Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(8):1019-1028. doi:10.7150/ijbs.19532 This issue Cite
Research Paper
Functional Analysis of a Novel FOXL2 Indel Mutation in Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type I
Peiwei Chai*, Fang Li*, Jiayan Fan*, Ruobin Jia*, He Zhang ![]() , Xianqun Fan
, Xianqun Fan ![]()
Department of Ophthalmology, Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, P.R. China
*These authors contributed equally to this report.
Received 2017-2-7; Accepted 2017-5-25; Published 2017-7-18
Abstract

Background: Blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) is an autosomal dominant disease with a low incidence rate. Indel mutations in the forkhead box L2 (FOXL2) gene cause two types of BPES that are distinguished by the presence (type I) or absence (type II) of premature ovarian failure (POF). The purpose of this study was to identify a possible deletion in FOXL2 in Chinese families with BPES and to clarify its relationship with POF.
Methods: An autosomal dominant Chinese BPES family with four generations was enrolled in this study. Peripheral venous blood was collected from all affected patients, and genomic DNA was extracted from leukocytes. The whole coding sequence and nearby 5' untranslated region (UTR) and 3'UTR of the FOXL2 gene were amplified using polymerase chain reaction (PCR) with three sets of overlapping primers, followed by sequencing analyses. The sequencing results were analysed using SeqMan software. Based on the patients' clinical manifestations and analysis of the identified indel mutation, we found that the mutation disturbed interactions between FOXL2 and the StAR gene. Furthermore, through subcellular localisation and functional studies, we observed significant mislocalisation of the mutant protein; the mutant protein was found in the cytoplasm, while the wild-type protein was found in the nucleus. Loss of function was confirmed by transcriptional activity assays, quantitative real-time PCR, and electrophoretic mobility shift assays.
Results: All affected patients presented with clinical features of BPES type I, including small palpebral fissures, ptosis, telecanthus, and epicanthus inversus with POF. A novel FOXL2 heterozygous indel mutation, c.19_95del, a 77-bp deletion that disrupts FOXL2 protein structure, was identified in all affected members of the family. In addition, this indel mutation significantly increased StAR mRNA expression by disrupting the ability of the FOXL2 protein to bind to the StAR promoter and act as a repressor of this gene.
Conclusions: A novel FOXL2 indel mutation was identified in Chinese families with BPES. Our results expand the spectrum of known FOXL2 mutations and provide additional insight into the structure-function relationships of the FOXL2 protein. Furthermore, this novel mutation resulted in the dysfunction of FOXL2 as a transcription factor, blocking its ability to bind to the promoter region of the StAR gene, resulting in POF in the affected patient.
Keywords: BPES, FOXL2, StAR, mutation
Background
Blepharophimosis-ptosis-epicanthus (BPES; OMIM#110100) exhibits autosomal dominant inheritance and is characterised by malformation of the eyelid. The disorder includes ptosis, epicanthus inversus, telecanthus and reduction of the horizontal fissure length [1] and has a prevalence of 1 in 50,000 [2]. BPES has been divided into two categories according to the presence or absence of infertility caused by premature ovarian failure (POF) in female patients: type I is characterised by eyelid malformation with POF, while POF is absent in type II [3]. The forkhead box L2 (FOXL2) gene (OMIM#605597) was mapped to the BPES critical region on chromosome 3q23, and mutations in this gene were subsequently associated with both types of BPES.
Human FOXL2 is an important member of the forkhead transcription factor family. The FOXL2 gene consists of a single exon of 2.7 kb located at chromosome 3q23. The gene encodes 376 amino acids, including a 100-amino-acid DNA-binding FKH domain and a poly-Ala tract [4]. The FOXL2 protein is expressed in foetal and adult granulosa cells in the ovary, in the mesenchyme of the developing eyelid and in embryonic and adult gonadotropic cells of the anterior pituitary, revealing that FOXL2 is a crucial factor in embryonic development of the ovaries and eyelids as well as in the growth of the female gonads [5]. Furthermore, in addition to its association with BPES, FOXL2 has been confirmed to be associated with the regulation of steroid metabolism, reactive oxygen species (ROS) detoxification, and inflammation in the female reproductive system [6].
