Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Role of the ROS-FOXO Axis in...
Role of FOXO in Cell Cycle Arrest
Role of Insulin/Growth Factors...
Crosstalk between FOXO and p53
FOXO Mutations in Cancer
Current Anti-Cancer Drugs...
Perspectives and Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(7):815-827. doi:10.7150/ijbs.20052 This issue Cite
Review
FOXO Signaling Pathways as Therapeutic Targets in Cancer
Mohd Farhan1, Haitao Wang2, Uma Gaur1, Peter J. Little3, Jiangping Xu2, Wenhua Zheng1 ![]()
1. Faculty of Health Sciences, University of Macau, Taipa, Macau, China;
2. School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, China;
3. School of Pharmacy, Pharmacy Australia Centre of Excellence, The University of Queensland, Woolloongabba, Queensland, 4102 Australia and Xin Hua College, Sun Yat- Sen University, China.
Received 2017-3-13; Accepted 2017-4-11; Published 2017-7-6
Abstract

Many transcription factors play a key role in cellular differentiation and the delineation of cell phenotype. Transcription factors are regulated by phosphorylation, ubiquitination, acetylation/deacetylation and interactions between two or more proteins controlling multiple signaling pathways. These pathways regulate different physiological processes and pathological events, such as cancer and other diseases. The Forkhead box O (FOXO) is one subfamily of the fork head transcription factor family with important roles in cell fate decisions and this subfamily is also suggested to play a pivotal functional role as a tumor suppressor in a wide range of cancers. During apoptosis, FOXOs are involved in mitochondria-dependent and -independent processes triggering the expression of death receptor ligands like Fas ligand, TNF apoptosis ligand and Bcl‑XL, bNIP3, Bim from Bcl-2 family members. Different types of growth factors like insulin play a vital role in the regulation of FOXOs. The most important pathway interacting with FOXO in different types of cancers is the PI3K/AKT pathway. Some other important pathways such as the Ras-MEK-ERK, IKK and AMPK pathways are also associated with FOXOs in tumorigenesis. Therapeutically targeting the FOXO signaling pathway(s) could lead to the discovery and development of efficacious agents against some cancers, but this requires an enhanced understanding and knowledge of FOXO transcription factors and their regulation and functioning. This review focused on the current understanding of cell biology of FOXO transcription factors which relates to their potential role as targets for the treatment and prevention of human cancers. We also discuss drugs which are currently being used for cancer treatment along with their target pathways and also point out some potential drawbacks of those drugs, which further signifies the need for development of new drug strategies in the field of cancer treatment.
Keywords: Apoptosis, nuclear translocation, FOXO, cell proliferation, anticancer drugs, transcription factors.
Introduction
Cancer is one of the major causes of morbidity in modern human populations. The leading cause of mortality in advanced communities has been cardiovascular diseases, but improved treatment of cardiovascular diseases is leading to an increased lifespan of the population. The prevalence of many diseases, including cancers, is age related so with the increase in average lifespan of populations, the occurrence, impact and burden of cancer is also increasing. Many human cancers are caused by the dysregulated activity of transcription factors. Transcription factors, also known as sequence specific DNA binding peptides, are polypeptides which bind to specific DNA sequences and regulate, (negatively and positively) the rate of transcription of genetic and cellular information from DNA to mRNA and ultimately lead to the altered expression of the index proteins. Transcription factors work in large complexes which are being more and more recognized for their heterogeneity and exquisite functional variability. Thus the roles and functions of transcription factors are commonly dysregulated in pathologies of human cancer which render them potential therapeutic targets [1, 2].
The remarkable molecular diversity of transcription factors as drivers of cellular transformation warrants the pursuit of them as therapeutic targets for drug discovery for prevention and modulation of the oncogenic processes underlying human cancers. Agents targeting transcription factors, for example, for proteolysis of their derived proteins, are among the few current curative strategies in cancer therapeutics [3].
One such class of transcription factors that may serve as a target for the modulation of cancer is the Forkhead box transcription factors (FOXO) family. FOXO proteins are growth factor and stress regulated transcription factors. FOXO proteins normally reside in the nucleus of quiescent or growth factor deprived cells where they act as regulators of gene transcription; in the presence of cell growth factors, FOXO proteins are relocalized to the cytosol and eventually subjected to degradation via the ubiquitin-proteasome pathway. In the absence of the cellular survival drive of growth factors, FOXO proteins translocate to the nucleus and upregulate a series of target genes, thereby promoting cell cycle arrest, stress resistance, and apoptosis [4]. This brief description points to the potential role of FOXO proteins in the regulation of expression of the adherent genes related to cancer and as a potential targets for the prevention and treatment of cancer.
FOXOs are involved and implicated in a broad range of cellular functions, including cellular differentiation, apoptosis, cell proliferation, DNA damage and repair and as mediators of oxidative stress [5, 6]. These are cellular properties, critical to the cell biology of cancer. Compelling evidence implicates dysregulation of the functioning of FOXO proteins with cancer progression and tumorigenesis [7, 8]. The intriguing cell biology of FOXO arises from their cellular location and translocation. Whereas most common regulators of transcription, for example, the extracellular signal-regulated kinase (ERK) are located in the cytoplasm and stimulation of cells through growth factor receptors results in target kinase phosphorylation and nuclear translocation, FOXO proteins are present in the nucleus and growth factor pathways promote the nuclear exclusion and translocation of phosphorylated FOXO to the cytoplasm where they are subjected to degradation [9].
The designation of FOXOs arose from the discovery of the Drosophila forkhead gene. The founding member and namesake of the FOX family is the fork head transcription factor in Drosophila, the mutation in which results in defective head involution. The forkhead box (FOX) family of genes have a conserved forkhead domain (the “forkhead box”) described as a “winged helix” as a result of the butterfly-like appearance on X-ray crystallography and nuclear magnetic resonance. The forkhead domain in FoxO proteins consists of three α-helices, three β-sheets, and two loops that are referred to as the wings [10, 11].
Of the mammalian forkhead transcription factors in the O class, FoxO1, FoxO3, FoxO4, and FoxO6 proteins play a significant role in normal cellular functioning as well as during progressive disease. The most recently cloned member is FoxO6 and progressive interest in FoxO1, FoxO3, and FoxO4 has shown that these transcription factors can promote cell proliferation as well as cell death [11]. The superfamily of forkhead contains a highly conserved DNA FOX domain. The FOXO transcription factors control tumor suppression pathways. FOXO6 mRNA is predominantly present in the adult and developing brain, and it also plays a key role in nervous system [12]. Majority of target genes activation or repression is regulated by the conserved FOX domain which is a 100 amino acid DNA binding domain of FOXO protein. Transcriptional functions and FOXO localization at the subcellular level are controlled by classic, post-translational modifications, including ubiquitination, phosphorylation and acetylation [12]. The FOXO1/FKHR, FOXO4/AFX and FOXO3/FKHRL1 genes were also detected during choromosomal translocation events in human tumors. This initial research suggested the important role of FOXO transcription factors in tumor development and progression [13, 14]. Individually FOXO1 mRNA is highly expressed in adipose tissue, FOXO4 mRNA is mostly found in the heart and FOXO3 mRNA is abundant in brain [15]. During development and carcinogenesis, the protein kinase B (AKT) pathway plays a prominent role in cell growth and cell survival. AKT is activated by AKT kinase PDK1/2, downstream target kinase of phospholipid kinase phosphatidylinositol 3-kinase (PI3K) and in the case of mammals it exists as three isoforms which are encoded by separate loci. Targeted deletion of all isoforms of AKT inactivates tissue functions [16] although only AKT2 and 3 isoforms are amplified in human cancer. FOXOs are key signaling proteins in growth factor signaling and are pathway positioned downstream of AKT. Growth-factor initiated kinase‑mediated phosphorylation of FOXOs promotes their interaction with the 14-3-3 protein, leading to export from the nucleus to the cytoplasm; this provides an example of the phenomena of nuclear exclusion in cell biology. FOXOs are also regulated by ubiqutin proteasome pathways.
