Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(8):961-975. doi:10.7150/ijbs.20074 This issue Cite
Research Paper
Vasopressin Mediates the Renal Damage Induced by Limited Fructose Rehydration in Recurrently Dehydrated Rats
Fernando E. García-Arroyo1, Edilia Tapia1,2, Mónica G Blas-Marron1, Guillermo Gonzaga1, Octaviano Silverio1, Magdalena Cristóbal1,2, Horacio Osorio1,2, Abraham S. Arellano-Buendía1,2, Cecilia Zazueta3, Omar Emiliano Aparicio-Trejo4, Juan G. Reyes-García5, José Pedraza-Chaverri4, Virgilia Soto6, Carlos Roncal-Jiménez7, Richard J. Johnson7, Laura G Sánchez-Lozada1,2 ![]()
1. Laboratory of Renal Physiopathology. INC Ignacio Chávez. Mexico City. Mexico
2. Dept. of Nephrology. INC Ignacio Chávez. Mexico City. Mexico
3. Dept. of Cardiovascular Biomedicine. INC Ignacio Chávez. Mexico City. Mexico
4. Dept. of Biology. Faculty of Chemistry, UNAM. Mexico City. Mexico
5. Sección de Estudios de Posgrado e Investigación, Escuela Superior de Medicina, IPN Mexico City. Mexico
6. Dept. of Pathology. INC Ignacio Chávez. Mexico City. Mexico
7. Div. of Renal Diseases, University of Colorado, Aurora CO, USA
Received 2017-3-14; Accepted 2017-5-26; Published 2017-7-18
Abstract

Recurrent dehydration and heat stress cause chronic kidney damage in experimental animals. The injury is exacerbated by rehydration with fructose-containing beverages. Fructose may amplify dehydration-induced injury by directly stimulating vasopressin release and also by acting as a substrate for the aldose reductase-fructokinase pathway, as both of these systems are active during dehydration. The role of vasopressin in heat stress associated injury has not to date been explored. Here we show that the amplification of renal damage mediated by fructose in thermal dehydration is mediated by vasopressin. Fructose rehydration markedly enhanced vasopressin (copeptin) levels and activation of the aldose reductase-fructokinase pathway in the kidney. Moreover, the amplification of the renal functional changes (decreased creatinine clearance and tubular injury with systemic inflammation, renal oxidative stress, and mitochondrial dysfunction) were prevented by the blockade of V1a and V2 vasopressin receptors with conivaptan. On the other hand, there are also other operative mechanisms when water is used as rehydration fluid that produce milder renal damage that is not fully corrected by vasopressin blockade. Therefore, we clearly showed evidence of the cross-talk between fructose, even at small doses, and vasopressin that interact to amplify the renal damage induced by dehydration. These data may be relevant for heat stress nephropathy as well as for other renal pathologies due to the current generalized consumption of fructose and deficient hydration habits.
Keywords: Vasopressin, chronic kidney disease
Introduction
Dehydration (preferential water loss over salt loss) has commonly been associated with a “pre-renal” type of acute kidney injury (AKI), but the condition has been viewed as completely reversible and not associated with permanent kidney damage. Recently, however, there has been increasing evidence that recurrent dehydration, especially when related to heat stress, can lead to chronic kidney disease (CKD)[1]. This fact has been an especial concern in Central America, where an epidemic of CKD has arisen among manual laborers in the sugarcane fields and other outside occupations (termed Mesoamerican Nephropathy) in which the primary risk factor is recurrent dehydration and heat stress.[2-4] Indeed, recently there has been recognition of epidemics of CKD with similar characteristics in other areas of the world, including India and Sri Lanka, leading to the suggestion that “heat stress nephropathy” might represent an emerging and major cause of CKD worldwide.[5]
Recently an animal model of CKD was developed by exposing mice to recurrent heat stress and dehydration.[6] Like the epidemics described in the literature, the primary histologic finding was tubulointerstitial fibrosis, although some glomerular ischemia was also observed.[6] The pathophysiologic mechanisms included the activation the polyol (aldose reductase-sorbitol dehydrogenase) pathway induced by heat dehydration [6]. In turn, the fructose generated from the polyol pathway can be metabolized by fructokinase which is present in the proximal tubule, leading to local adenosine triphosphate (ATP) depletion, oxidative stress, chemokine release, and uric acid generation [7]. In the mouse model of dehydration associated CKD, endogenously produced fructose was produced in the renal cortex, and proximal tubular injury was documented, and this was not observed in fructokinase deficient mice [6]. These studies led to the conclusion that activation of the polyol-fructokinase pathway is likely one of the mediator systems driving heat stress, dehydration-associated CKD.
Dehydration is also a potent stimulus for the release of vasopressin, which in turn aids urinary concentration [8]. While vasopressin was initially viewed as strictly a beneficial hormone to help prevent water loss, it also has been implicated as a mediator of renal injury, and acute effects including glomerular hyperfiltration and albuminuria have been reported.[8-10] Indeed, there is also evidence that suppression of vasopressin can slow the progression of CKD in a rodent model [11] leading to the recognition that vasopressin may also be an important mediator for heat stress-associated CKD.
Interestingly, there is emerging literature linking fructose metabolism with vasopressin.[12] For example, infusion of hypertonic fructose is known to stimulate vasopressin release in humans, whereas this is not observed with equimolar glucose.[13]. Recently we found that heat stress induces activation of the aldose reductase-fructokinase pathway in the hypothalamus and that blocking fructose metabolism can blunt the rise in vasopressin to heat stress [14]. Indeed, fructose can directly stimulate vasopressin synthesis in hypothalamic explants, but this is not observed if fructose metabolism is blocked [14]. Moreover, a sweetened beverage containing fructose markedly exacerbated the renal damage induced by mild heat dehydration in rats [15].
Here we investigated the effect of blocking vasopressin receptors V1a and V2 under conditions of mild but recurrent heat stress and limited rehydration with water or fructose. We report that blocking vasopressin receptors V1a and V2 protected against renal injury when fructose was provided as a rehydration fluid. Moreover, vasopressin also regulated the polyol-fructokinase pathway. As far as we know, this is the first study to show that vasopressin may have a causal role in heat stress nephropathy.
Methods
All the experiments were conducted according to the “Guide for the Care, and Use of Laboratory Animals” (National Academy Press, Washington, DC, 1996) and were approved by the Instituto Nacional de Cardiología Ignacio Chávez Research Committee.
Animal model
Heat-induced dehydration procedure: Male Wistar rats (200-225 g BW) were exposed to 37°C for one hour during the morning hours, after receiving a dose of vehicle (water) or conivaptan (≈3 mg/kg BW/day by intragastric gavage)[16]. Fluid for rehydration was provided ad libitum for two hours immediately after dehydration. The rest of the day and on the weekend the rats received tap water and food ad libitum. Rats were allocated to the following experimental groups (6-8 for renal hemodynamics evaluation and 5 additional animals for renal cortex mitochondrial and tissue studies):
NC= Control (no dehydration, water ad libitum)
W+V= Dehydration, tap water + vehicle
W+C= Dehydration, tap water + conivaptan
F+V= Dehydration, 10% fructose water + vehicle
F+C= Dehydration, 10% fructose water + conivaptan
Rats were weighed before and after dehydration procedure. The volume of fluid ingested during 2 h of rehydration was measured. The food and water intake the rest of the day were also measured.
Plasma testing
Creatinine (SpinReact. Girona, Spain), and urea (Wienner Lab. Rosario, Argentina) were analyzed by enzymatic based commercial kits, and sodium concentration was analyzed by an autoanalyzer. Plasma copeptin was measured by ELISA (Peninsula Laboratories. San Carlos, CA, USA), previous reverse phase extraction (SepPak cartridges, Waters. Milford, MA). Plasma uric acid was measured by an enzymatic based kit (Sekisui Diagnostics. Kent, UK). Markers of systemic inflammation included IL-1, IL-6, and TGF-beta and were evaluated by ELISA (Abcam. Cambridge, MA, USA).
Urine testing
Sixteen-hour urine was collected in metabolic cages starting during the rehydration period at 4 weeks. After the 2 h of rehydration period with the experimental fluid (water or 10% fructose), bottles containing tap water were replaced. The food and fluids were provided at libitum during urine collection. Urine was analyzed for creatinine, urea, and sodium using the same methods as for plasma samples.