To date, over 110 BPES-related mutations in over 210 families have been reported. Intragenic mutations of the FOXL2 gene make up the greatest proportion of the genetic defects detected in BPES, representing 71% of the group [7]. Frameshift mutations (most frequent), in-frame mutations, nonsense mutations and missense mutations are all observed in the FOXL2 gene. Additionally, approximately 17% of indel mutations detected in the FOXL2 gene are located outside its transcription unit [8]. Some authors claim that mutations that truncate the protein before the poly-Alanine tract usually result in type I, while mutations that extend the protein are usually associated with type II [9]. However, to date, evidence regarding the mechanism of FOXL2 dysregulation in the ovaries of BPES patients has been lacking. Previous studies have claimed that the use of a transactivation reporter system such as FLRE-luc or SIRT1-luc provides a better means of classifying BPES and enables one to test whether a mutation gives rise to POF [10]. Steroidogenic acute regulatory protein (StAR) plays a crucial role in acute regulation of steroid hormone synthesis by cleaving cholesterol into pregnenolone. StAR activity is found in granulosa cells during follicular differentiation and functional maturation of ovarian antral follicles [11]. Furthermore, FOXL2 represses StAR by binding to its promoter [12]. One mutation that results in FOXL2 dysfunction was found to disrupt FOXL2 binding with the StAR promoter and give rise to POF [13].
Here, we report a novel heterozygous FOXL2 mutation, c.19_95del, identified in Chinese families with BPES type I. This BPES type I-associated mutation, which results in a 77-bp deletion of the 15th-95th bases in the open reading frame (ORF) region of the FOXL2 gene, produced two truncated proteins: FOXL2-mutated type-1 (FOXL2-MT-1) and FOXL2-mutated type-2 (FOXL2-MT-2). FOXL2-MT-1 disrupted the overall structure of the protein, including the FKH domain and poly-Ala tract, and had only 8 amino acids in common with the wild-type (WT) FOXL2. FOXL2-MT-2 contained part of the FKH domain and the entire poly-Ala tract. In our experiment, FOXL2-MT-1 had no functional domain, whereas FOXL2-MT-2 retained its transcriptional activity; thus, two questions arose: 1) Does the FOXL2-MT-2 protein exist? 2) If the FOXL2-MT-2 protein does exist, does FOXL2 retain its transcriptional activity as the WT? Using Western blot and subcellular location analyses, we determined that FOXL2-MT-2 is expressed within both the nucleus and cytoplasm. We also found that this mutation disrupts FOXL2-MT-2 binding to the StAR promoter, an observation that clarifies the mechanism underlying POF in the female patients of BPES families.
Methods
Patients
Chinese families with BPES type I were ascertained through the Shanghai Ninth People's Hospital of Shanghai Jiaotong University School of Medicine. Nine individuals, including 5 affected individuals, were recruited (Figure 1A). An ophthalmologist performed detailed examinations of the patients and diagnosed BPES based on the following criteria: blepharophimosis, ptosis, epicanthus inversus, and telecanthus (Figure 1B). We examined DNA samples from these 9 patients. In addition, to determine the type of BPES, the clinical data of the female patients were examined in detail (Table 1). The clinical features of the patients are listed below (Table 2). The proband of the family (IV:3), a 9-year-old boy, acquired the pathogenic gene from his father. All affected patients presented with typical features of BPES, including small palpebral fissure, ptosis of the eyelids, epicanthus inversus, and telecanthus.