These FOXO regulatory pathways are triggered by the PI3K/AKT pathway which mediates cellular functions of proliferation and cell growth [17]. The balance between degradation of FOXOs and nuclear exclusion could be related to signal intensity which is responsible for the nuclear export [18]. While AKT phosphorylates FOXO1, 3, 4 on three sites and leads to their cytoplasmic translocation, AKT phosphorylates FOXO6 on only two residues. Phosphorylation of these two residues on FOXO6 not only inactivates FOXO6 but also prevents its export from nucleus to cytoplasm [19]. In addition to AKT, there are other negative regulators of FOXOs, such as DYRK1A (dual-specificity tyrosine-phosphorylation-regulated kinase 1A), CK1 (casein kinase 1), SGK (serum and glucocorticoid-regulated kinase), IκB kinase (IKK) and the ubiquitous MAP kinase, ERK [20]. Other kinases, which regulate FOXOs are JNK (c-Jun N-terminal kinase) and MST1 (Mammalian Ste20-like kinase) which act under conditions of elevated oxidative stress. JNK and MST1 phosphorylate FOXO4 under cellular stress conditions and promote its movement from cytoplasm to the nucleus where it regulates gene transcription. AMPK (AMP-activated protein kinase) phosphorylates and activates FOXOs under conditions of nutrient stress and triggers the expression of genes which play an important role in stress resistance and energy metabolism [21]. FOXOs and their role in the different type of diseases are summarized in Table 1.
Types of FOXO and their involvement in various diseases
| Types of FOXOs | Pathological Problems | Key roles of FOXOs | References |
|---|---|---|---|
| FoxO3a, FOXO proteins | Cancer | Transcriptional Activity of FOXO3a required to avert chronic myelogenous leukemia and B-chronic lymphocytic leukemia | [22,23] |
| FOXO proteins are involved in cell cycle regulation and proliferation process | [24, 25] | ||
| Lack of functional FOXO proteins lead to prostate, breast and thymic tumors | [26-30] | ||
| FOXO1, FOXO3a | Alzheimer's disease and aging | FOXO protein translational activity prevention decreased the cell loss during neurodegeneration and oxidative stress | [31,56] |
| Amyloid caused phosphorylation of FOXO3a and FOXO1 | [32] | ||
| β-catenin had ability to modulate transcriptional activity of FOXO | [33] | ||
| FOXO1, FOXO3a | Diabetes mellitus | FOXO3a and FOXO1 linked with increased mortality and increased HbA1c | [34] |
| FOXO3a and FOXO1 associated with high risk of Stroke and diabetes | [35] | ||
| In several animal model of diabetes, FOXO3a activity loss may lead to disease complications | [36] |
FOXOs have been considered to be tumor suppressors due to their pro-apoptotic and anti-proliferative actions. Although its activity and expression are unaltered in different cancers, recent studies have revealed new and unexpected functions of FOXOs in the promotion of cancer and in modulating responses to cancer treatment. In this review we address the prospects of targeting FOXOs in regulating cancer and the underlying mechanisms involved.
Role of the ROS-FOXO Axis in Cancer
Role of FOXO in oxidative stress and oxidation-triggered apoptosis
Apoptotic genes are not only related to cell killing but also play an essential role in replication and transcription. Cellular apoptosis is an essential process in tissue and organ development and it can also be involved in disease processes related to diabetes mellitus (DM), neurodegenerative diseases and cardiovascular injury [37]. However, most importantly, by regulating apoptotic pathways, cellular signaling pathways and transcription factors control unregulated cell proliferation and tumor growth [38].
Apoptotic cell death is a self‑generated process involving both early and late events. Microglias have a phosphatidylserine receptor (PSR) which is expressed during oxidative stress. Membrane phosphatidylserine (PS) binds to these receptors and directs microglia to target phagocytosis of cells [39, 40] which is an early event in cell apoptosis [41-43]. This blockage of PSRs working in microglia inhibits microglial activation [44, 45] leading to externalization of PS residues in cells at the time of oxidative stress. This externalization of PS leads to anoxia [46] and reactive oxygen species (ROS) exposure [47]. Some agents that form ROS, such as 6‑hydroxydopamine [48], are involved in late events during the apoptotic process including fragmentation of genomic DNA [49]. Enzymes which mediate fragmentation and degradation of DNA are called endonucleases, and they consist of the acidic and cationic independent endonucleases (DNase II), 97 kDa magnesium—dependent endonucleases and cyclophilins.
The initiation of apoptosis within cells by FOXO proteins engages pathways associated with oxidative stress. Oxidative stress occurs when ROS are released into the cellular environment. Mutations of mitochondrial DNA and the generation of oxygen free radicals leads to tissue toxicity at the organ level and it underpins the aging process [37]. ROS include multiple chemical entities, for example, superoxide free radicals, singlet oxygen, hydrogen peroxide, peroxynitrite and nitric oxide [49]. During normal physiological conditions, these reactive species are formed at low levels and are scavenged by endogenous antioxidants such as glutathione peroxidase, superoxide dismutase and a diverse array of small molecules (like vitamin B3, D3, C, E,) and catalase [50, 51]. Forkhead transcription factors like FOXO1 and FOXO3a commonly present during oxidative stress cause apoptotic cell injury [52, 53]. Under other kinds of oxidative stresses, FOXO3a and the MAP kinase, JNK, lead to modification of apoptotic ligands, which activate a cell death pathway mediated by Fas protein [54] causing the pro-apoptotic action of p53 [55]. When FOXO activity is compromised throughout the oxidative stress process then it becomes advantageous for cell survival. For example, protein or gene knockdown of FOXO3a or FOXO1 can reduce ischemia‑induced infarct size in brain to some extent [56], intervene to shield metabotropic glutamate receptors in periods of vascular injury [57], increase neuronal survival or pancreatic β-cell with the assistance of NAD+ precursors throughout oxidative stress [58] and give trophic factor protection with neurotrophins and erythropoietin (EPO) [59,60]. Thus, FOXO proteins come to the fore as a potential target to control tumor growth, not only by engaging with pro-apoptotic pathways, but also by blocking cell cycle progression.
Interaction between ROS and FOXO and its contribution to cancer
Hyper activation of the PI3K/AKT pathway is a characteristic of many different cancers and is often seen together with the absence of tumor suppressor gene phosphatase and tensin homolog (PTEN) [61]. Therefore, FOXO proteins could be considered as tumor suppressors, as inactivation of FOXO proteins is an early event in the tumorigenic process. In support of this proposal, mice lacking FOXO1, FOXO3 and FOXO4 developed lymphoproliferative diseases and hemangiomas, which are circumstances linked with new neoplasms. It is worth mentioning that hemangiomas are a characteristic of Cowden's syndrome where the tumor suppressor gene PTEN is found inactivated because of mutation [62, 63]. In cells which are not transformed, ROS are formed at normal levels due to basal levels of cell metabolism and at these lower levels ROS are successfully inactivated by the effective cellular antioxidant defense mechanisms. ROS are also generated during active growth factor-receptor signaling [64]. As compared to untransformed cells, most cancer cells generate high levels of ROS indicating an increased metabolic rate, which confers a permanent state of increased oxidative pressure [65]. As ROS production remains persistently elevated, the cancer progresses in association with genomic instability, a high amount of DNA damage and decreased mismatch repair [66]. Additionally, the disturbed redox conditions lead to the formation of signaling components necessary for cell survival and proliferation and reduced involvement of FOXO3a [67]. Compatible with a role for ROS at the time of tumorigenesis, Ras-transformed fibroblasts exhibit pronounced ROS [68] and superoxide overproduction producing oxidase Mox1 which is sufficient to change immortalized NIH3T3 fibroblasts [69]. So, continuous oxidative stress can bestow a survival and growth benefit to cancer cells by triggering signaling pathways that encourage cellular proliferation and transformation [70]. Along with this, the induction of a number of proapoptotic FOXO stimulated genes, including B-cell lymphoma-2-interacting mediator of cell death (Bim) and B-cell lymphoma 6 as a response against hydrogen peroxide, can be inactivated by either expressing the dominant-negative mutant or inhibition of endogenous FOXO3a [70,71]. Cancer cells have developed mechanisms that potentially break the nexus between ROS aggregation and Bim-induced apoptosis by FOXO. Colorectal carcinoma cells are linked to ERK mediated suppression of Bim which is achieved by FOXO3a degradation by ERK [72].
