclasts play a key role in regulating bone structure and function throughout the life of vertebrates [35]. Previous studies indicated that deletion of SR-A resulted in decreased osteoclastogenesis via inhibition of the expression of RANK and related molecules [8, 12]. However, the signaling pathways involved in attenuation of osteoclastogenesis still remain to be elucidated.
Most of the craniofacial bones originate from neural crest cells, while all other bones in the body originate from the mesoderm. Each bone lineage has different properties [36, 37]. Therefore, two animal models representing the head and the extremities were included in this study. These experimental models are the TM and the ACLT OA model, which are often used for analyzing bone homeostasis. We demonstrated in both models, and also in vitro, that SR-A gene disruption attenuated osteoclastogenesis, and we also showed that SR-A activates the ERK/JNK pathway in osteoclasts mediating production of IL-6.
Our micro-CT study demonstrated significantly increased bone volume, trabecular number, and decreased trabecular separation in SR-A-/- mice compared to SR-A+/+ mice. These results were likely due to uncompensated osteoblast activity in SR-A-/- mice. We then examined the number of osteoblasts and osteoclasts, and found that osteoclastogenesis decreased significantly in SR-A-/- mice. However, the number of osteoblasts was virtually the same in the two groups. This suggests that SR-A affects bone turnover by promoting osteoclastogenesis, but not osteoblastogenesis, which is consistent with previous reports [8].
Osteoclasts secrete factors such as IL-1, IL-6, insulin-like growth factor-1 (IGF-1), and tumor necrosis factor- α (TNF-α), and these factors play a vital role in osteoclastogenesis [26-30]. In addition, some of these factors can regulate the function of SR-A. For example, IL-6 promotes expression of SR-A on macrophages in rheumatoid arthritis [31], and blockade of IL-6 and TNF-α can inhibit the expression of SR-A in human arterial endothelial cells [32]. Therefore, we investigated how SR-A affects the production of these factors. Interestingly, we found that SR-A-/- osteoclasts produced lower levels of IL-1 and IL-6 than SR-A+/+ cells, as determined by analysis of both mRNA expression and secretion of IL-1 and IL-6 protein. In order to characterize the effect of IL-1 and IL-6 on SR-A-/- osteoclasts, we added IL-1 and IL-6 to these cells and found that IL-6 can revert the reduced osteoclastogenesis caused by the loss of SR-A function more obviously. Moreover, tooth movement-induced osteoclastogenesis in SR-A+/+ mice was prevented by blocking of IL-6. These results are in agreement with previous studies that showed that IL-6 causes bone resorption by inducing osteoclast formation [38, 39]. Because IL-6 can also promote the expression of SR-A in macrophages [31], our data suggest a positive feedback, with SR-A inducing IL-6 expression in osteoclasts and IL-6 inducing the expression of SR-A in the same cells. However, the overall effect of IL-6 on osteoclastogenesis is still not completely clear. There is some evidence of negative effects of IL-6 on osteoclasts. Yoshitake et al. [40] showed that IL-6 directly acts on osteoclast progenitors and suppresses their differentiation. The apparent discrepancy observed in these two studies may arise because the effect of IL-6 in the study by Yoshitake et al. was evaluated in a different culture system with a high IL-6 concentration (100 ng/ml). IL-6 was reported to induce the differentiation of osteoclasts in a concentration-dependent manner [41], and that there was no inhibition at low IL-6 concentration. Moreover, there are also studies suggesting that the effect of IL-6 on osteoclasts might be related to the age of the animal [42]. Compared with prepubertal rats, adult rats showed a decrease in osteoclastogenesis after IL-6 treatment. The function of IL-6 on osteoclasts appears to be a double-edged sword that can influence osteoclast differentiation through complex and sometimes seemingly contradictory mechanisms. This particular issue will require further investigation that is clearly outside the scope of the current study.
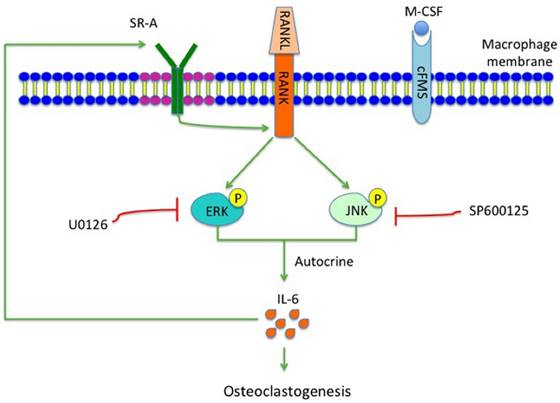
During the early stage of osteoclast differentiation, RANK activation in preosteoclasts leads to increased expression and activation of MAPKs, including ERK, JNK and p38, which is important for the regulation of osteoclast formation [5]. Takemura et al. [8], reported that SR-A deletion resulted in reduced expression levels of RANK and RANK-related osteoclast-differentiation molecules such as NFATc1 and MITF. In the present study, we showed that phosphorylation of ERK/JNK decreased significantly after deletion of SR-A. When RANKL-induced SR-A+/+ bone marrow derived-macrophages (BMMs) were treated with ERK or JNK inhibitor, but not with p38 inhibitor, the number of osteoclasts decreased and osteoclast size was smaller than the control cells. In addition, the expression of IL-6 declined in ERK or JNK inhibitor-treated cells. In conclusion, our results strongly suggest that SR-A promotes osteoclastogenesis, in addition to the previously reported increase of expression of RANK, by activating ERK and JNK signaling and inducing IL-6 expression, followed by induction of SR-A expression (Figure 8). The central role of SR-A suggests that SR-A could be a new therapeutic target for the regulation of bone homeostasis in craniofacial and other bones.