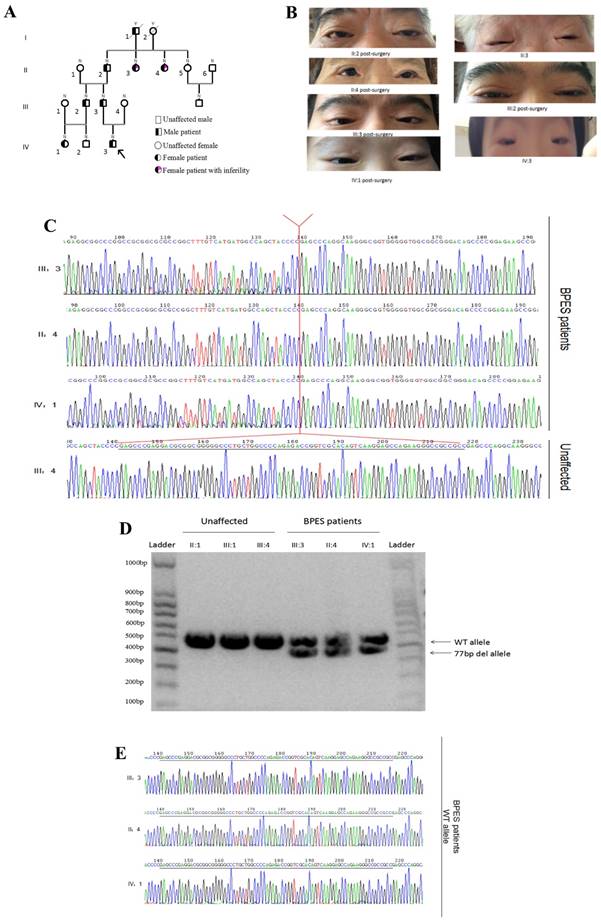
(A) Four-generation BPES type I pedigree. (B) Photographs of the faces of the probands post-surgery. (C) Genomic analysis of the cloned PCR products of the FOXL2 gene, which was inserted into the multiple cloning site (EcoRI) of the pGEM-T Easy vector. This analysis reveals the indel FOXL2 mutation [c.19_95del] found in affected members of this BPES type I family. This variant was absent in 100 control individuals, including 3 relatives of the affected families. (D) The PCR products amplified by the FOXL2-1F and FOXL2-1R sequences from samples from 3 BPES patients and unaffected family members. Gel electrophoresis (2% agarose) of the PCR products from the BPES patients revealed two fragments of 433 and 356 bp. The unaffected individuals contained a single fragment of 433 bp. The left and right lane is the DNA marker (100 bp). (E) The WT allele of BPES patients, without the c.19_95 mutation.
Clinical details of the female patients, including patient age, age at time of menarche, age at time of menopause, and number of pregnancies.
| Patient | Age | Menarche Age | Menopause Age | No. of Pregnancies |
|---|---|---|---|---|
| II-3 | 76 | 14~16 | 37 | 0 |
| II-4 | 74 | 14 | 37~38 | 0 |
| IV-1 | 8 | N/A | N/A | 0 |
Clinical features of the Chinese families with BPES.
| Patient | Age (years) | IICD (mm) | IPFH (mm) | HPFL (mm) | Levator function (mm) | |||
|---|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | RE | LE | |||
| II-2 | 77 | 33 | 4 | 4 | 24 | 25 | 2 | 2 |
| II-3 | 76 | 31 | 4 | 3 | 25 | 24 | 2 | 2 |
| II-4 | 74 | 36 | 4 | 4 | 24 | 25 | 2 | 2 |
| III-2 | 35 | 34 | 4 | 4 | 23 | 24 | 2 | 2 |
| III-3 | 33 | 34 | 2 | 3 | 23 | 24 | 2 | 2 |
| IV-1 | 8 | 33 | 3 | 3 | 22 | 23 | 2 | 2 |
| IV-3 | 4 | 31 | 2 | 3 | 22 | 22 | 2 | 2 |
Abbreviations: HPFL: horizontal palpebral fissure length; IICD: inner intercanthal distance; IPFH: vertical interpalpebral fissure height; LE: left eye; RE: right eye.