The oxidative stress defense response is induced through FOXO-dependent manganese-dependent superoxide dismutase (MnSOD) induction, and catalase and GADD45 activity in nonmalignant cells. Relevant to the role of FOXO proteins in decreasing ROS levels, FOXO1, FOXO3 and FOXO4 triple-knockout mice hematopoietic stem cells showed evidence of increased intracellular ROS levels [73]. When the PI3K/AKT pathway is activated during tumorigenesis, it down regulates the ability of FOXO to detoxify ROS, leading to an increase in intracellular ROS and augmented cancer progression. However, increased cellular ROS levels could also trigger kinases activated by stress, as well as JNK which amplify the FOXO protein transcriptional activation potential [74]. Collectively, these findings indicate that there is a tightly synchronized feedback loop between FOXO proteins and ROS. FOXO modulates the transcription of genes involved in redox regulation, while ROS regulates the activity of FOXO factors.
Role of FOXO in Cell Cycle Arrest
There are many studies which have investigated the tumor suppressor activity of FOXOs to control the expression of genes implicated in cell death (FasL, Bim, Trail) and cell cycle arrest (CDKN1A/p21, p27KIP1). FOXO4 and FOXO3a can mediate cell cycle arrest in murine myoblastic cell lines by processes involving modulation of growth arrest and expression of new protein 45 formed by the DNA damage process [75]. Control of the cell cycle by transcription factor E2F-1 enhanced the expression of FOXO3a and FOXO1 leading ultimately to cell cycle arrest [76]. Importantly, the loss of FOXO3a expression in conjunction with the oncogene c-myc, nuclear factor‑κB (NF-κB) and p27, leads to cell cycle initiation and alteration of malignant mouse cells and oncogenic activation. Some data indicated that the FOXO proteins made use of the p53 upstream regulator p19 (Arf) by the oncogene myc to obstruct lymphoma progression and cell cycle induction [77].
Role of Insulin/Growth Factors In the Regulation of FOXO
AKT/FOXO is a canonical downstream target of growth factor receptor signaling
The serine/threonine protein kinase AKT, targets FOXOs as a direct substrate. AKT is a ubiquitous mediator of cellular signaling pathways initiated by insulin and many growth factors in a wide variety of cell types [Fig.1 and Fig.2]. Growth factor or more specifically insulin initiated activation of their cognate protein tyrosine kinase receptors often stimulates the recruitment and activation of PI3K, which further activates serine/threonine kinases, including SGK and AKT protein kinase family members [78]. The conservation of the PI3K/AKT/SGK pathway from worms to mammals supports the contention that this pathway is important in multicellular organisms. Preliminary genetic research in worms revealed that the FOXOs are very important downstream targets of the PI3K/AKT pathway in the context of longevity and development [79]. Our previous studies also demonstrated that insulin growth factor-1 (IGF-1) is capable of inducing the phosphorylation of FOXO3a (FKHRL1) in PC12 cells and Uveal Melanoma cells by activating the PI3K/AKT kinase pathway [65, 66, 80, 81]. Recently, we also described the involvement of the PI3K/AKT/FOXO3a pathway in the IGF-1 induced protective action in RGC-5 retinal cells against cell death caused by the anti-arrythmic drug, amiodarone [82]. Another study performed on PC12 cells showed the protective effect of venlafaxine against cell death caused by corticosterone, by involvement of PI3K/AKT/FOXO3a signaling pathway [83]. FOXO1, FOXO4 and FOXO3 are phosphorylated through AKT at three regulatory sites -Thr32, Ser315 and Ser253. These specific regulatory sites, being consensus sequences for AKT phosphorylation, are conserved from C. elegans to mammals [84]. SGK and AKT phosphorylate a combination of sites present in FOXOs [85]; SGK preferentially phosphorylates Ser315 and AKT mediates the phosphorylation of Ser253 whereas Thr32 is phosphorylated by both the kinases. IGF-I [86], interleukin 3 [87], insulin [88], nerve growth factor [89], erythropoietin [90] and epidermal growth factor [91] are the growth factors which phosphorylate all three FOXO regulatory sites. This indicates that FOXO transcription factors integrate a wide range of phosphorylation pathways by external stimuli utilizing all three conserved residues which are phosphorylated by SGK and AKT. Thus, by the phosphorylation of these three residues, SGK and AKT integrate FOXO transcription factor responses from a broad range of external stimuli. The selective phosphorylation of residues of FOXO by several protein kinases may permit FOXOs to selectively react to various closely associated stimuli, like IGF-I and insulin [92]. Other sites on FOXOs are targeted by growth factor and insulin signaling pathways, including the residues Ser322 and Ser325 in FOXO1 which are also phosphorylated by growth factor stimulation [93]. The phosphorylation of residues Ser322 and Ser325 in FOXO1 is 'primed' by Ser329 phosphorylation and is also mediated by CK1 [93]. Phosphorylation of Ser329 can also occur by the dual tyrosine (Y) which is controlled by kinase 1 (DYRK1) [94]. Collectively, all these phosphorylation processes are involved in the control and regulation of the subcellular localization of FOXO which is the most important determinant of its role in cell biology.
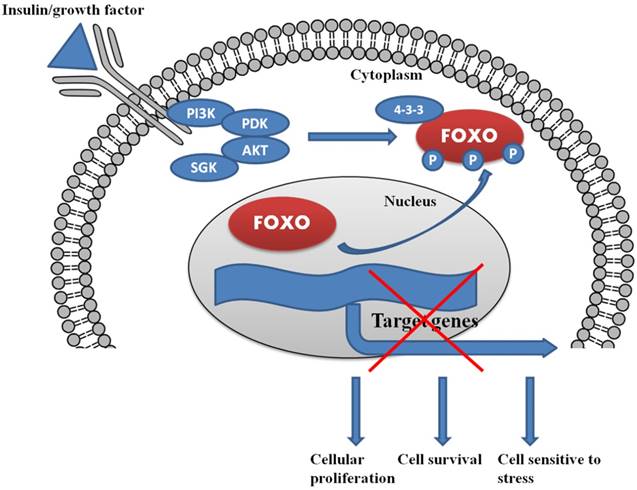
Insulin and growth factor control the complete regulation of FOXO within the cells. The lack of insulin/growth factor results in the localization of FOXO in the nucleus and triggers the process of cell cycle arrest and cell death.
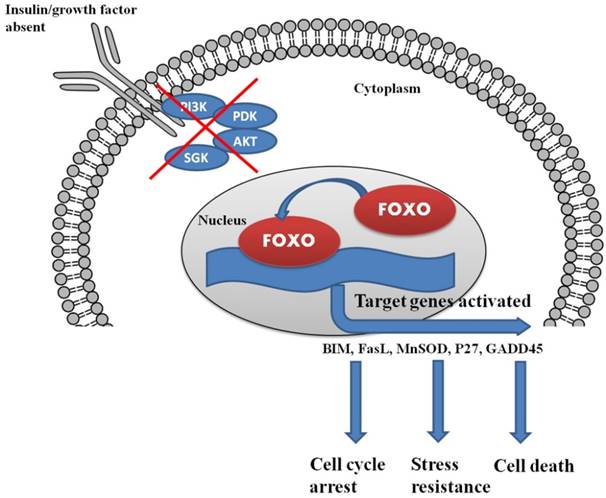
In the presence of insulin/growth factor, all the growth factor binds with the receptor and activates the PI3K/AKT/SGK pathway. AKT and SGK phosphorylates the FOXO at different sites, and upon phosphorylation, it binds with 14-3-3 protein. The binding of 14-3-3 protein with phosphoryated FOXO helps FOXO translocate form nucleus to cytoplasm.