N-Acetyl-β-D glucosaminidase (NAG) activity in urine samples: NAG activity was measured accordingly to published methods [17, 18]. In brief, for the determination NAG activity, 4-nitrophenyl-N-acetyl-β-D-glucosaminide was used as substrate. One unit of enzymatic activity (U) represents the amount of enzyme, which hydrolyses one µmol of substrate per min at 37°C. The results were expressed as U/min.
Blood pressure
Systolic blood pressure (SBP) was measured in conscious rats by tail-cuff sphygmomanometer (XB-1000 Kent Scientific) at week 4[17]. For each rat, 5 measurements were made, and the average was reported. Animals were trained for the procedure before starting the experiments.
Renal hemodynamics
Rats were anesthetized with sodium pentobarbital (30 mg/kg, i.p.), and placed on a homeothermic table to maintain the corporal temperature at 37°C. Trachea, both jugular veins, both femoral arteries and left ureter were catheterized with polyethylene tubing PE-240, PE-50, and PE-10, respectively. Euvolemic conditions were maintained by the infusion of 10 ml/kg of body weight of isotonic bovine serum albumin (BSA, 5 mg/dl) during surgery, followed by an infusion of isotonic saline. Mean arterial pressure (MAP) was continuously monitored with a pressure transducer (model MLT844, AD Instruments, Colorado Springs, CO, USA) and recorded on a data acquisition system (Power Lab, AD Instruments. Colorado Springs, CO, USA). A 1-mm ultrasound transit-time flow probe (TS420, Transonic Systems. Ithaca, NY, USA) filled with ultrasonic coupling gel was placed around the left renal artery to record renal blood flow (RBF). Renal vascular resistance (RVR) was calculated according to the formula RVR = MAP/RBF, and renal plasma flow (RPF) was calculated according to the formula: RPF = RBF X (1 - hematocrit). Capillaries with blood were centrifuged (MICRO-MB, Thermo Fisher Scientific Inc. Waltham, MA, USA), and the hematocrit was obtained in a microcapillary reader. At the end of the study, the kidney was washed and fixed by perfusion for ketohexokinase (KHK) immunohistochemistry.
In three additional rats of F+V and F+C groups, glomerular filtration rate (GFR) was evaluated. In those animals, experimental set up was similar as for the RBF experiments, but urine samples were taken from a bladder cannula (PE-90). In brief, a basal plasma sample was obtained, and then rats were infused with 5% polyfructosan (Inutest, Fresenius-Kabi, Austria), A 60-minute period was allowed to reach equilibrium, and plasma and urine samples were obtained. After 30 minutes, samples of plasma and urine were obtained. Polyfructosan was measured colorimetrically at 450 nm according to Davidson and Sackner[18] in plasma and urine samples to calculate GFR.
Renal cortex tissue analysis
Sorbitol, fructose, and uric acid: Sorbitol (Biovision. Milpitas, CA, USA), fructose (colorimetric assay) and uric acid (Sekisui Diagnostics. Kent, UK), were measured in cortical lysates. Sorbitol was measured directly from the lysates, fructose was further extracted with perchloric acid, and uric acid was further extracted as previously described [7].
Markers of oxidative stress: In renal cortex homogenates, lipid peroxidation (4-HNE) and protein carbonyls were evaluated by colorimetric methods.
Western-blotting: Western blot analyses for neutrophil gelatinase-associated lipocalin (NGAL, Santa Cruz Biotechnology, Dallas, TX, USA) aldose reductase (GeneTex, Irvine, CA, USA), KHK (GeneTex, Irvine, CA, USA), xanthine oxidase (Santa Cruz Biotechnology, Dallas TX, USA), vasopressin receptors V1a (Santa Cruz Biotechnology, Dallas, TX, USA) and V2 (Abcam, Cambridge, MA, USA), p22phox (Genetex, Irvine, CA, USA), gp91phox (Nox2, Genetex- Irvine, CA, USA), Nox4 (Novus, Saint Charles, MO, USA) were performed and corrected for β-actin (GeneTex, Irvine, CA, USA) for renal cortex lysates, and voltage- dependent anion carrier (VDAC, Genetex. Irvine, CA, USA) for isolated mitochondrial lysates.
KHK immunohistochemistry: A private independent Laboratory of Pathology processed the immunohistochemistry, morphometry analysis was done by one of the authors (V. Soto, Nephropathologist) in a blinded fashion. The renal expression of KHK was evaluated using an anti-KHK antibody (Genetex, Irvine, CA, USA) by morphometry. For each slide 30 non-overlapping fields of renal cortex were digitalized (X400) using a microscope (Olympus BX51). Each area was captured by a digital camera (Vr-Evolution, Media Cybernetics). The images were processed using Image-Pro-Plus, version 7.2 (Image-Pro INC, Media Cybernetics). Taking advantage of the software color recognition capabilities and the positive signal in a brown color that provided the indirect immunoperoxidase technique, the number of total brown pixels was quantified as the percentage of the total area of each field. A mean of 30 fields, evaluated in each slide, was obtained.
Respiration measurements and complexes I, II, III and IV activities determinations in isolated mitochondria obtained from renal cortex
Rats were euthanized at the end of the 4 weeks by deep anesthesia and abdominal aortae exsanguination. Both kidneys were washed by perfusion with cold PBS and the right kidney excised, divided into cortex and medulla, frozen in liquid nitrogen, and stored until further processing. The left kidney was used to prepare renal cortical mitochondria for measuring mitochondrial respiration, mitochondrial complexes, and aconitase activities and for western-blotting of Nox4, p22phox and gp91phox subunits using the constitutive mitochondrial protein VDAC1 as load control.
Mitochondria isolation: Mitochondria were isolated from one-third of the renal cortex of the right kidney that was sectioned with surgical scissors and washed with 20 mM sucrose buffer. Tissue was placed in a cold glass Dounce homogenizer with a tight-fitting B pestle, and tissue homogenate was generated by several strokes of the B pestle. The homogenate was then centrifuged at 3,000 rpm (rotor radius 8 cm), 4°C to clean off remaining blood and fat. Mitochondria were subsequently isolated by differential centrifugation at 10,000 rpm (rotor radius 8 cm) with 12% and 24% percoll (Sigma-Aldrich. St Louis, MO, USA). Mitochondrial button was suspended in sucrose buffer without EDTA for protein quantification and assay of mitochondrial respiration
Mitochondrial oxygen consumption: Was determined using a micro Clark-type oxygen electrode (Strathkelvin Instruments. North Lanarkshire, Scotland). The experiments were carried out in 1.5ml of the basic medium, containing 125 mM KCl,10 mM HEPES and 3 mM KH2PO4, pH=7.3). To assess mitochondrial respiration driven by malate-glutamate substrates (10 mM), to produce NADH and feed the respiration chain in Complex I, 15 μg of freshly obtained mitochondria were added to the reaction mixture. State 4 respiration (not coupled to ATP synthesis) was then measured. To evaluate State 3 (respiration coupled to ATP synthesis), 2 μL of adenosine diphosphate (ADP, 4.4 mg/mL) was added. Oxygen consumption was also evaluated using succinate plus rotenone (10mM succinate, 1 μg/ml rotenone) and 30 μg of freshly obtained mitochondria. Respiratory control indexes (RC) were calculated as the ratio between the State 3 and the State 4 rates.
Mitochondrial complexes I (NADH-ubiquinone oxidoreductase), II (succinate-ubiquinone oxidoreductase), III (ubiquinol-cytochrome C oxidoreductase) and IV (cytochrome c oxidase) activities were also evaluated in renal cortex isolated frozen and thawed mitochondria accordingly to methods previously reported [19].
ATP content: Renal cortical ATP levels were measured by bioluminescence using a commercial kit (Roche Molecular Biochemicals, Mannheim, Germany) accordingly to the manufacturer instructions.
Mitochondrial western-blotting: Western-blotting of Nox4, p22phox and gp91phox subunits using the constitutive mitochondrial protein VDAC1 as load control.