DNA extraction and sequencing
One hundred randomly healthy volunteers were evaluated using an Automatic Nucleic Acid Isolation System (QuickGene-610L; Fujifilm Life Science, Tokyo, Japan). The patients' genomic DNA was extracted from peripheral blood leukocytes (51206; QIAGEN, Hilden, Germany). The region of the FOXL2 gene was divided into three segments, and genomic fragments encompassing the FOXL2 coding sequence were amplified using the following primers: FOXL2-1F: 5#-TTGAGACTTGGCCGTAAGCG-3#; FOXL2-1R: 5#-CTCGTTGAGGCTGAGGTTGT-3# FOXL2-2F: 5#-ACAACCTCAGCCTCAACGAG-3#; FOXL2-2R: 5#-CCAGGCCATTGTACGAGTTC-3#; FOXL2-3F: 5#-GCTTCCTCAACAACTCGTGGC-3#; and FOXL2-3R: 5#-CTGCATCCTCGCATCCGTCT-3#. Polymerase chain reaction (PCR) was performed with 100 ng genomic DNA, 25 μl 2× GC-rich buffer, 8 μl of a dNTP mixture (2.5 mmol/L), 1 U LA Taq (Takara Biotechnology (Dalian) Co., Ltd), 2 μl (10 μmol/L) of each primer, and ddH2O to a final volume of 50 μl. The PCR amplification was protocol was as follows: 95 °C for 3 min; 40 cycles at 94 °C for 60 s, 63 °C or 65 °C for 30 s, and 72 °C for 30 s; and a final elongation step at 72 °C for 5 min. The PCR products were sequenced as previously described.
Western blotting
Cells were grown in 10-cm dishes and then washed twice with cold PBS when the cells covered the dish. The cells were then lysed in 200 µl RIPA lysis buffer (Beyotime Institute of Biology, Shanghai, China) and phenyl methane sulfonyl fluoride (1:100). The protein concentrations were determined using a BCA Protein Assay Kit (Beyotime Institute of Biology). SDS protein loading buffer was added to the samples, followed by boiling for 10 min. A total of 5 µg of protein from all of the samples was resolved using 10% SDS/PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were then incubated in 5% BSA in Tris-buffered saline (TBS) for 60 min to block non-specific binding and subsequently incubated at 4°C overnight with primary antibodies, followed by washing three times with TBS Tween (0.1%). The membranes were finally incubated for 60 min with the corresponding secondary antibodies, and the immunoreactive proteins were detected using an Odyssey Infrared Imaging System (LICOR Biosciences, Lincoln, NE, USA). Immunoblotting was performed using primary antibodies against EGFP (1:800, ab184601, Abcam, USA), FOXL2-1 (1:1000, 1-100 Aa, ab188584, Abcam, USA), or FOXL2-2 (1:1000; 364-376 Aa, ab5096, Abcam, USA).
Construction of the expression vector
Complementary DNA (cDNA) encoding WT FOXL2 was prepared by PCR using primers incorporating restriction enzyme sites (EcoRI-BamH1). The DNA fragment amplified from the WT gene was cloned into digested pcDNA 3.1 and EGFP-N1 plasmids, producing pcDNA3.1-FOXL2-WT and N1-FOXL2-EGFP-WT. The mutant vector was synthesized by PCR using the WT plasmids as templates with the following primers: FOXL2-MT-F: 5#-TCGAGCTCAAGCTTCGAATTCGCCACCATGATGGC-3# and FOXL2-MT-R: 5#-GCCCTTCTCGAACATGTCTTCGCAGGCCGGGTC-3#. To construct the reporter plasmid PGL3-StAR expression vector, PCR products were synthesized and inserted into the multiple cloning site of PGL3-basic (KpnI-Bg1II). The primers prepared to amplify the StAR gene promoter were as follows: StAR, sense: 5#-GGGGTACCCCCA-CAGGTTTCTGAGCCTCAATTCC-3#; and StAR, antisense: 5#-GAAGATCTTCAGCATTGCTTTGGCCGTTTG-3#. Plasmids were isolated and purified using anion exchange columns (QIAGEN, Hilden, Germany), and all constructs were sequenced to confirm that no additional mutations had been incorporated.