Correlation of FOXO and other pathways in cancer
The PI3K/AKT pathway promotes cell survival by phosphorylating and thus inhibiting the action of multiple signaling proteins like FOXO transcription factors [95]. Multiple studies supported the oncogenic role of the PI3K/AKT pathway in cancer cells including gain-of-function of tyrosine kinases by PI3K or RAS by mutations in encoded genes or mutations of PTEN leading to loss of FOXOs functions [96]. In cancers, FOXOs mostly remain in the inactive phosphorylated state in the cytosol thus maintaining normal cellular proliferation and survival. This conclusion has been supported by a number of studies which show that pre-B lymphocyte transformation with BCR‑ABL requires the PI3K/AKT pathway and concurrent repression of FOXO3-induced apoptosis is also required [97]. In the same manner for transfected Ba/F3 cells, the activity of FLT3-ITD, a mutant receptor which is a common resident of acute myeloid leukemia, resulted in the activation of PI3K/AKT pathway with consequent inactivation of FOXO3 and phosphorylation. As a result, FLT3-ITD reduced FOXO3-induced p27KIP1 (CDKN1B) and Bim (BCL2L11) expression, which assists in maintaining cell survival and proliferation [98]. The PI3K pathway is also dysregulated and activated in breast, thyroid and cervical cancer. FOXO1 is reactivated in a breast cancer cell line due to targeted depletion of PI3K with the help of siRNA and the activation of FOXO1, 3 and 4 lead to apoptosis and cell cycle arrest [99]. FOXO1 when reactivated in cervical cancer cells treated with the PI3K inhibitor, LY294002, promotes an anti‑proliferative and pro-apoptotic response which is similar to the down regulated effects mentioned above [100]. In a murine lung cancer model, when PI3K is inhibited, it promotes the activation of FOXOs and reactivates p27KIP1 expression [101]. Other available agents which inhibit the PI3K pathway restore FOXO activity and thus induce apoptosis and cell cycle arrest. When the PI3K/AKT pathway in thyroid cancer cells is blocked by non-steroidal, anti-inflammatory drugs, such as sulindac sulfide, FOXO3 is activated which triggers the expression of growth arrest and DNA-damage-inducible protein 45α, Bim and p27KIP1 [102]. In B lymphocytes, FOXO1 is expressed at a high level, but its expression is reduced in Hodgkin's lymphomas. The FOXO1 repression in these cells could be ascribed to different mechanisms, including constitutive activation of ERK and AKT and when FOXO1 is reintroduced then cell proliferation is reduced but apoptosis increased [103].
The MAPK, ERK, phosphorylates FOXO3 on specific residues including Ser294, Ser425 and Ser344 which are notably different from the AKT target sites; ERK‑mediated FOXO3 phosphorylation leads to an interaction with the MDM2 (E3-ubiquitin ligase) [104]. MDM2 polyubiquitinates FOXO3 rendering it a substrate for proteasomes and proteolysis. In human breast cancer tissue, a correlation between FOXO3 and MDM2 expression was observed and a high tumor rate was associated with FOXO3- negative and MDM2-positive tissues, showing the pathological importance of this relationship [104]. ERK and AKT were shown to control FOXO3 in glioblastomas [105]. FOXO3 depletion could thus partially result in ERK-mediated tumorigenesis.
The IKK/NF-κB pathway was first recognized for its role in inflammation and the innate immune response. This pathway has more recently been recognized as a signaling pathway associated with the development of different types of cancers. IKKα and β directly target FOXO3 by inducing the phosphorylation of the Ser644 residue which starts its degradation through proteasomes and triggers nuclear exclusion [106]. FOXO3 is often restricted to the cytoplasm in acute myeloid leukemia (AML) because of phosphorylation by active IKK, but not by AKT. As a result, IKK stimulates proliferation and cell survival and FOXO favors tumorigenesis [107].
A very important member of the IKK family, IKKε, is also proficient in the phosphorylation of FOXO3 on Ser644 residue, thereby blocking apoptosis. The phosphorylation of FOXO3 on the Ser644 residue is associated with IKKε expression in human lung cancer [108]. These results implied that the regulation of FOXO3 by various IKK family members might be a key mechanism in promoting tumorigenesis.
Role of Micro-RNAs in the regulation of FOXO in cancer
Micro‑RNAs (miRs) regulate FOXO levels in cancers. Some micro-RNAs including miR‑183, miR‑182 and miR‑96 act as regulators of FOXO expression in various cancer types. Recent studies in breast cancer suggested that miR-96 overexpression leads to proliferation of tumor cells by targeting FOXO3 and FOXO1 in bladder cancer [109, 110]. The microRNA miR-182 is up-regulated and targets FOXO3 in melanoma cells and MITF, leading to enhanced invasion characteristics [111]. As well as in the MCF7 human breast cancer cell line, miR-27a along with miR‑182 and miR‑96 targeted FOXO1 and enhanced tumor cell growth [112]; in classical Hodgkin's lymphoma, FOXO1 is down-regulated, in part through the mutual action of miR-182, miR-183 and miR-96 [103]. In endometrial cancer, micro-RNAs, including miR-182, miR-183 and miR-96 down-regulate FOXO1, which guides cancer cell survival and proliferation [113]. Based on studies in cell lines, there are many other micro-RNAs which down-regulate FOXO1 and support cancer cell proliferation and cell survival for example, miR-135b in osteosarcoma cells [114], miR-370 in prostate cancer [115], miR-411 in lung cancer [116] and miR-1269 in hepatocellular carcinoma [117].
FOXOs and the regulation of angiogenesis
Angiogenesis is a process which involves the growth of new capillaries from pre-existing blood vessels. Angiogenesis plays a key role in the growth of solid tumors by providing the vasculature and perfusion required for tissue expansion [118]. The mechanism of angiogenesis involves stimulation of endothelial cells by angiogenic factors (e.g., vascular endothelial growth factor (VEGF)) to promote their migration, proliferation, invasion and tube formation [119]. FOXOs are involved in these processes as both pro- and anti‑angiogenic factors. The main supporting evidence for the pro‑angiogenic function of FOXO1 arises from embryonic development studies. Foxo1-/- mice died at E11.5 due to impaired vascular development of embryos and yolk sacs. In mature endothelial cells, FOXOs promote the expression of proangiogenic genes like vascular cell adhesion molecule‑1 (VCAM‑1) which are regulated by VEGF [120]. Additionally, Angiopoietin‑1 inactivated FOXO1, and in endothelial cells FOXO1 triggered the genes which are especially involved in vascular remodeling, destabilization of blood vessel integrity and apoptosis. In a murine hind limb ischemia model, FOXO3-/- mice had a higher capillary density 14 days after induction of ischemia compared to wild type mice. This study suggested that FOXO3 regulates vessel formation in the postnatal stage [121]. The role of FOXOs has been studied in tumor angiogenesis. Analysis of two hundred and seventy two clinical samples from patients with gastric cancer showed that FOXO1 is constitutively phosphorylated and inactivated in 85 per cent of tumors. Phosphorylation of FOXO1 increases HIF-1 and VEGF levels and promotes angiogenesis in the cancer tissue. These reports suggested that FOXO1 inactivation promotes angiogenesis in gastric cancer [122].
Crosstalk between FOXO and p53
FOXO3 interacts with the tumor suppressor p53 at different levels showing a signal transduction crosstalk between FOXO3 and p53. This crosstalk is a physical interaction [122, 123] because FOXO3 can stabilize p53 [124] or activate it indirectly via the up-regulation of p19ARF (CDKN2A), an upstream regulator of p53 [125]. Also, in fibroblasts, p53 binds to a site in the second intron of FOXO3 to induce its expression during DNA damage. In these cells, FOXO3 is not essential for p53-mediated cell cycle arrest, possibly because of damage by other factors or other FOXO isoforms. FOXO3 is required, at least partially, for p53-induced apoptosis. Furthermore, FOXO3 loss does not increase the rate of tumor development in p53 deficient mice but affects the tumor spectrum, since tumors that do not often appear in p53-/- mice (adenocarcinomas and angiolipomas) developed when both p53 and FOXO3 were deleted [126].