Aconitase activity determination in mitochondria: Aconitase activity inhibition is a reliable marker of superoxide levels both in vivo and in-vitro. Aconitase activity was measured in frozen and thawed mitochondria using a commercial kit (Cayman Chemical, Ann Arbor, MI, USA).
Statistical Analysis
Statistical analysis was performed using one-way ANOVA followed by multiple comparisons of selected pairs by T-tests with the Bonferroni correction. The following comparisons were performed: NC vs. W+V; W+V vs. W+C; W+V vs. F+V and F+V vs. F+C. All values presented were expressed as the mean ±SD. Significance was defined as P ≤ 0.05.
Results
Fluid consumption, caloric intake, and body weight loss induced by heat exposure (Table 1): Rats rehydrated with fructose drank more fluid compared to water rehydrated rats. Caloric intake from fructose amounted to 8% of overall calories, which is similar or lower than that observed among subjects ingesting a typical Western diet [20]. Conivaptan treatment increased total fluid intake regardless of the fluid used for rehydration. Overall weight loss from the procedure was 4% of BW.
Mean body weight loss induced by heat exposure and mean fluids consumption.
| Parameter/Group | NC | W+V | W+C | F+V | F+C |
|---|---|---|---|---|---|
| Mean total water intake (mL) | 38±3 | 40±4 | 46±5b | 46±5b | 52±7c |
| DBW loss (% BW) | NA | 3.6±0.3 | 3.6±0.5 | 3.7±0.5 | 4.3±0.4c |
| Mean rehydration fluid (mL/2 h) | NA | 8.8±1.7 | 10.1±1.8 | 14.7±5.8b | 14.4±5.9 |
| Total Kcal/d | 81±8 | 69±9 | 67±4 | 74±5 | 72±3 |
| Mean Kcal fructose (Kcal/d) | 6±0.3 | 6±0.2 | |||
| Final BW | 318±21 | 291±23 | 282±17 | 273±11 | 273±16 |
NC:Normal control , W+V: Water+Vehicle, W+C: Water+CVN , F+V: Fructose+Vehicle , F+C: Fructose+CVN,. DBW: Delta body weight, . a=p<0.05 vs NC; b= p<0.05 vs W+V; c=p<0.05 vs F+V.
Plasma vasopressin (copeptin) levels and V1a and V2 receptors expression (Fig 1): Heat stress was associated with an increase in plasma copeptin (vasopressin). Rehydration with fructose resulted in higher plasma copeptin levels compared to water rehydrated rats. Blockade of V1a and V2 receptors with conivaptan had no effect on copeptin levels.
Markers of vasopressin activation. A) Plasma copeptin (a stable vasopressin surrogate) measured at 4 weeks of follow-up in blood samples took in fully-hydrated state. B) Renal cortex expression of V1a and V2 vasopressin receptors. NC:Normal control , W+V: Water+Vehicle, W+C: Water+ Conivaptan , F+V: Fructose+Vehicle , F+C: Fructose+Conivaptan. *= p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
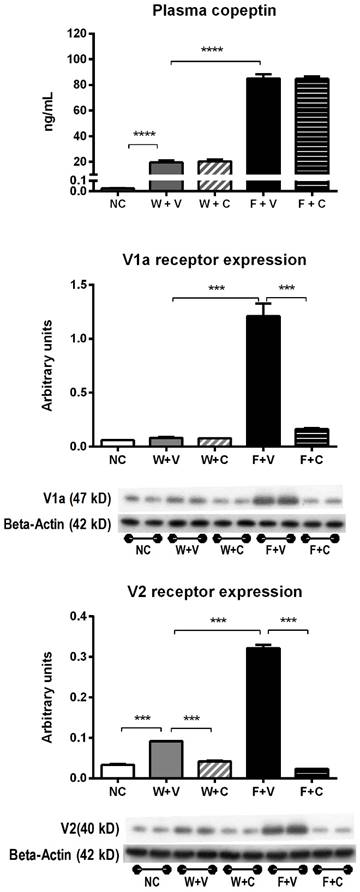
On the other hand, renal cortex expression of vasopressin receptors V1a and V2 was increased by thermal dehydration and amplified by fructose. Conivaptan was able to prevent the overexpression of vasopressin receptors in both water and fructose rehydrated animals (Fig 1).
Conivaptan treatment blocked renal damage induced by thermal dehydration
Renal functional parameters (Fig 2A) and blood pressure (Fig 2B): Decreased renal blood flow with increased renal vascular resistance was observed only in F+V rats. Those parameters were corrected by the treatment with conivaptan. Creatinine clearance (CrCl) showed a significant decrement in W+V compared to NC; this effect was further amplified in F+V group. Conivaptan treatment prevented this effect only in F+C group. We also observed similar values of CrCl in the groups subjected to renal hemodynamic studies: NC=1.31±0.07 mL/min; W+V= 1.02±0.16 mL/min, p=0.0001; W+C= 1.03±0.11 mL/min, p=ns vs W+V; F+V= 0.64±0.07 mL/min, p=0.0001 vs W+V; F+C= 0.99±0.16 mL/min, p=0.0001 vs F+V. Also, when whole GFR was measured using the marker polyfructosan in both groups of fructose rehydrated rats (n= 3, each), we observed a similar tendency: F+V = 1.4±1 mL/min, and F+C = 2.6±0.6.
Conivaptan treatment blocked renal damage induced by thermal dehydration. A) Renal hemodynamic parameters (renal blood flow and renal vascular resistance) and creatinine clearance (CrCl) evaluated at the end of 4 weeks of follow-up. B) Systolic blood pressure measured at the end of the follow-up.C) Markers of tubular damage. N-acetyl-beta-D-glucosaminidase (NAG) urine excretion and neutrophil gelatinase-associated lipocalin (N-GAL) renal cortex expression. NC:Normal control , W+V: Water+Vehicle, W+C: Water+ Conivaptan, F+V: Fructose+Vehicle , F+C: Fructose+ Conivaptan. *= p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
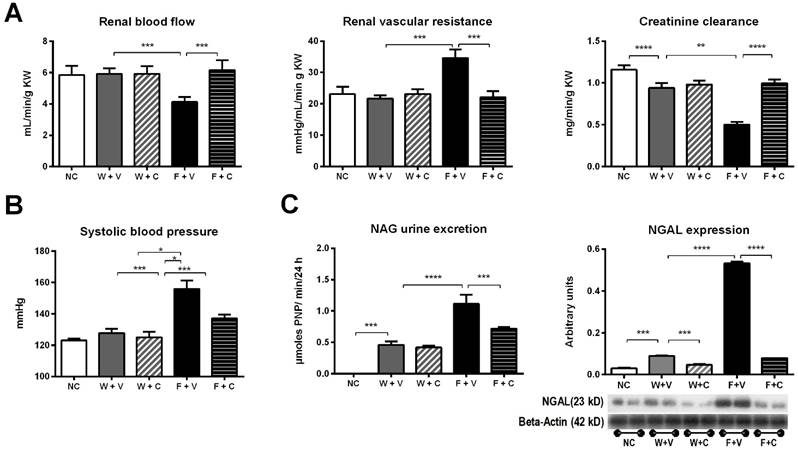
A significant increment in systolic blood pressure was noticed only in F+V group; this effect was partially alleviated by conivaptan treatment (NC= 123±3 mmHg; W+V= 128±9 mmHg; W+C= 125±12 mmHg; F+V= 156±12 mmHg; F+C= 137±6 mmHg; p<0.05 W+V vs F+V and p<0.05 F+V vs F+C). Similar findings were observed in plasma sodium concentration (NC= 138±2 mEq/L; W+V= 141±2 mEq/L; W+C= 140±2 mEq/L; F+V= 147±2 mEq/L; F+C= 143±2 mEq/L; p<0.05 W+V vs F+V and p<0.05 F+V vs F+C)
Markers of tubular injury (Fig. 2C): Urine N-Acetyl-β-D glucosaminidase (NAG) activity was increased in W+V compared to NC. This alteration was amplified in F+V group. Treatment with conivaptan partially prevented this effect. Also, we found an increased expression of neutrophil gelatinase-associated lipocalin (NGAL) in the renal cortex of W+V rats in comparison to NC. This marker of proximal tubule injury was also further increased in F+V group. Conivaptan treatment in fructose rehydrated rats prevented this effect.