Subcellular location of FOXL2-WT and FOXL2-MT
Twenty-four hours before transfection, Murine leydig tumour cell line 1 (MLTC-1) cells were seeded into 6 cm dishes (1*105 cells/dish) in Dulbecco's modified Eagle's medium (DMEM; Gibco, CA, USA) containing 10% foetal calf serum (Gibco-Invitrogen, Grand Island, NY, USA) and 1% penicillin/streptomycin and maintained at 37 °C in a 5% CO2 atmosphere. The MLTC-1 cells were transfected with N1-FOXL2-EGFP, N1-FOXL2-WT-EGFP or N1-FOXL2-MT-EGFP using 3 μl Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). Subcellular localisation/aggregation was observed after 72 h of transfection by confocal laser scanning microscopy. Statistical analysis of co-localization of DAPI and EGFP are calculated by using Image J.
Electrophoretic mobility shift assay
The proteins were synthesized using a prokaryotic expression system. WT and MT encoding genes were cloned into the expression vector pET28b and confirmed using Sanger sequencing. The WT and MT proteins were induced by adding 0.5 mM IPTG when the OD600=0.8-1.0 and E. coli BL21(DE3) was cultured at 20°C overnight. The cells were collected by centrifugation at 10,000 rpm for 5 min and disrupted using high pressure crushing. The cell lysates were incubated with Ni-NTA at 4°C for 1 h and washed with 30 mM and 50 mM imidazole to remove the unspecific binding proteins. The WT and MT proteins were eluted with 300 mM imidazole and dialyzed with storage buffer (50 mM Tris-Hcl, 150 mM NaCl, 1 mM DTT, and 10% glycerol pH 8.0). The WT and MT FOXL2 proteins were identified using electrophoresis.
To prepare the fluorescent (FAM)-labelled probe STAR-1, STAR-1-F (FAM) and STAR-1-R primers were annealed and then purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, USA). Similarly, (FAM)-labelled probe STAR-2 was prepared by annealing the STAR-2-F (FAM) and STAR-2-R primers and then purified with the Wizard® SV Gel and PCR Clean-Up System (Promega, USA). FAM-labelled probes were quantified with NanoDrop 2000C (Thermo, USA). The primer sequences were as follows: StAR-1-F-FAM:
5#-FAM-TCTAAGCTGCCCCTGCTTCTCTCCCCTCCATCCCTCGCCTGGCCCTGTCCTCCCTACTCTCCCCTGC-3#; StAR-1-R: 5#-GCAGGGGAGAGTAGGGAGGACAGGGCCAGGCGAGGGATGGAGGGGAGAGAAGCAGGGGCAGCTTAGA-3#; StAR-2-F-FAM: 5#FAM-CTGTCCTCCCTACTCTCCCCTGCACCCTCCCCCGCCCCAAGCTCCCCACAAACGGCCAAAGCA-3#; and StAR-2-R: 5#-TGCTTTGGCCGTTTGTGGGGAGCTTGGGGCGGGGGAGGGTGCAGGGGAGAGTAGGGAGGACAG-3#. The electrophoretic mobility shift assay (EMSA) was performed in a 20 µl reaction volume that contained 40 ng probe and various proteins in a reaction buffer of 50 mM Tris-HCl [pH 8.0], 100 mM KCl, 2.5 mM MgCl2, 0.2 mM DTT, 2 μg salmon sperm DNA and 10% glycerol. After the reaction system was incubated for 30 min at 37°C, samples were loaded into 10% PAGE gels buffered with 0.5× TBE. Gels were scanned with an ImageQuant LAS 4000 Mini (GE Healthcare).