FOXO Mutations in Cancer
Somatic alterations in FOXO genes, along with chromosomal translocations and somatic point mutations, have been observed in few tumor cases. FOXO fusion proteins act as oncogenes. The first altered FOXO1 forkhead domain gene was seen in alveolar rhabdomyosarcoma (ARMS) a domain of forkhead gene that attaches to PAX3 resulting in an a t (2;13) translocation. Forkhead was the gene named in rhabdomyosarcoma (FKHR) and later named FOXO1 [127]. A blending between FOXO1 and PAX7 was also described previously [t (1;13) translocation] [13]. These two blended proteins hold an integral PAX DNA-binding domain (DBD) attached to the branched domain of forkhead box and the transactivation domain of FOXO1 [127, 13]. Many models have been used to recognize the oncogenic mechanism of cell transformation by the fusion protein of PAX3-FOXO1. shRNA targeting of PAX3-FOXO1, showed that this fusion protein is crucial for proliferation and transformation of the ARMS cells. However, even though the fusion protein helps in oncogenesis, it is not a strong oncogene and a high expression level is required to support tumorigenesis [128].
PAX3- FOXO1 complexes alone are not sufficient to induce tumors, but additional genetic lesions are needed as revealed through studies performed on transduced cells, transgenic or knock-in mice [129]. For illustration, conditional knock-in PAX3-FOXO1 mice did not develop tumors unless there was also anchorage restricted inactivation of p53 or CDKN2A pathway. The interruption of these two pathways had previously been observed in human ARMS and seems to work with FOXO fusion proteins to encourage tumorigenesis [130, 131].
Due to the existence of an unbroken PAX DBD, these chimeric proteins have the ability to transactivate genes from PAX-binding sites, but their transcriptional activity is higher when compared to PAX7 proteins or wild-type PAX3 [132]. The improved activity of this fusion protein could clarify tumorigenesis, at least in part, by disturbed transcription of target genes. For example, PAX3-FOXO1 up-regulated N-Myc, and this guides the fusion protein to transform cells [13,129]. BCL-XL, a type of anti-apoptotic factor, is also up-regulated via a PAX3‑FOXO1 mechanism, and is very important for survival of ARMS cells [128, 131]. However many of the genes identified in these studies still need to be validated. The reduction in one FOXO1 allele because of chromosomal translocation was also predicted to add to tumorigenesis. However, haplo insufficiency of FOXO1 does not speed up tumor expansion in mice with PAX3-FOXO1 expressed in mortally dividing muscle cells [131].
FOXO4 and FOXO3 were next recognized in fusion proteins along with MLL (mixed lineage leukemia, encoded by the KMT2A gene) in acute leukemia [t(X;11) and t(6;11) translocations, respectively]. The C-terminal part of FOXO is present in both PAX3/7- FOXO1 fusion proteins and transactivation domain of MLL-FOXO3/4 proteins [133, 134]. MLL translocations are associated with acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). Many other branches of MLL in fusion proteins have been identified. It was hypothesized that shortened MLL contributed to leukemogenesis regardless of the fusion branch as explained with mice expressing the MLL-LacZ fusion protein, which lead to hematological tumors. However, the breach could either provide a transactivation domain to MLL or alleviate the shortened MLL protein [135].
Activation of the PI3K/AKT signaling pathway, leads to decreased activity of FOXO and decreased levels of FOXO target genes like catalase and MnSOD [136]. Both MnSOD and catalase belong to a huge, miscellaneous family of antioxidant enzymes. As discussed earlier, cellular ROS levels can be modulated by PI3K/AKT/FOXO downstream of the insulin receptor. Up-regulation of MnSOD by increased FOXO expression resulted in a decrease in cellular ROS [136] and an increase in ROS improved FOXO transcriptional activity, and thus behaved as a feedback mechanism. Small GTPase Ral is activated by increased ROS, which resulted in phosphorylation of the stress kinase JNK. Activated JNK resulted in phosphorylation of the T451 and T447 sites of FOXO4. These specific phosphorylations are necessary for FOXO4 transcriptional activity as revealed by mutational analysis. H2O2 treatment increased FOXO transcriptional activity and initiated the activation and transportation of FOXO4 from the cytoplasm to the nucleus. T447/451 phosphorylation by FOXO4 can mediate transcription of catalase and MnSOD, which leads to a decrease in ROS levels. Thus, establishment of FOXO4 by oxidative stress is an element of a negative feedback loop to decrease the levels of oxidative stress within the cell, by avoiding damage to DNA, proteins and lipids [137]. The homeostatic mechanism to manage ROS levels is controlled by signaling pathways that can have both positive (Ras/Ral/ JNK) and negative (PI3K/AKT) effects on FOXO. Growth factors, including insulin, have the capacity to regulate both pathways concurrently. Cells maintain a basal level of AKT activity in the presence of serum and H2O2 treatment, even at fairly low concentrations and can activate Ral/JNK signaling which results in the activation of FOXO. Thus, AKT action is fairly low, for example, as matched to insulin-induced AKT activity, the Ral/JNK signaling pathway acts in a conflicting manner. TNFα increases FOXO transcriptional activity in A14 cells and this occurs with a marked activation of JNK and weak activation of PKB. If AKT activity dominates the JNK activity, this will result in relocation of FOXO to the cytosol. Additionally, PKB-mediated phosphorylation of FOXO also leads to ubiquitin attachment and degradation of FOXO protein [138]. The effects of acetylation and deacetylation on FOXO transcriptional activity are conflicting. During periods of low oxidative stress, FOXOs are at first activated by JNK-mediated phosphorylation, but later or at higher concentrations FOXOs are inactivated by acetylation and/or ubiquitination. Deacetylation of FOXO by Sir2 can thus extend the FOXO action induced by H2O2 treatment in array to ensure full activation of the antioxidant targets of FOXO [137].
Current Anti-Cancer Drugs Impacting FOXO Functions and Signaling Pathways
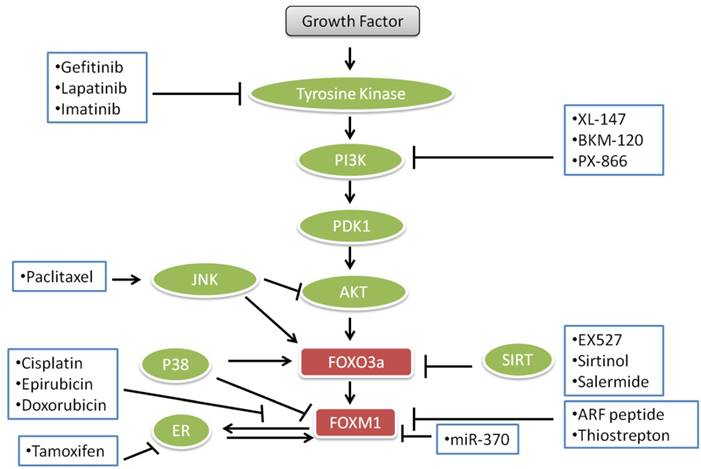
FOXO transcription factors and proteins are involved in cell death and survival and multiple signal transduction pathways which are essential for cell proliferation, angiogenesis and tumor suppressor gene functions. These roles are dysregulated in human cancers rendering FOXOs as potential targets for the treatment of cancers. Some anticancer agents like lapatinib, imatinib and paclitaxel, gefitinib, epirubicin, cisplatin, doxorubicin and different endocrine agents modulate the FOXO3a-FOXM1 transcriptional axis (Fig. 3). Lapatinib, imatinib and gefitinib, known as “tinibs” were developed and introduced as specific tyrosine kinase (TK) inhibitors. Tinibs act by binding and competing with ATP on the catalytic domain of tyrosine kinases and ultimately block the autophosphorylation of receptor-tyrosine kinases and downstream signaling. Tinibs block a small number of different TKs and their clinical action might arise from these multiple targets [139]. An array of new drugs are biologicals including monoclonal antibodies (“mabs”) such as trastuzumab, which is directed against the ERBB2 receptor extracellular domain and interferes with the PI3K-AKT -FOXO3a-FOXM1 axis signaling. Paclitaxel is a drug, which is mostly used to treat pancreatic, breast, ovarian, lung, and other cancers [140]. Paclitaxel interferes with microtubule assembly and triggers JNK, which among other actions leads to the accumulation of FOXO3a. Cisplatin, also used for the treatment of cancer, binds to DNA, leading to production of platinum-DNA adducts and ultimately inducing apoptosis [141]. Epirubicin and doxorubicin, anthracycline antibiotics, intercalate with DNA and cause DNA damage and cell death. Epirubicin, doxorubicin and cisplatin down-regulate FOXM1 expression. Doxorubicin causes nuclear translocation of FOXO3a by inducing the phosphorylation of p38 MAP kinase in a manner analogous to other drugs used for the inhibition of PI3K-AKT -FOXO3a-FOXM1 pathways for the treatment of cancer as shown in Fig. 3.