Conivaptan treatment prevented the overactivation of polyol-fructokinase pathway induced by thermal dehydration
Recurrent heat-induced dehydration induces an abnormal activation of the polyol-fructokinase pathway [6, 21]. Therefore, we studied the effects of conivaptan treatment on the activation of this pathway (Figure 3). Renal cortex sorbitol concentration was significantly greater in W+V chronically dehydrated rats compared to NC rats, and conivaptan treatment did not modify this response (Fig 3A). Aldose reductase expression was also increased in those groups, and conivaptan partially prevented this effect (Fig 3B). Fructose rehydration further increased sorbitol concentration in renal cortex and significantly increased aldose reductase expression. In this condition, conivaptan partially prevented those effects (Figs 3A and 3B).
Conivaptan treatment prevented the overactivation of polyol-fructokinase pathway induced by thermal dehydration. Renal cortex sorbitol (A), fructose (C) and uric acid (G) concentrations were significantly increased by dehydration (W+V); rehydration with 10% fructose amplified these effects (F+V). Conivaptan treatment prevented the rise in cortical sorbitol in fructose-rehydrated rats (A). In parallel, aldose reductase (B) sorbitol dehydrogenase (D), KHK (E) and xanthine oxidase (F) renal cortex expressions were significantly increased by fructose rehydration. Conivaptan treatment in fructose rehydrated rats (F+C) fully prevented KHK overexpression and had a partial effect on aldose reductase, sorbitol dehydrogenase and xanthine oxidase. NC:Normal control , W+V: Water+Vehicle, W+C: Water+ Conivaptan , F+V: Fructose+Vehicle , F+C: Fructose+ Conivaptan *= p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
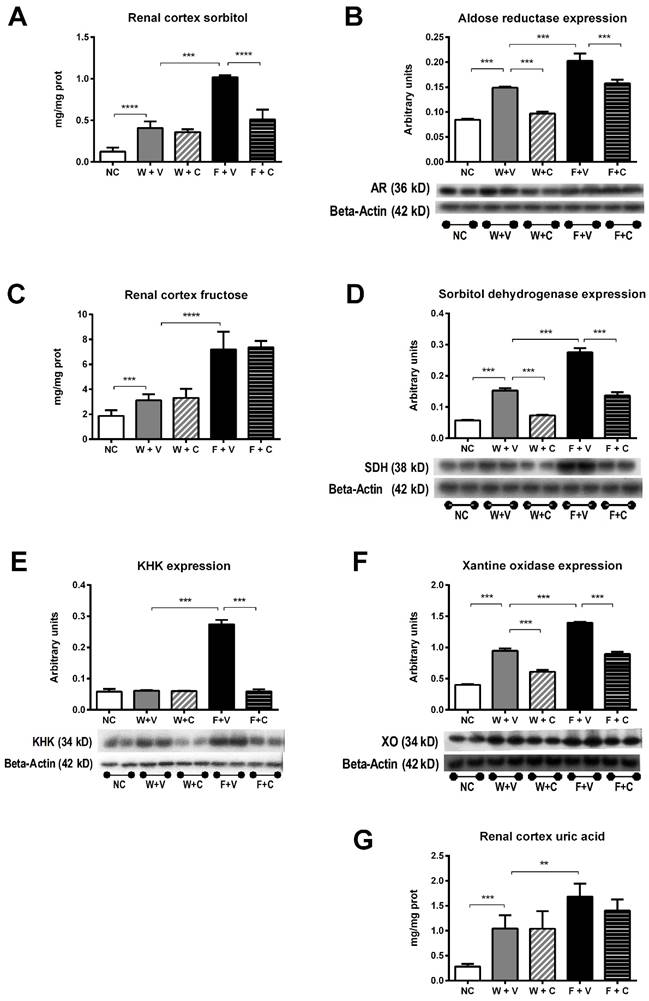
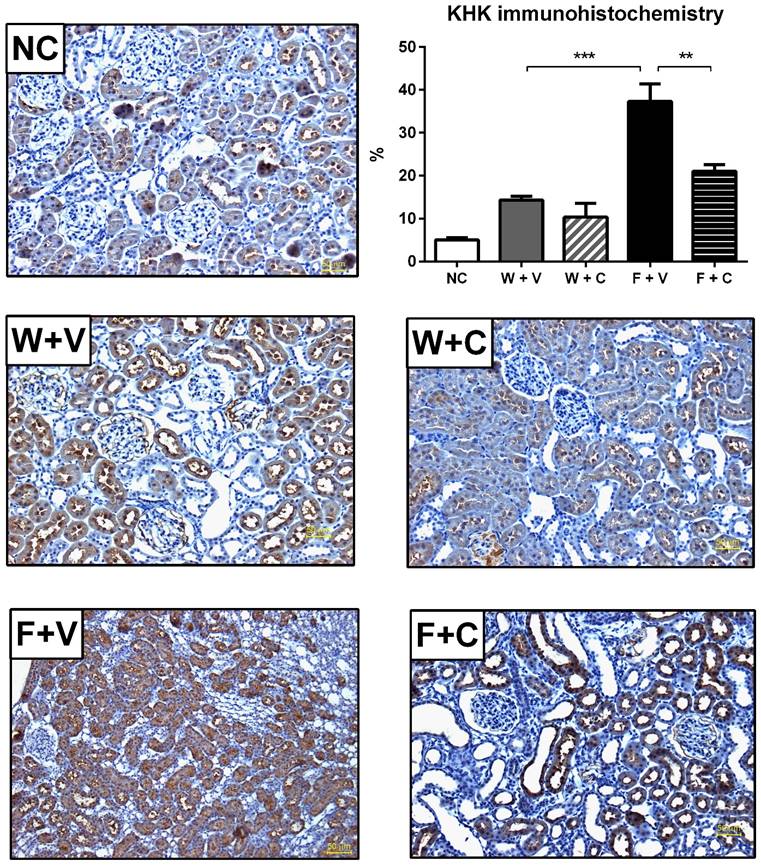
Renal cortex fructose concentration had a mildly, but significant, increment in W+V compared to NC group, in agreement with an increase in sorbitol dehydrogenase (SDH) expression and despite that KHK expression remained without changes. Conivaptan treatment partially prevented SDH overexpression and did not modify the other effects. In both groups that received fructose as rehydration fluid, there was a further increment in the concentration of renal cortex fructose. In F+V group a significant increment in SDH and KHK expression was also observed, and conivaptan treatment prevented this effect (Figs 3C and 3D). Moreover, KHK immunohistochemistry provided similar results in independent rats (Fig 4).
Conivaptan treatment prevented the overexpression of KHK by immunohistochemistry (magnification X200). It was confirmed by IMHC that thermal dehydration induces the overexpression of KHK in renal proximal tubules (identified by the presence of luminal brush border), and this effect is amplified by rehydration with fructose. Conivaptan treatment prevented this overactivation. NC:Normal control , W+V: Water+Vehicle, W+C: Water+ Conivaptan , F+V: Fructose+Vehicle , F+C: Fructose+ Conivaptan. **=p<0.01, ***=p<0.001.
Renal cortex uric acid and xanthine oxidase expression were also increased in W+V group in comparison to NC. Conivaptan treatment had a partial effect since it did not modify uric acid concentration but it prevented the overexpression of xanthine oxidase. Fructose rehydration induced a further increment in uric acid and xanthine oxidase expression in the renal cortex. Again, conivaptan treatment had a partial effect and significantly reduced xanthine oxidase overexpression without modifying uric acid concentration (Figs 3E and 3F).
Potential mechanisms by which conivaptan exerted protection in the renal damage induced by thermal dehydration
We next analyzed the mechanism likely involved in the protective effect exerted by conivaptan. There is evidence that fructose and uric acid induce renal damage through the increment of oxidative stress mediated by Nox enzymes [22]. This effect has been demonstrated to alter mitochondrial function, therefore inducing greater oxidative stress in a vicious circle.