Hypothetical model of the role of SR-A and IL-6 signaling in osteoclastogenesis. SR-A regulates IL-6-induced osteoclastogenesis by activating ERK and JNK signaling pathways via RANK and amplification involving IL-6 signaling.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81371179), Natural Science Foundation of Jiangsu Province (BK20150048), and Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (2014-037).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sims NA, Vrahnas C. Regulation of cortical and trabecular bone mass by communication between osteoblasts, osteocytes and osteoclasts. Arch Biochem Biophys. 2014;561:22-8
2. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. 2013;92:860-7
3. Hayashibara T, Hiraga T, Sugita A, Wang L, Hata K, Ooshima T, Yoneda T. Regulation of osteoclast differentiation and function by phosphate: potential role of osteoclasts in the skeletal abnormalities in hypophosphatemic conditions. J Bone Miner Res. 2007;22:1743-51
4. Fan X, Biskobing DM, Fan D, Hofstetter W, Rubin J. Macrophage colony stimulating factor down-regulates MCSF-receptor expression and entry of progenitors into the osteoclast lineage. J Bone Miner Res. 1997;12:1387-95
5. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
6. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251-64
7. Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. 2009;50(Suppl):S282-6
8. Takemura K, Sakashita N, Fujiwara Y, Komohara Y, Lei X, Ohnishi K. et al. Class A scavenger receptor promotes osteoclast differentiation via the enhanced expression of receptor activator of NF-kappaB (RANK). Biochem Biophys Res Commun. 2010;391:1675-80
9. Platt N, Haworth R, Darley L, Gordon S. The many roles of the class A macrophage scavenger receptor. Int Rev Cytol. 2002;212:1-40
10. Ben J, Zhang Y, Zhou R, Zhang H, Zhu X, Li X. et al. Major vault protein regulates class A scavenger receptor-mediated tumor necrosis factor-alpha synthesis and apoptosis in macrophages. J Biol Chem. 2013;288:20076-84
11. Greaves DR, Gordon S. Thematic review series: the immune system and atherogenesis. Recent insights into the biology of macrophage scavenger receptors. J Lipid Res. 2005;46:11-20
12. Fraser I, Hughes D, Gordon S. Divalent cation-independent macrophage adhesion inhibited by monoclonal antibody to murine scavenger receptor. Nature. 1993;364:343-6
13. Lin YL, de Villiers WJ, Garvy B, Post SR, Nagy TR, Safadi FF. et al. The effect of class a scavenger receptor deficiency in bone. J Biol Chem. 2007;282:4653-60
14. Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K. et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292-6
15. Ben J, Jin G, Zhang Y, Ma B, Bai H, Chen J. et al. Class A scavenger receptor deficiency exacerbates lung tumorigenesis by cultivating a procarcinogenic microenvironment in humans and mice. Am J Respir Crit Care Med. 2012;186:763-72
16. Taddei SR, Moura AP, Andrade I Jr, Garlet GP, Garlet TP, Teixeira MM, da Silva TA. Experimental model of tooth movement in mice: a standardized protocol for studying bone remodeling under compression and tensile strains. J Biomech. 2012;45:2729-35
17. Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061-9
18. Chen W, Ma J, Zhu G, Jules J, Wu M, McConnell M. et al. Cbfbeta deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfbeta required for skeletal development. Proc Natl Acad Sci U S A. 2014;111:8482-7
19. Yadav S, Assefnia A, Gupta H, Vishwanath M, Kalajzic Z, Allareddy V, Nanda R. The effect of low-frequency mechanical vibration on retention in an orthodontic relapse model. Eur J Orthod. [Epub ahead of print]
20. Taddei SR, Queiroz-Junior CM, Moura AP, Andrade I Jr, Garlet GP, Proudfoot AE. et al. The effect of CCL3 and CCR1 in bone remodeling induced by mechanical loading during orthodontic tooth movement in mice. Bone. 2013;52:259-67
21. Zhen G, Wen C, Jia X, Li Y, Crane JL, Mears SC. et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19:704-12
22. Chambers MG, Bayliss MT, Mason RM. Chondrocyte cytokine and growth factor expression in murine osteoarthritis. Osteoarthritis Cartilage. 1997;5:301-8