Current important anticancer drugs, their therapeutic targets and interactions with FOXO3a pathways.
Perspectives and Conclusion
Cancer is a major cause of human morbidity and mortality. Extensive research over several decades has revealed that the dysregulated cellular processes in cancer cover a wide spectrum of cellular signaling and cell biology. There have been some major advances in the treatment of a few cancers, but many remain particularly difficult to understand and treat. There is a clear need to discover and develop new agents and new delivery modalities for the treatment of a broad range of cancers. Novel drugs addressing target(s) should be efficacious with limited side effects and a low propensity for the development of resistance. The PI3K/PTEN, AKT and FOXO3a networks are hormone and growth factor regulated pathways that are dysregulated in cancer and thus represent an opportunity for the development of new drugs.
As a tumor suppressor gene, we propose in this review that activation of FOXO promotes cell cycle arrest and cancer cell apoptosis, which is beneficial for the treatment of cancer. Activation of FOXOs is constantly associated with resistance to stress and lifespan extension [142]. However, emerging data have challenged the inhibitory role of FOXOs in cancer growth. FOXOs may also enhance the proliferation, survival and invasion of cancer cells [143, 144]. These findings indicate the complicated functions of FOXOs in the process of carcinogenesis. Hence, further studies are needed to identify the specific and contextual roles of FOXOs in specific tumors. FOXO transcription factors include FOXO1, FOXO3a, FOXO4 and FOXO6. Whether the target genes for these factors are different or similar is still not well understood. FOXO transcription factors may play different, even opposite functions under different conditions [145]. Therefore, more work should be carried out to investigate the relationship between different isoforms of FOXOs, their roles, function and regulation and specific cancers. Activating FOXOs is viewed as a promising strategy for blocking the development of cancer. However, currently, there are no compounds available which target FOXOs directly and hinder the progress of cancer. Notably, as FOXOs are implicated in multiple physiological processes, chronic activation or inhibition of FOXOs might cause intolerable side effects, such as effects on glucose metabolism.
In this review, we have addressed FOXOs, FOXO proteins and their roles in different pathways involved in cancer. FOXOs are intimately involved in the very cellular processes which are dysregulated in cancer. Interfering with the role of FOXOs has the potential to block the neoplastic progression and metastasis. In the regulation of cell death and survival, there are several pathways which are involved in apoptosis and also in cancer. So, more research and enhanced knowledge of these pathways and their aberrant status in cancers may provide targets that could be used for the development of novel agents for the treatment of human cancers.
Acknowledgements
This research was financially supported by the Guangdong Provincial Project of Science and Technology (2011B050200005), the National Natural Science Foundation of China (31371088), SRG2015-00004-FHS and MYRG2016-00052-FHS from University of Macau, and the Science and Technology Development Fund (FDCT) of Macao (FDCT 021/2015/A1 and FDCT016/2016/A1).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lee TI, Richard A. Young Transcriptional Regulation and its Misregulation in Disease. Cell. 2013;152(6):1237-1251
2. Sinha S, Shukla S, Khan S. et al. Telomeric Repeat Containing RNA (TERRA): Aging and Cancer. CNS & Neurological Disorders - Drug Targets. 2015;14(7):936-946
3. Yeh JE, Toniolo PA, Frank DA. Targeting transcription factors: promising new strategies for cancer therapy. Curr Opin Oncol. 2013;25(6):652-658
4. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410-7425
5. Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med. 2012;52:7-18
6. Gomes AR, Brosens JJ, Lam EW. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle. 2008;7:3133-3136
7. Ho KK, Myatt SS, Lam EW. Many forks in the path: cycling with FoxO. Oncogene. 2008;27:2300-2311
8. Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847-859
9. Wang X, Chen WR, Xing D. A pathway from JNK through decreased ERK and Akt activities for FOXO3a nuclear translocation in response to UV irradiation. J Cell Physiol. 2012;227(3):1168-78
10. Tuteja G, Kaestner KH. Forkhead transcription factors I. Cell. 2007;130:1160
11. Maiese K, Chong ZZ, Shang YC. et al. A “FoxO” in Sight: Targeting FoxO Proteins from Conception to Cancer. Med Res Rev. 2009;29(3):395-418
12. Fu Z, Tindall DJ. FOXOs. Cancer and regulation of apoptosis. Oncogene. 2008;27:2312-2319
13. Davis RJ, D'Cruz CM, Lovell MA. et al. Fusion of PAX7 to FKHR by the variant t(1;13) (p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869-2872
14. Parry P, Wei Y, Evans G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer. 1994;11:79-84
15. Papanicolaou KN, Izumiya Y, Walsh K. Forkhead Transcription Factors and Cardiovascular Biology. Circ. Res. 2008;102:16-31
16. Franke TF, Hornik CP, Segev L. et al. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983-8998
17. Matsuzaki H, Daitoku H, Hatta M. et al. A Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl Acad. Sci. U.S.A. 2003;100:11285-11290
18. Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276-2288
19. Jacobs FM, vander HLP, Wijchers PJ. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278(38):35959-35967
20. Vander HLP, Hoekman MF, Smid MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380(Pt 2):297-309
21. Wang Y, Zhou Y, Graves DT. FOXO transcription factors: their clinical significance and regulation. BioMed Res Int. 2014:925350
22. Kikuchi S, Nagai T, Kunitama M. et al. Active FKHRL1 overcomes imatinib resistance in chronic myelogenous leukemia-derived cell lines via the production of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Sci. 2007;98(12):1949-1958
23. Ticchioni M, Essafi M, Jeandel PY. et al. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene. 2007;26(50):7081-7091
24. Furukawa-Hibi Y, Yoshida-Araki K, Ohta T. et al. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277(30):26729-26732
25. Nowak K, Killmer K, Gessner C. et al. E2F-1 regulates expression of FOXO1 and FOXO3a. Biochim Biophys Acta. 2007;1769(4):244-252
26. Paik JH, Kollipara R, Chu G. et al. FoxOs are lineagerestricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309-323
27. Sunters A, Madureira PA, Pomeranz KM. et al. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer. cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006;66(1):212-220
28. Eddy SF, Kane SE, Sonenshein GE. Trastuzumab-resistant HER2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007;67(19):9018-9023
29. Lynch RL, Konicek BW, McNulty AM. et al. The progression of LNCaP human prostate cancer cells to androgen independence involves decreased FOXO3a expression and reduced p27KIP1 promoter transactivation. Mol Cancer Res. 2005;3(3):163-169
30. Li Y, Wang Z, Kong D. et al. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J Biol Chem. 2007;282(29):21542-21550
31. Min Cui, Yunlong Huang, Yong Zhao. et al. New insights for FOXO and cell-fate decision in HIV-infection. Adv Exp Med Biol. 2009;665:143-159
32. Smith WW, Norton DD, Gorospe M. et al. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J Cell Biol. 2005;169(2):331-339
33. Hoogeboom D, Essers MA, Polderman PE. et al. Interaction of FOXO with {beta}-Catenin Inhibits {beta}-Catenin/T Cell Factor Activity. J Biol Chem. 2008;283(14):9224-9230