Markers of renal cortex oxidative stress (Fig 5): Dehydrated rats that received water as rehydration fluid developed mild renal cortex oxidative stress as shown by the increase in lipid peroxidation and protein carbonylation (W+V vs. NC, p= 0.001). Conivaptan treatment prevented lipid peroxidation in this condition (W+V vs W+C, p= 0.01) (Fig 5A). Also, the expression of gp91phox was increased in W+V (p=0.01 vs. NC), and conivaptan treatment reduced the expression of gp91phox and Nox4 (W+C vs. W+V, p=0.05) (Fig 5B).
Renal cortex markers of oxidative stress. A) Thermal dehydration was associated with increased renal oxidative stress that was further augmented by fructose rehydration. Conivaptan prevented the rise in oxidative stress. B) Fructose rehydration also significantly increased the expression of p22-phox, gp91-phox and Nox4 and conivaptan treatment prevented those changes as well. NC:Normal control , W+V: Water+Vehicle, W+C: Water+ Conivaptan , F+V: Fructose+Vehicle , F+C: Fructose+ Conivaptan. *= p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
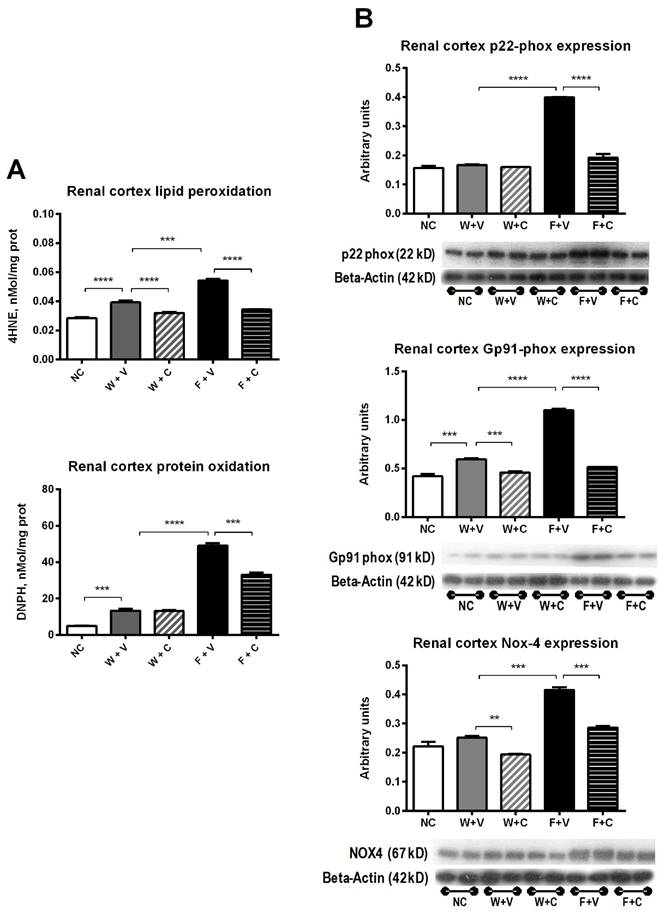
Fructose rehydration induced a further increment in renal cortex oxidative stress. Therefore, lipid peroxidation and protein carbonylation were significantly increased (F+V vs. W+V, p=0.001). Conivaptan treatment prevented this effect (F+V vs F+C, p=0.001) (Fig 5A). In agreement with those data, fructose consumption also induced a significant overexpression of p22phox, gp91phox, and Nox4 in the renal cortex. Conivaptan treatment prevented the overexpression of pro-oxidant enzymes (Fig 5B).
Markers of systemic inflammation (Table 2): Thermal dehydration induced a significant increment of plasma IL-1 beta, IL-6, and TNF-alpha in both groups that were rehydrated with water. Rehydration with fructose further increased the cytokines plasma concentration. In this case, conivaptan treatment prevented the additional increment of plasma cytokines to values similar to water rehydrated groups.
Markers of systemic inflammation.
| Parameter/Group | NC | W+V | W+C | F+V | F+C |
|---|---|---|---|---|---|
| IL-1 beta pg/mL | 11±0.6 | 19±1.5a | 18±2.2 | 60±2.7b | 24±1.3c |
| IL-6 pg/mL | 188±7 | 270±14a | 256±21 | 585±17b | 320±11c |
| TNF-alpha pg/mL | 35±1 | 60±1a | 58±3 | 79±1b | 64±1c |
NC:Normal control , W+V: Water+Vehicle, W+C: Water+CVN , F+V: Fructose+Vehicle , F+C: Fructose+CVN. IL-1: Interleukine 1, IL-6: Interleukine 6 , TNF-alpha: Tumor necrosis factor alpha. a=p<0.05 vs NC; b= p<0.05 vs W+V; c=p<0.05 vs F+V.
Mitochondrial function (Table 3): No changes in RC were noticed between W+V, W+C and NC mitochondria oxidizing either malate/glutamate or succinate plus rotenone, in spite of the substantial decrement in complex I and complex II activities in W+V rats compared to NC (-23%, p<0.001 and -28%, p<0.001, respectively). Conivaptan treatment did not modify complex I activity compared to W+V group, but significantly reduced the activity of complex II (74% of W+V, p<0.001). Fructose used as rehydration fluid affected further mitochondrial function, reducing respiratory control index when using malate-glutamate or succinate plus rotenone. Accordingly, the activities of complex I and II were significantly reduced compared to W+V (Complex I, -71%, p<0.001; complex II, -79%, p<0.001). Conivaptan treatment partially rescued the activity of both complexes. Complex III activity remained unchanged in all the experimental groups, whereas complex IV activity was equally susceptible to dehydration (W+V and F+V) with or without conivaptan (W+C and F+C). In agreement with previous results, ATP renal content was significantly decreased in heat-exposed rats, and further decreased in rats rehydrated with fructose. Conivaptan treatment prevented the fall of ATP content induced both by dehydration and fructose rehydration.
Mitochondrial function.
| Parameter/Group | NC | W+V | W+C | F+V | F+C |
|---|---|---|---|---|---|
| RCR M/G | 4.3±0.2 | 3.9±0.3a | 4.0±0.2 | 3.6±0.3b | 3.6±0.3 |
| RCR S/R | 3.4±0.3 | 3.1±0.1 | 3.2±0.3 | 2.5±0.2b | 3.4±0.3c |
| Complex I activity nM/min/mg prot | 98±7 | 76±9a | 69±9 | 22±3b | 70±7c |
| Complex II activity nM/min/mg prot | 348±39 | 251±26a | 186±36b | 52±4b | 136±15c |
| Complex III activity nM/min/mg prot | 23±3 | 20±3 | 20±4 | 22±1 | 21±6 |
| Complex IV activity nM/min/mg prot | 35±3 | 9±1a | 9±2a | 9±2 | 9±1 |
| [ATP] renal cortex nmol/mg prot | 21±6 | 14±3a | 14±2 | 10±0.3a | 18±5c |
NC:Normal control , W+V: Water+Vehicle, W+C: Water+CVN , F+V: Fructose+Vehicle , F+C: Fructose+CVN. RCR: Respiratory control rate, M/G: Malate/Glutamate substrates, S/R: Succinate/rotenone substrates, ATP: Adenosin triphosphate. a=p<0.05 vs NC; b= p<0.05 vs W+V; c=p<0.05 vs F+V.
Markers of mitochondrial oxidative stress (Fig 6): As the increase in oxidative stress negatively impacts mitochondrial function, we also evaluated aconitase activity and the expression of various Nox enzymes in mitochondrial fractions.
Renal cortex isolated mitochondria markers of oxidative stress. Thermal dehydration decreased the activity of the mitochondrial aconitase, and this was further reduced by fructose rehydration. Conivaptan prevented the decrement in aconitase activity in fructose-rehydrated rats. In parallel, fructose rehydration significantly increased the protein expression of mitochondrial p22-phox and Nox-4. Conivaptan treatment also prevented the overregulation of these proteins as well.