Target gene promoter activity assay
CHO cells were maintained in F-12K medium supplemented with 10% foetal calf serum and 1% penicillin/streptomycin. They were plated in 24-well plates at a concentration of 30,000 cells in 500 μl of culture medium per well. After 24 h, the cells were transfected with PGL3-StAR (0.5 μg/well) plus the pcDNA3.1 expression vector with WT or mutant FOXL2 cDNAs using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). The PRL-TK vector constitutively expresses Renilla luciferase and thus served as an indicator to estimate transfection efficiency. The total DNA concentration was maintained at 1 μg/200 μl culture medium by the inclusion of the empty pcDNA3.1 vector in each well. The control group was incubated with the pcDNA3.1 vector only. The cells were incubated with plasmids in Opti-MEM media (Gibco-Invitrogen) for 5 h. Then, the media was replaced with fresh media containing 10% foetal calf serum, and the cells were incubated for another 48 h. Luciferase assays were conducted using the Dual- Luciferase Reporter System (Promega). Transfected cells were solubilised in 200 μl lysis buffer. After a brief centrifugation to remove cell debris, 20 μl of cell lysate was used to measure luciferase activity on a Lumimark luminometer (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's instructions.
Real-time PCR
To evaluate StAR gene expression, quantitative SYBR Green real-time (RT) PCR was performed on an ABI 7300 system. MLTC-1 cells in six-well plates were transfected with 4 μg of pcDNA3.1 expression vector with WT or mutant FOXL2 cDNAs and an empty pcDNA3.1 vector using Lipofectamine 2000 reagent. After 48 h, the cells were cultured in DMEM supplemented with 2% bovine serum albumin for 2 h. Total mRNA was then extracted from the cells using TRIzol (Invitrogen) according to the manufacturer's instructions. cDNA was then synthesized in a 20 μl mixture. RT-PCR was performed with 2 μg RNA using Superscript II (Invitrogen). The housekeeping gene GAPDH was used as an endogenous control. The following primers were used: StAR-F: 5#-CAGACTTCGGGAACATGCCT-3#; StAR-R: 5#-CCCTTGAGGTCGATGCTGAG-3#; GAPDH-F: 5#-CTGCCAACGTGTCAGTGGTG-3#; and GAPDH-R: 5#-TCAGTGTAGCCCAGGATGCC-3#.
RESULTS
Identification of a novel indel mutation (c.19_95del) in a Chinese family with BPES type I
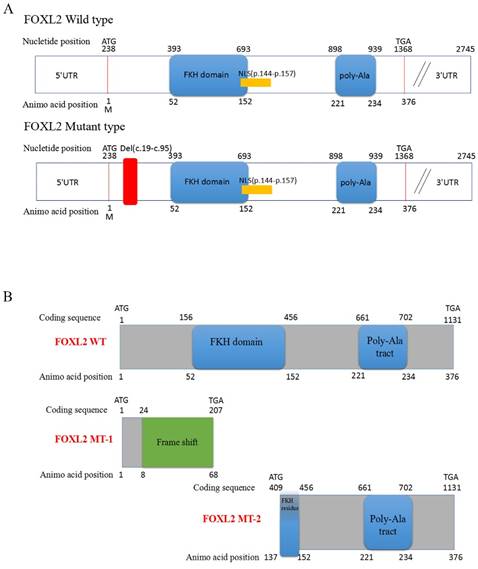
Sequencing of the coding sequence and nearby 5' untranslated region (UTR) and 3'UTR of the FOXL2 gene derived from genomic DNA revealed a novel heterozygous indel mutation (c.19_95del) that disturbs the amino acid sequence by replacing the glutamic acid (Glu) at the 7th amino acid position with alanine (Ala) (Figure 1C). As a result, patients with BPES presented with two alleles: a pathogenic allele and a WT allele (Figure 1D, 1E). This mutation was not present in 100 normal subjects or in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP). The mutation produced 2 truncated proteins, FOXL2-MT-1 and FOXL2-MT-2 (Figure 2A, 2B). FOXL2-MT-1 comprised 69 amino acids due to the introduction of a premature stop codon at 207 bp, with only 8 amino acids that were similar to the WT FOXL2 N' terminal sequence; the remaining 61 frameshift amino acid sequence was completely different from the WT FOXL2 and was terminated by a TGA base sequence after the 68th amino acid. FOXL2-MT-1 lost its function as a transcription factor, resulting from a lack of the FOXL2 functional domains (FKH domain and poly-Ala tract). Therefore, in our further experiments, we did not determine the expression of FOXL2-MT-1 or assess its function as a transcription factor. However, FOXL2-MT-2 retained 240 amino acids that were consistent with the full-length WT FOXL2 and encoded a 376 amino acid protein. FOXL2-MT-2 appeared to also possess some of its transcriptional activity due to the presence of the FKH domain (137th-152th amino acids in the FOXL2 WT sequence) and the entire poly-Ala structure (Figure 2B). Thus, it is important to determine 1) whether FOXL2-MT-2 can be expressed and 2) whether FOXL2-MT-2 retains its transcriptional activity.