34. Kim JR, Jung HS, Bae SW. et al. Polymorphisms in FOXO gene family and association analysis with BMI. Silver Spring, Md. 2006:188-193
35. Kuningas M, Magi R, Westendorp RG. et al. Haplotypes in the human Foxo1a and Foxo3a genes; impact on disease and mortality at old age. Eur J Hum Genet. 2007;15(3):294-301
36. Kato M, Yuan H, Xu ZG. et al. Role of the Akt/FoxO3a pathway in TGF-beta1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J Am Soc Nephrol. 2006;17(12):3325-3335
37. Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207-46
38. Maiese K, Chong ZZ, Shang YC. et al. Rogue proliferation versus restorative protection: where do we draw the line for Wnt and forkhead signaling? Expert opinion on therapeutic targets. 2008;12:905
39. Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64:557-69
40. Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003;24:228-32
41. Chong ZZ, Kang J, Li F, Maiese K. mGluRI Targets Microglial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr Neurovasc Res. 2005;2:197-211
42. Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin and nuclear factor-kappaB. Curr Neurovasc Res. 2006;3:187-201
43. Lin SH, Maiese K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J Cereb Blood Flow Metab. 2001;21:262-75
44. Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad and caspase-mediated pathways. Br J Pharmacol. 2003;138:1107-18
45. Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003;74:37-51
46. Chong ZZ, Li F, Maiese K. Cellular demise and inflammatory microglial activation during betaamyloid toxicity are governed by Wnt1 and canonical signaling pathways. Cell Signal. 2007;19:1150-62
47. Chong ZZ, Lin SH, Kang JQ, Maiese K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol Neurobiol. 2003;23:561-78
48. Salinas M, Diaz R, Abraham NG. et al. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J Biol Chem. 2003;278:13898-904
49. Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation and injury. Histol Histopathol. 2007;22:1251-67
50. Maiese K. Diabetic stress: new triumphs and challenges to maintain vascular longevity. Expert Rev Cardiovasc Ther. 2008;6:281-4
51. Slomka M, Zieminska E, Lazarewicz J. Nicotinamide and 1-methylnicotinamide reduce homocysteine neurotoxicity in primary cultures of rat cerebellar granule cells. Acta Neurobiol Exp (Wars). 2008;68:1-9
52. Maiese K, Chong ZZ, Hou J. et al. Erythropoietin and oxidative stress. Curr Neurovasc Res. 2008;5:125-42
53. Nakamura T, Sakamoto K. Forkhead transcription factor FOXO subfamily is essential for reactive oxygen species-induced apoptosis. Mol Cell Endocrinol. 2007;281:47-55
54. Barthelemy C, Henderson CE, Pettmann B. Foxo3a induces motoneuron death through the Fas pathway in cooperation with JNK. BMC Neurosci. 2004;5:48
55. You H, Yamamoto K, Mak TW. Regulation of transactivation-independent proapoptotic activity of p53 by FOXO3a. Proc Natl Acad Sci USA. 2006;103:9051-6
56. Won CK, Ji HH, Koh PO. Estradiol prevents the focal cerebral ischemic injury-induced decrease of forkhead transcription factors phosphorylation. Neurosci Lett. 2006;398:39-43
57. Chong ZZ, Li F. Maiese K Group I Metabotropic Receptor Neuroprotection Requires Akt and Its Substrates that Govern FOXO3a, Bim and beta-Catenin During Oxidative Stress. Curr Neurovasc Res. 2006;3:107-17
58. Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24:728-43
59. Chong ZZ, Maiese K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br J Pharmacol. 2007;150:839-50
60. Caporali A, Sala-Newby GB, Meloni M. et al. Identification of the prosurvival activity of nerve growth factor on cardiac myocytes. Cell Death Differ. 2008;15:299-311
61. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-562
62. Paik JH, Kollipara R, Chu G. et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309-323
63. Yakubov E, Ghoochani A, Buslei R. et al. Hidden association of Cowden syndrome, PTEN mutation and meningioma frequency. Oncoscience. 2016;3(5-6):149-155
64. Catarzi S, Degl'Innocenti D, Iantomasi T. et al. The role of H2O2 in the platelet-derived growth factor-induced transcription of the gammaglutamylcysteine synthetase heavy subunit. Cell Mol Life Sci. 2002;59:1388-1394
65. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794-798
66. Singh KK. Mitochondria damage checkpoint, aging, and cancer. Ann N Y Acad Sci. 2006;1067:182-190
67. Petros JA, Baumann AK, Ruiz-Pesini E. et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719-724
68. Irani K, Xia Y, Zweier JL. et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649-1652
69. Suh YA, Arnold RS, Lassegue B. et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79-82
70. Nakamura T, Sakamoto K. Forkhead transcription factor FOXO subfamily is essential for reactive oxygen species-induced apoptosis. Mol Cell Endocrinol. 2008;281:47-55
71. Kajihara T, Jones M, Fusi L. et al. Differential expression of FOXO1 and FOXO3a confers resistance to oxidative cell death upon endometrial decidualization. Mol Endocrinol. 2006;20:2444-2455
72. Wickenden JA, Jin H, Johnson M. et al. Colorectal cancer cells with the BRAF(V600E) mutation are addicted to the ERK1=2 pathway for growth factorindependent survival and repression of BIM. Oncogene. 2008;27:7150-7161
73. Tothova Z, Kollipara R, Huntly BJ. et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325-339
74. Essers MA, de Vries-Smits LM, Barker N. et al. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181-1184
75. Maiese K, Chong ZZ, Li F. et al. Erythropoietin: Elucidating new cellular targets that broaden therapeutic strategies. Prog Neurobiol. 2008;85:194-213
76. Nowak K, Killmer K, Gessner C. et al. E2F-1 regulates expression of FOXO1 and FOXO3a.Biochim Biophys Acta. 2007. 1769:244-52
77. Bouchard C, Lee S, Paulus-Hock V. et al. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 2007;21:2775-87
78. Cantley L. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-1657
79. Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319-1322
80. Fengxia Y, Rifang L, Farhan M. et al. Elucidating the role of the FoxO3a transcription factor in the IGF-1-induced migration and invasion of uveal melanoma cancer cells. Biomedicine & Pharmacotherapy. 2016;84:1538-1550
81. Zheng WH, Kar S, Quirion R. Insulin-like Growth Factor-1-induced Phosphorylation of the Forkhead Family Transcription Factor FKHRL1 Is Mediated by Akt Kinase in PC12 Cells. J. Biological Chem. 2000;275:39152-39158
82. Liao R, Yan F, Zeng Z. et al. Amiodarone-Induced Retinal Neuronal Cell Apoptosis Attenuated by IGF-1 via Counter Regulation of the PI3k/Akt/FoxO3a Pathway. Mol Neurobiol. 2016:1-13
83. Wang H, Zhou X, Huang J. et al. The role of Akt/FoxO3a in the protective effect of venlafaxine against corticosterone-induced cell death in PC12 cells. Psychopharmacology. 2013;228:129
84. Alessi DR, Caudwell FB, Andjelkovic M. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα Curr. Biol. 1997;7:261-269
85. Brunet A, Park J, Tran H. et al. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001;21:952-965
86. Rena G, Guo S, Cichy S. et al. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol.Chem. 1999;274:17179-17183
87. Dijkers PF, Medema RH, Pals C. et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol. 2000;20:9138-9148
88. Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J. Biol. Chem. 1999;274:15982-15985
89. Zheng WH, Kar S, Quirion R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J. Neurochem. 2002;80:1049-1061
90. Kashii Y, Uchida M, Kirito K. et al. A member of Forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3-kinase-Akt activation pathway in erythropoietin signal transduction. Blood. 2000;96:941-949