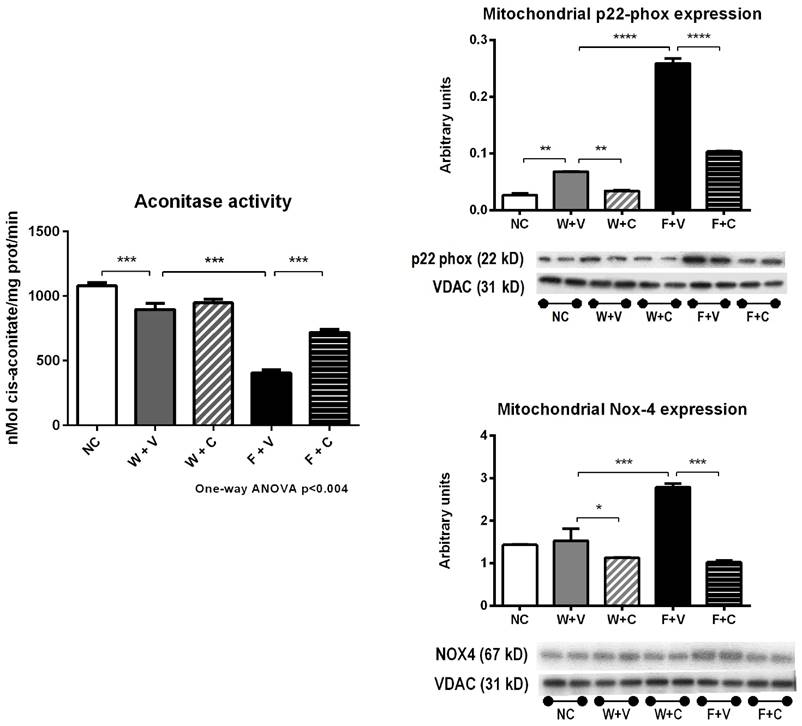
In support with the data previously presented, we found a significant reduction in aconitase activity in W+V group compared to NC (p<0.01) (Fig 6A). In parallel, p22phox expression was also increased; gp91phox and Nox4 expressions were not altered. Conivaptan treatment in rats that received water as rehydration fluid significantly reduced Nox4 expression (p<0.001) (Fig 6B).
Fructose rehydration further decreased aconitase activity (p<0.001 vs. W+V) (Fig 6A) and in concurrence, significantly increased the expression of p22phox, gp91phox, and NOX4 in mitochondrial fractions obtained from renal cortex. Conivaptan treatment suppressed the increment in these mitochondrial oxidative stress markers (Fig 6B).
Discussion
Here we demonstrated that reduced CrCl, tubular injury, oxidative stress and mitochondrial dysfunction could be induced by mild chronic recurrent heat stress and limited rehydration with a fructose-containing beverage in physiologically relevant amounts. The renal injury associated with fructose rehydration resulted in higher serum vasopressin (copeptin) levels and could be prevented by conivaptan, an inhibitor of the V1a and V2 receptors. Conivaptan also blocked the upregulation of the polyol-fructokinase pathway and V1a and V2 receptors induced by heat stress and fructose rehydration. In contrast, water rehydrated animals had less renal injury, and in this situation, conivaptan treatment was only partially effective. These studies suggest a synergistic effect of fructose with vasopressin pathways.
Role of vasopressin in renal injury induced by thermal dehydration when fructose beverage was provided as a rehydration fluid
Fructose-containing beverages produced greater renal damage in comparison to rehydration with tap water in a model of mild dehydration induced by thermal stress [21]. Here we document similar findings. A strength of this model is that the dehydration is mild (representing ≈4 % of total body weight) and is in the range observed in humans with modest exercise in hot environments. We document that vasopressin is an important mediator in this pathological process since conivaptan prevented the decrement in CrCl and also the increment in markers of tubular damage (NAG and NGAL). Moreover, renal damage was documented after only 3 cycles of thermal dehydration and rehydration with fructose in parallel with the rise in plasma copeptin (vasopressin) and KHK overexpression. Conivaptan prevented these early changes, suggesting that renal injury is slow and likely cumulative (Supplementary materials).
In addition, we found a significant decrement of RPF accompanied by an increase in RVR and a significant decrement of GFR in the F+V group. These changes were prevented by conivaptan, documenting a pivotal role for vasopressin in mediating heat stress-induced renal disease when fructose is provided as a rehydration fluid. We hypothesized that the fructose-dependent renal vasoconstriction has a perpetuating and aggravating effect on renal function. A key aspect is that the amount of fructose administered was relatively low (≈8% of daily kCal) and was limited to 2 hours each day following the dehydration procedure in contrast to other studies where rats also developed renal cortical vasoconstriction but received higher concentrations of fructose ad libitum [23, 24]. Given that one 12 oz serving of sweetened beverage (150 kCal) provides approximately 6-8% of daily kCal requirements in normal adults [25], our studies may be relevant to potential injurious mechanisms induced by sweetened drinks in the dehydrated state.
We also confirmed a rise in blood pressure in dehydrated rats rehydrated with fructose and this effect was prevented by conivaptan (Fig 2B). The effect of vasopressin receptors dual inhibition on blood pressure was previously reported in spontaneously hypertensive rats (SHR) rats, in which this treatment prevented the rise in blood pressure when initiated before the onset of hypertension[26]. Moreover, chronic activation of the V2 receptor induces increased sodium reabsorption that eventually may contribute to developing systemic hypertension [27]. Fructose rehydration was associated with a significantly higher vasopressin secretion, a higher expression of the V2 receptor in kidney cortex and a rise in plasma sodium in comparison to water rehydrated rats, suggesting that sodium reabsorption was enhanced in this group. In W+V rats, the increase in systemic vasopressin was 75% lower in comparison to fructose rehydration (F+V). Accordingly, the elevation in plasma sodium was of less magnitude in this group (W+V=141 vs. F+V=147 mEq/L, p<0.05). In addition, fructose intake is also associated with the development of systemic hypertension by mechanisms dependent and independent of its metabolism by fructokinase (such as oxidative stress and inflammation) [28]. Treatment with conivaptan in fructose rehydrated rats resulted in a significant reduction in V2 receptor expression and plasma sodium (F+V=147 vs F+C=143 mEq/L, p<0.05), suggesting that V2 receptor blockade prevented the stimulation of sodium reabsorption. Moreover, conivaptan treatment also significantly reduced fructokinase overregulation and oxidative stress. Thus, the attenuation of sodium reabsorption and the prevention of fructokinase overregulation afforded by conivaptan likely conferred an antihypertensive effect in fructose rehydrated rats.
As we showed [21], rehydration with water also was associated with a lower CrCl in comparison to non-dehydrated control rats, but this was not related to renal vasoconstriction suggesting an increase in efferent resistance mediated by a local vasoconstrictor probably.
Crosstalk between vasopressin and aldose reductase-fructokinase pathways
Fructose stimulates vasopressin secretion directly [13, 14], and we recently showed that this effect is mediated through fructokinase in hypothalamic explants [14]. Consistent with those findings, F+V rats had significantly higher levels of copeptin in comparison to W+V.
While rehydration with fructose amplified the effects of vasopressin, we also found evidence for cross-talk between vasopressin and the endogenous polyol-fructokinase pathway. Indeed, thermal dehydration induced activation of the polyol-fructokinase pathway in the kidney and this was increased when fructose was provided as part of the rehydration fluid. Moreover, the blockade of vasopressin V1a and V2 receptors has an inhibitory effect on the activation of this pathway. Therefore, conivaptan prevented the overexpression of aldose reductase, sorbitol dehydrogenase and xanthine oxidase in water rehydrated animals and in fructose rehydrated rats KHK overexpression was also suppressed by conivaptan treatment. This latter effect was noteworthy since the increase in renal fructose load induces the upregulation of its metabolic pathway[29]; thus, the fact that the blockade of vasopressin receptors was able to suppress this response suggest a strong link between both metabolic pathways, which likely is also present in other pathological situations.
Sorbitol concentration in renal tissue mirrored the effect on aldose reductase. However, fructose and uric acid concentrations remained high even in the setting of vasopressin receptors blockade. This apparent paradox might be related to the fact that exogenous fructose could have accumulated in renal tissue, and then further metabolized to uric acid. Another possibility is that fructokinase and xanthine oxidase had a compensatory increase in their enzymatic activities since conivaptan effectively prevented their overexpression.
So far it is not clear how vasopressin can regulate the polyol pathway. However, long-term systemic vasopressin might increase plasma osmolarity at least through two different pathways: 1) Increased sodium reabsorption [27], and 2) Preferential partition of extracellular water in the interstitial compartment [30]. As aldose reductase, the rate-limiting enzyme in the polyol pathway is particularly regulated by the transcription factor activated by tonicity TonEBP/NFAT5; then chronic vasopressin activation might indirectly overregulate this pathway by increasing plasma osmolarity.