Computational prediction of the c.19_95 del of FOXL2 resulting in 2 truncated proteins. (A) The deleted region of FOXL2 in the BPES patients is labelled in red. (B) Computational prediction of the 2 truncated proteins, FOXL2-MT-1 and FOXL2-MT-2.
Mutated FOXL2-MT-2 is expressed within the nucleus and cytoplasm, whereas WT FOXL2 is primarily localised within the nucleus
To further confirm that FOXL2-MT-2 could be translated into a truncated protein after FOXL2-MT-1 is pre-terminated, we performed Western blotting using the anti-EGFP tag, FOXL2-1 antibody (anti-N'terminal 1st-100th Aa), or FOXL2-2 antibody (anti-N'terminal 364th-376th Aa) in MLTC-1 cells transfected with N1-FOXL2-WT-EGFP or N1-FOXL2-MT-EGFP in Figure 3A. The FOXL2-1 and FOXL2-2 antibody targeting sequence is shown in Figure 3B. The results showed that there was a clear 72 kDa band using either the EGFP, FOXL2-1 or FOXL2-2 antibodies after the cells were transfected with N1-FOXL2-WT-EGFP. However, a smaller 51 kDa band was observed with the EGFP and FOXL2-2 antibodies after the cells were transfected with N1-FOXL2-MT-EGFP, whereas no band was observed with the FOXL2-1 antibody. These results reconfirmed that 1) FOXL2-MT-2 could be translated into a protein, even after FOXL2-MT-2 was pre-terminated and 2) FOXL2-MT-2 retained some of the FOXL2 functional structure.
(A) Western blotting showing the expression of FOXL2-WT and FOXL2-MT using two different FOXL2 antibodies and the anti-EGFP tag antibody. GAPDH was used as a reference gene.(B)The target peptide of anti-FOXL2-1 and anti-FOXL2-2.(C) Subcellular localisation of WT and indel mutant FOXL2 proteins. The middle panel corresponds to the most representative subcellular localisation of FOXL2 as a fusion protein with green fluorescent protein (EGFP-tagged). The left panel corresponds to Hoechst nuclear staining. The right panel is a merged image of the previous two images. Nuclear fluorescence obtained with the WT protein N1-FOXL2-WT-EGFP. Cytoplasmic mislocalisation is observed in the cells transfected with N1-FOXL2-MT-EGFP. Scale bar: 5 µM. (D) Statistical analysis of the nuclear and cytoplasmic distribution of FOXL2-WT and FOXL2-MT.