91. Jackson JG, Kreisberg JI, Koterba AP. et al. Phosphorylation and nuclear exclusion of the forkhead transcription factor FKHR after epidermal growth factor treatment in human breast cancer cells. Oncogene. 2000;19:4574-4581
92. Nakae J, Barr V, Accili D. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000;19:989-996
93. Rena G, Woods YL, Prescott AR. et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 2002;21:2263-2271
94. Woods YL, Rena G, Morrice N. et al. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 2001;355:597-607
95. Brunet A, Bonni A, Zigmond MJ. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857-868
96. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signaling controls tumour cell growth. Nature. 2006;441(7092):424-430
97. Kharas MG, Deane JA, Wong S. et al. Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood. 2004;103(11):4268-4275
98. Scheijen B, Ngo HT, Kang H. et al. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene. 2004;23(19):3338-3349
99. Reagan-Shaw S, Ahmad N. RNA interference-mediated depletion of phosphoinositide 3-kinase activates forkhead box class O transcription factors and induces cell cycle arrest and apoptosis in breast carcinoma cells. Cancer Res. 2006;66(2):1062-1069
100. Prasad SB, Yadav SS, Das M. et al. Down regulation of FOXO1 promotes cell proliferation in cervical cancer. J Cancer. 2014;5(8):655-662
101. Kelly-Spratt KS, Philipp-Staheli J, Gurley KE. et al. Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of Kras-driven lung cancer. Oncogene. 2009;28(41):3652-3662
102. Weidinger C, Krause K, Mueller K. et al. FOXO3 is inhibited by oncogenic PI3K/Akt signaling but can be reactivated by the NSAID sulindac sulfide. J Clin Endocrinol Metab. 2011;96(9):E1361-E1371
103. Xie L, Ushmorov A, Leithauser F. et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119(15):3503-3511
104. Yang JY, Zong CS, Xia W. et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10(2):138-148
105. Sunayama J, Sato A, Matsuda K. et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells. 2011;29(9):1327-1337
106. Hu MC, Lee DF, Xia W. et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117(2):225-237
107. Chapuis N, Park S, Leotoing L. et al. IkappaB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood. 2010;116(20):4240-4250
108. Guo JP, Tian W, Shu S. et al. IKBKE phosphorylation and inhibition of FOXO3a: a mechanism of IKBKE oncogenic function. PLoS One. 2013;8(5):e63636
109. Lin H, Dai T, Xiong H. et al. Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS One. 2010;5(12):e15797
110. Guo Y, Liu H, Zhang H. et al. miR-96 regulates FOXO1-mediated cell apoptosis in bladder cancer. Oncol Lett. 2012;4(3):561-565
111. Segura MF, Hanniford D, Menendez S. et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci USA. 2009;106(6):1814-1819
112. Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284(35):23204-23216
113. Myatt SS, Wang J, Monteiro LJ. et al. Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res. 2010;70(1):367-377
114. Pei H, Jin Z, Chen S. MiR-135b promotes proliferation and invasion of osteosarcoma cells via targeting FOXO1. Mol Cell Biochem. 2015;400(1-2):245-52
115. Wu Z, Sun H, Zeng W. et al. Upregulation of MircoRNA-370 induces proliferation in human prostate cancer cells by downregulating the transcription factor FOXO1. PLoS One. 2012;7(9):e45825
116. Zhao Z, Qin L, Li S. miR-411 contributes the cell proliferation of lung cancer by targeting FOXO1. Tumour Biol. 2016;37(4):5551-60
117. Yang XW, Shen GZ, Cao LQ. et al. MicroRNA-1269 promotes proliferation in human hepatocellular carcinoma via downregulation of FOXO1. BMC Cancer. 2014;14(1):909
118. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182-6
119. Yoo SY, Kwon SM. Angiogenesis and its therapeutic opportunities. Mediators Inflamm. 2013:127170
120. Abid MR, Shih SC. et al. A novel class of vascular endothelial growth factor-responsive genes that require forkhead activity for expression. J Biol Chem. 2006;281(46):35544-35553
121. Potente M, Urbich C, Sasaki K. et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Investig. 2005;115(9):2382-2392
122. Kim SY, Yoon J, Ko YS. et al. Constitutive phosphorylation of the FOXO1 transcription factor in gastric cancer cells correlates with microvessel area and the expressions of angiogenesis- related molecules. BMC Cancer. 2011;11:264
123. Wang F, Marshall CB, Yamamoto K. et al. Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J Mol Biol. 2008;384(3):590-603
124. You H, Yamamoto K, Mak TW. Regulation of transactivation- independent proapoptotic activity of p53 by FOXO3a. Proc Natl Acad Sci USA. 2006;103(24):9051-9056
125. Bouchard C, Lee S, Paulus-Hock V. FoxO transcription factors suppress Mycdriven lymphomagenesis via direct activation of Arf. Genes Dev. 2007;21(21):2775-2787
126. Renault VM, Thekkat PU, Hoang KL. et al. The pro-longevity gene FoxO3 is a direct target of the p53 tumor suppressor. Oncogene. 2011;30(29):3207-3221
127. Galili N, Davis RJ, Fredericks WJ. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5(3):230-235
128. Xia SJ, Holder DD, Pawel BR. et al. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol. 2009;175(6):2600-2608
129. Mercado GE, Xia SJ, Zhang C. et al. Identification of PAX3-FKHRregulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosomes Cancer. 2008;47(6):510-520
130. Lagutina I, Conway SJ, Sublett J. Pax3- FKHR knock-in mice show developmental aberrations but do not develop tumors. Mol Cell Biol. 2002;22(20):7204-7216
131. Keller C, Arenkiel BR, Coffin CM. et al. Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18(21):2614-2626
132. Fredericks WJ, Galili N, Mukhopadhyay S. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15(3):1522-1535
133. Borkhardt. A, Repp R, Haas OA, et al. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23). Oncogene. 1997;14(2):195-202
134. Hillion J, Le Coniat M, Jonveaux P. et al. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997;90(9):3714-3719
135. Dobson CL, Warren AJ, Pannell R. et al. Tumorigenesis in mice with a fusion of the leukaemia oncogene Mll and the bacterial lacZ gene. EMBO J. 2000;19(5):843-851
136. Kops GJ, Dansen TB, Polderman PE. et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316-32
137. Essers MAG, Weijzen S, Vries-Smits AMM. et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. The EMBO Journal. 2004;23:4802-4812
138. Matsuzaki H, Daitoku H, Hatta M. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA. 2003;100:11285-11290
139. Bernard R, Getachew R, Kamato D. et al. Evaluation of the potential synergism of imatinib-related poly kinase inhibitors using growth factor stimulated proteoglycan synthesis as a model response. J Pharm Pharmacol. 2016;68(3):368-78
140. Paclitaxel. The American Society of Health-System Pharmacists. Retrieved January, 2. 2015
141. Apps MG, Choi EHY, Wheate NJ. The state-of-play and future of platinum drugs. Endocrine-related Cancer. 2015;22(4):219-233
142. Klotz LO, Sánchez-Ramos C, Prieto-Arroyo I. et al. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51-72
143. Storz P, Döppler H, Copland JA. et al. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol Cell Biol. 2009;29(18):4906-17
144. Sisci D, Maris P, Cesario MG. et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle. 2013;12(21):3405-20
145. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410-25
Author contact
![]() Corresponding author: Prof. Wenhua Zheng, Ph.D, MD., Faculty of Health Sciences, University of Macau, Room 4021, Building E12, Avenida de Universidade, Taipa, Macau Email: wenhuazhengmo
Corresponding author: Prof. Wenhua Zheng, Ph.D, MD., Faculty of Health Sciences, University of Macau, Room 4021, Building E12, Avenida de Universidade, Taipa, Macau Email: wenhuazhengmo