23. Albers J, Keller J, Baranowsky A, Beil FT, Catala-Lehnen P, Schulze J. et al. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol. 2013;200:537-49
24. Lee YD, Yoon SH, Park CK, Lee J, Lee ZH, Kim HH. Caveolin-1 Regulates Osteoclastogenesis and Bone Metabolism in a Sex-dependent Manner. J Biol Chem. 2015;290:6522-30
25. Bai H, Li N, Zhou X, Wang C, Zhang Y, Zhu X. et al. GRP78 inhibits macrophage adhesion via SR-A. J Biomed Res. 2014;28:269-74
26. Yao Z, Xing L, Qin C, Schwarz EM, Boyce BF. Osteoclast precursor interaction with bone matrix induces osteoclast formation directly by an interleukin-1-mediated autocrine mechanism. J Biol Chem. 2008;283:9917-24
27. Tani-Ishii N, Tsunoda A, Teranaka T, Umemoto T. Autocrine regulation of osteoclast formation and bone resorption by IL-1 alpha and TNF alpha. J Dent Res. 1999;78:1617-23
28. Roodman GD, Kurihara N, Ohsaki Y, Kukita A, Hosking D, Demulder A. et al. Interleukin 6. A potential autocrine/paracnne factor in Paget's disease of bone. J Clin Invest. 1992;89:46-52
29. Nakao A, Fukushima H, Kajiya H, Ozeki S, Okabe K. RANKL-stimulated TNFalpha production in osteoclast precursor cells promotes osteoclastogenesis by modulating RANK signaling pathways. Biochem Bioph Res Co. 2007;357:945-50
30. Fiorelli G, Formigli L, Zecchi Orlandini S, Gori F, Falchetti A, Morelli A. et al. Characterization and function of the receptor for IGF-I in human preosteoclastic cells. Bone. 1996;18:1269-76
31. Hashizume M, Mihara M. Atherogenic effects of TNF-α and IL-6 via up-regulation of scavenger receptors. Cytokine. 2012;58:424-30
32. Hashizume M, Mihara M. Blockade of IL-6 and TNF-α inhibited oxLDL-induced production of MCP-1 via scavenger receptor induction. Eur J Pharmacol. 2012;689:249-54
33. Liu W, Yin Y, Zhou Z, He M, Dai Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflamm Res. 2014;63:33-43
34. Nikolic DM, Vadali S, He B, Ware J, Kelly T, Post SR. Prostaglandins produced during class A scavenger receptor-mediated macrophage adhesion differentially regulate cytokine production. J Leukoc Biol. 2015;97:901-8
35. Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504-8
36. Reichert JC, Gohlke J, Friis TE, Quent VM, Hutmacher DW. Mesodermal and neural crest derived ovine tibial and mandibular osteoblasts display distinct molecular differences. Gene. 2013;525:99-106
37. Kobayashi T, Kronenberg HM. Overview of skeletal development. Methods Mol Biol. 2014;1130:3-12
38. Hashizume M, Mihara M. The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis. 2011;2011:765624
39. Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I. et al. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res. 1996;11:88-95
40. Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J Biol Chem. 2008;283:11535-40
41. Duplomb L, Baud'huin M, Charrier C, Berreur M, Trichet V, Blanchard F. et al. Interleukin-6 inhibits receptor activator of nuclear factor kappaB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of Serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology. 2008;149:3688-97
42. De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M. et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006;54:3551-63
Author contact
![]() Corresponding authors: Junqing Ma, MD, PhD, Jiangsu Key Laboratory of Oral Diseases, Nanjing Medical University, Nanjing, Jiangsu 210029, China, Phone: 0086-25-85031976, E-mail: jmaedu.cn; Qi Chen, PhD, Atherosclerosis Research Center, Key Laboratory of Cardiovascular Disease and Molecular Intervention, Nanjing Medical University, Nanjing, Jiangsu 210029, China, Phone: 0086-25-86862610, E-mail: qichenedu.cn
Corresponding authors: Junqing Ma, MD, PhD, Jiangsu Key Laboratory of Oral Diseases, Nanjing Medical University, Nanjing, Jiangsu 210029, China, Phone: 0086-25-85031976, E-mail: jmaedu.cn; Qi Chen, PhD, Atherosclerosis Research Center, Key Laboratory of Cardiovascular Disease and Molecular Intervention, Nanjing Medical University, Nanjing, Jiangsu 210029, China, Phone: 0086-25-86862610, E-mail: qichenedu.cn