Mechanisms involved in the protective effect of conivaptan
Conivaptan treatment conferred more benefit in fructose rehydrated rats: Vasopressin secretion is stimulated by dehydration due to osmotic mechanisms [31]. Accordingly, we observed that in dehydrated rats rehydrated with water (W+V) there was a significant rise in systemic copeptin (vasopressin). The importance of the present studies is that we confirmed that fructose is an additional and potent stimulus for vasopressin secretion [6, 13, 14]. An intriguing finding was that conivaptan conferred more benefit in fructose rehydrated rats, in which systemic vasopressin levels were markedly high. An explanation for this apparent paradox is that although conivaptan blocks both V1a and V2 receptors, it more potently inhibits the V2 receptor [32]. Moreover, it has been regarded that V1a receptor activation attenuates V2 receptor actions, therefore providing a protective effect in the long term [27]. As fructose rehydration induced a stronger vasopressin secretion (copeptin) compared to water (this and other studies [6, 13, 14]), it is possible that the higher levels of systemic vasopressin were able to activate V1a receptor partially, therefore conferring a superior protection in comparison to rats that received water as rehydration fluid, in which the levels of systemic vasopressin were much lower, likely allowing conivaptan to inhibit both receptors fully.
Oxidative stress: Conivaptan was able to prevent the further increase in renal oxidative stress caused by rehydration with fructose in dehydrated rats. It has been shown that increased levels of vasopressin-stimulated vascular superoxide production through activation of the V1a receptor [33, 34]. Fructose and UA also increase renal oxidative stress via activation of NOX-4 [22]. In our experimental model, conivaptan in addition to inhibiting V1a receptor also induced its downregulation. Also, conivaptan prevented KHK and XO overexpression; these effects might contribute to maintaining NOX4 expression in normal levels. Therefore, the combination of these effects likely provided an antioxidant effect.
As expected, the effect in W+V rats was less evident. Thus, we found a mild increase in oxidative stress lesser in comparison to F+V rats. Also, conivaptan had a partial effect on oxidative stress markers, thus, normalized the lipid peroxidation and decreased the expression of Nox4 and Gp91-phox.
Systemic inflammation: We documented a significant increase of systemic inflammatory cytokines in all dehydrated groups, and this effect was accentuated in fructose rehydrated rats. Thermal exposure induces an acute and delayed inflammatory response in which IL-1beta, IL-6, and TNF-alpha are consistently high in the circulation [35, 36]. The observation that cytokines were higher with fructose rehydration also implicates direct effects of fructose [37]. On the other hand, vasopressin has an immunomodulatory role [38], and it is plausible that might contribute to preserving a systemic inflammatory state. Thus, in animals rehydrated with fructose we found higher levels of plasma copeptin and systemic cytokines. The observation that conivaptan also blocked the amplification of cytokines by fructose again identifies an important cross-talk between the fructose and vasopressin pathways
Mitochondrial alterations: Mitochondria are both, the primary sources of ROS and also susceptible to oxidative stress [39]. Moreover, fructose and UA augment mitochondrial oxidative stress [40, 41]. Therefore, we determined if mitochondria were involved in the increased oxidative stress due to dehydration and rehydration with the different beverages. Fructose rehydration decreased respiratory control rate when using either malate-glutamate or succinate plus rotenone. In agreement with this data, complexes I and II activities were significantly reduced. The electrons that leak at different sites from the respiratory chain react with O2, producing superoxide radical. The main sites of physiological ROS synthesis remain to be definitively determined, but it is agreed that superoxide generated by complexes I and II is released on the matrix side [42]. Previous reports have described similar alterations in mitochondria isolated from tissues exposed acutely and chronically to hyperthermia [43-45]. In the present studies, although we did not measure the activity of mitochondrial ATP-synthase, and ATP content results from the sum of mitochondrial synthesis and glycolysis, the cortical content of ATP showed parallel changes in respiratory control rate values, which indicate that defective substrate oxidation might account for diminished ATP synthesis at the level of complexes I and II.
Formerly it was reported that the mitochondrial stress induced by hyperthermia, in addition, to producing similar responses to oxidative stress, is also a stimulus to enhance oxidative stress [46, 47]. Therefore, we evaluated aconitase activity since this enzyme is very sensitive to oxidative stress-induced damage [48]. We found that aconitase activity was decreased in dehydrated rats, and this was further reduced in fructose rehydrated animals, implicating inactivation of the enzyme mediated by oxidative stress. In our studies, we found a significant increment in renal tissue oxidative stress in heat-exposed rats, evaluated as lipid peroxidation (4-HNE) and protein carbonylation, and this effect was enhanced by fructose rehydration. In this regard, 4-HNE has been shown to be toxic to mitochondrial function and significantly reduces the activity of aconitase [49].
Increased expression of mitochondrial p22phox and Nox4enzymes are found in states of increased oxidative stress [50] therefore, we evaluated the expression of these enzymes in mitochondrial lysates. We found increased expression of both proteins in mitochondrial lysates of heat-dehydrated rats that received fructose as a rehydration fluid. Confocal microscopy has shown significant co-localization of Nox4 and p22phox with mitochondrial F1F0-ATP synthase [50]. Moreover, upregulation of mitochondrial Nox4 promotes oxidative stress and mitochondrial dysfunction in cardiomyocytes and renal cells [50, 51], and part of this effect likely is mediated by Nox4 inactivation of complex I [52]. In the present study, we found a decreased activity of complex I and increased expression of NOX4 in mitochondria; those findings were in concert with increased oxidative stress. Collectively these results suggest that increased oxidative stress induced functional alterations in renal cortex mitochondria, therefore the reduction of oxidative stress accomplished by conivaptan provided benefit in fructose rehydrated rats.
In summary, we document the importance of the type of rehydration fluid in a mild model of heat stress-induced renal damage. Specifically, rehydration with even small amounts of fructose can induce impaired renal function and tubular injury associated with oxidative stress, systemic inflammation, and mitochondrial dysfunction. The mechanism by which fructose induces renal injury is by stimulating vasopressin, and blocking vasopressin receptors with conivaptan protects against the renal injury largely. We also document a major crosstalk between vasopressin and fructose metabolism. These studies have important implications for the mechanisms by which heat stress induces renal injury, of the importance of the type of rehydration, and also on the growing recognition of the interaction of fructose and vasopressin in disease.
Supplementary Material
Supplementary information and figure.
Acknowledgements
This study was supported by generous funds from Danone Research, Palaiseau, France.
This paper is considered a contribution by the University of Colorado Climate Change and Health Consortium.
Competing Interests
RJJ and CARJ have patent applications related to blocking fructose metabolism as a means to prevent acute and chronic kidney disease. RJJ also has funding from Amway and is on the Scientific Board of Amway. RJJ also has grants from Questcor, the NIH, and the State of Colorado and is a member of Colorado Research Partners. The other authors have no conflict of interest to declare.
References
1. Johnson RJ, Rodriguez-Iturbe B, Roncal-Jimenez C, Lanaspa MA, Ishimoto T, Nakagawa T. et al. Hyperosmolarity drives hypertension and CKD-water and salt revisited. Nat Rev Nephrol. 2014;10:415-20
2. Wesseling C, Crowe J, Hogstedt C, Jakobsson K, Lucas R, Wegman DH. Resolving the enigma of the mesoamerican nephropathy: a research workshop summary. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2014;63:396-404
3. Weiner DE, McClean MD, Kaufman JS, Brooks DR. The Central American epidemic of CKD. Clinical journal of the American Society of Nephrology: CJASN. 2013;8:504-11
4. Johnson RJ, Sanchez-Lozada LG. Chronic kidney disease: Mesoamerican nephropathy-new clues to the cause. Nat Rev Nephrol. 2013;9:560-1
5. Glaser J, Lemery J, Rajagopalan B, Diaz HF, Garcia-Trabanino R, Taduri G. et al. Climate Change and the Emergent Epidemic of Chronic Kidney Disease from Heat Stress in Rural Communities: The Case for Heat Stress Nephropathy. CJASN. 2016 in press
6. Roncal Jimenez CA, Ishimoto T, Lanaspa MA, Rivard CJ, Nakagawa T, Ejaz AA. et al. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int. 2014;86:294-302
7. Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L. et al. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J Am Soc Nephrol. 2009;20:545-53
8. Bouby N, Fernandes S. Mild dehydration, vasopressin and the kidney: animal and human studies. European journal of clinical nutrition. 2003;57(Suppl 2):S39-46
9. Bardoux P, Bichet DG, Martin H, Gallois Y, Marre M, Arthus MF. et al. Vasopressin increases urinary albumin excretion in rats and humans: involvement of V2 receptors and the renin-angiotensin system. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2003;18:497-506
10. Bardoux P, Martin H, Ahloulay M, Schmitt F, Bouby N, Trinh-Trang-Tan MM. et al. Vasopressin contributes to hyperfiltration, albuminuria, and renal hypertrophy in diabetes mellitus: study in vasopressin-deficient Brattleboro rats. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10397-402
11. Bouby N, Bachmann S, Bichet D, Bankir L. Effect of water intake on the progression of chronic renal failure in the 5/6 nephrectomized rat. Am J Physiol. 1990;258:F973-9
12. Johnson RJ, Stenvinkel P, Jensen T, Lanaspa M, Roncal C, Song Z. et al. Metabolic and kidney diseases in the setting of climate change, water shortage, and survival factors. JASN. 2016 in press
13. Wolf JP, Nguyen NU, Dumoulin G, Berthelay S. Influence of hypertonic monosaccharide infusions on the release of plasma arginine vasopressin in normal humans. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 1992;24:379-83
14. Song Z, Roncal-Jimenez CA, Lanaspa-Garcia MA, Oppelt SA, Kuwabara M, Jensen T. et al. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J Neurophysiol. 2017;117:646-54
15. García-Arroyo FE, Cristobal M, Arellano-Buendia AS, Osorio H, Tapia E, Soto V. et al. Rehydration with Soft Drink-like Beverages Exacerbates Dehydration and Worsens Dehydration-associated Renal Injury. American journal of physiology Regulatory, integrative and comparative physiology. 2016 in press
16. Tomura Y, Tahara A, Tsukada J, Yatsu T, Uchida W, Iizumi Y. et al. Pharmacological profile of orally administered YM087, a vasopressin antagonist, in conscious rats. Clinical and experimental pharmacology & physiology. 1999;26:399-403
17. Tapia E, Cristobal M, Garcia-Arroyo FE, Soto V, Monroy-Sanchez F, Pacheco U. et al. Synergistic effect of uricase blockade plus physiological amounts of fructose-glucose on glomerular hypertension and oxidative stress in rats. Am J Physiol Renal Physiol. 2013;304:F727-36
18. Davidson WD, Sackner MA. Simplification of the anthrone method for the determination of inulin in clearance studies. JLab ClinMed. 1963;62:351-6
19. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235-46
20. Rizkalla SW. Health implications of fructose consumption: A review of recent data. Nutr Metab (Lond). 2010;7:82
21. Garcia-Arroyo FE, Cristobal M, Arellano-Buendia AS, Osorio H, Tapia E, Soto V. et al. Rehydration with soft drink-like beverages exacerbates dehydration and worsens dehydration-associated renal injury. Am J Physiol Regul Integr Comp Physiol. 2016;311:R57-65
22. Lanaspa MA, Tapia E, Soto V, Sautin Y, Sanchez-Lozada LG. Uric acid and fructose: potential biological mechanisms. Seminars in nephrology. 2011;31:426-32
23. Sanchez-Lozada LG, Tapia E, Jimenez A, Bautista P, Cristobal M, Nepomuceno T. et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. AmJPhysiol Renal Physiol. 2007;292:F423-F9
24. Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, Soto V, vila-Casado C, Vega-Campos IP. et al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. AJP - Renal Physiology. 2008;294:F710-F8
25. Malik VS, Schulze MB, Hu FB. Intake of sugar-sweetened beverages and weight gain: a systematic review. The American journal of clinical nutrition. 2006;84:274-88
26. Sladek CD, Blair ML, Sterling C, Mangiapane ML. Attenuation of spontaneous hypertension in rats by a vasopressin antagonist. Hypertension. 1988;12:506-12
27. Perucca J, Bichet DG, Bardoux P, Bouby N, Bankir L. Sodium Excretion in Response to Vasopressin and Selective Vasopressin Receptor Antagonists. Journal of the American Society of Nephrology. 2008;19:1721-31
28. Madero M, Perez-Pozo SE, Jalal D, Johnson RJ, Sanchez-Lozada LG. Dietary fructose and hypertension. Current hypertension reports. 2011;13:29-35
29. Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez-Lozada LG, Lanaspa MA. et al. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. AmJPhysiol Renal Physiol. 2010;298:F712-F20
30. Kunes J. Vasopressin and water distribution in rats with DOCA-salt hypertension. Journal of Hypertension. 1989;7(Suppl 6):S204-S5
31. Robertson GL. Physiology of Vasopressin Secretion and Thirst. Translational Endocrinology & Metabolism. 2012:3
32. Ingelfinger JR, Berl T. Vasopressin Antagonists. The New England journal of medicine. 2015;372:2207-16
33. Li L, Galligan JJ, Fink GD, Chen AF. Vasopressin induces vascular superoxide via endothelin-1 in mineralocorticoid hypertension. Hypertension. 2003;41:663-8
34. Faraco G, Wijasa TS, Park L, Moore J, Anrather J, Iadecola C. Water deprivation induces neurovascular and cognitive dysfunction through vasopressin-induced oxidative stress. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:852-60
35. Welc SS, Phillips NA, Oca-Cossio J, Wallet SM, Chen DL, Clanton TL. Hyperthermia increases interleukin-6 in mouse skeletal muscle. Am J Physiol Cell Physiol. 2012;303:C455-66
36. Welc SS, Clanton TL, Dineen SM, Leon LR. Heat stroke activates a stress-induced cytokine response in skeletal muscle. J Appl Physiol (1985). 2013;115:1126-37
37. Cirillo P, Sautin YY, Kanellis J, Kang DH, Gesualdo L, Nakagawa T. et al. Systemic inflammation, metabolic syndrome and progressive renal disease. Nephrol DialTransplant. 2009
38. Mavani GP, DeVita MV, Michelis MF. A review of the nonpressor and nonantidiuretic actions of the hormone vasopressin. Front Med (Lausanne). 2015;2:19
39. Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free radical biology & medicine. 2003;35:1-8
40. Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA. et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. The Journal of biological chemistry. 2012;287:40732-44
41. Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. A possible link between hepatic mitochondrial dysfunction and diet-induced insulin resistance. European journal of nutrition. 2016;55:1-6
42. Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free radical biology & medicine. 2011;51:1289-301
43. Huang C, Jiao H, Song Z, Zhao J, Wang X, Lin H. Heat stress impairs mitochondria functions and induces oxidative injury in broiler chickens. Journal of animal science. 2015;93:2144-53
44. Mujahid A, Sato K, Akiba Y, Toyomizu M. Acute heat stress stimulates mitochondrial superoxide production in broiler skeletal muscle, possibly via downregulation of uncoupling protein content. Poultry science. 2006;85:1259-65
45. Qian L, Song X, Ren H, Gong J, Cheng S. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell stress & chaperones. 2004;9:281-93
46. Slimen IB, Najar T, Ghram A, Dabbebi H, Ben Mrad M, Abdrabbah M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. International journal of hyperthermia: the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 2014;30:513-23
47. Kikusato M, Toyomizu M. Crucial role of membrane potential in heat stress-induced overproduction of reactive oxygen species in avian skeletal muscle mitochondria. PloS one. 2013;8:e64412
48. Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11168-72
49. Galam L, Failla A, Soundararajan R, Lockey RF, Kolliputi N. 4-Hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget. 2015
50. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation research. 2010;106:1253-64
51. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14385-90
52. Koziel R, Pircher H, Kratochwil M, Lener B, Hermann M, Dencher NA. et al. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. The Biochemical journal. 2013;452:231-9
Author contact
![]() Corresponding author
Corresponding author