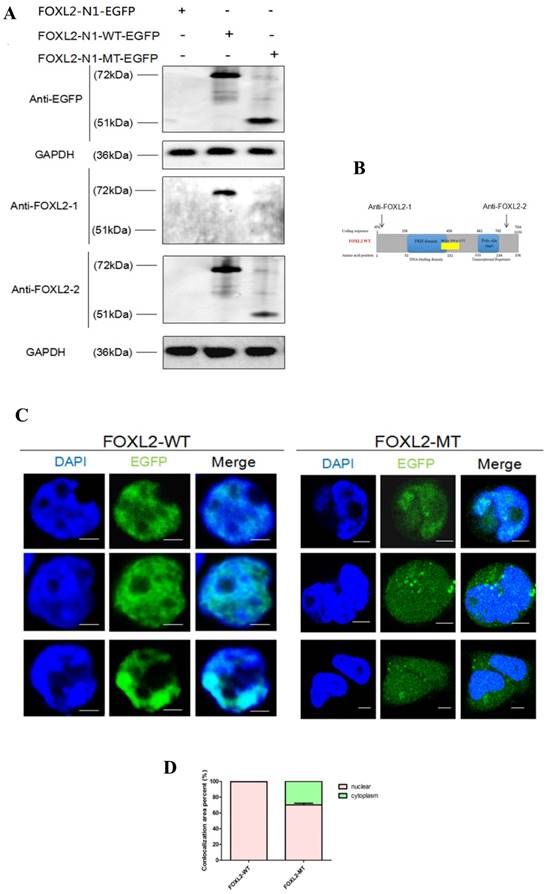
To further investigate the effect of the mutation (c.19_95del) on the subcellular localisation of the FOXL2 protein, we conducted localisation studies in MLTC-1 cells. MLTC-1 cells were confirmed to lack FOXL2 expression, eliminating the effect of endogenous expression. The cells were transfected with N1-FOXL2-WT-EGFP or N1-FOXL2-MT-EGFP. The EGFP tag was attached with FOXL2-WT and FOXL2-MT-2 (FOXL2-MT-1 is pre-terminated). Consistent with its function as a transcription factor, WT FOXL2 was diffusely distributed only in the nucleus. In contrast, MLTC-1 cells transfected with the mutant construct displayed expression in both the cytoplasm and nucleus (Figure 3C, 3D). The result indicted that the indel mutation of FOXL2 may disrupt its function as a transcription factor.
Mutated FOXL2 fails to regulate downstream StAR
We next performed dual reporter assays to measure the effects of the FOXL2 mutation (c.19_95del) on the transactivation capacity of the protein by evaluating its binding to the StAR promoter. These reported assays showed that FOXL2 exhibits strong repressor activity and that stable repression of the StAR promoter occurs. An analysis of luciferase activity clearly showed that WT FOXL2 repressed StAR promoter activity in a dose-dependent manner. In contrast, cells transfected with the mutant FOXL2 protein (c.19_95del) did not inhibit the luciferase activity of the StAR promoter reporter vector (Figure 4A). When the dose of mutant FOXL2 protein was gradually increased, luciferase activity did not show a decreasing trend.
StAR promoter activity, as determined using reporter gene assays, RT-PCR and EMSA. (A) As reflected by the luciferase activity, WT FOXL2 represses StAR promoter activity in a dose-dependent manner. However, there is no such change in luciferase activity in the presence of mutant FOXL2 or empty vector. Statistically significant differences are indicated by * P<0.05, ** P<0.01, NS, P>0.05. (B) Relative StAR mRNA expression when cells were transiently transfected with the pcDNA3.1 vector, WT FOXL2 and mutant FOXL2. Statistically significant differences are indicated by * P, 0.05 and ** P, 0.01. (C) Western blotting showing the basal expression of FOXL2 when CHO and MLTC-1 was expressed. (D) Western blotting showing the expression of FOXL2 using the anti-FOXL2-2 antibody when MLTC-1 cells were transfected with pcDNA3.1-FOXL2-WT, pcDNA3.1-FOXL2-MT or the empty vector. The WT FOXL2 protein expresses a 50 kDa band, whereas the mutated FOXL2 protein shows a 29 kDa band. (E) Electrophoresis showing the purified WT and MT-2 FOXL2 proteins. (F) The position of the FOXL2-StAR promoter complex is indicated with the arrow. EMSA indicates that the WT FOXL2 protein interacted with the StAR promoter but that the mutant-type FOXL2 abrogates these interactions. A 100-fold excess of unlabelled probe was used to demonstrate specific binding between FOXL2 and the StAR promoter. Non-specific protein (IgG) and empty probe were used as negative controls.
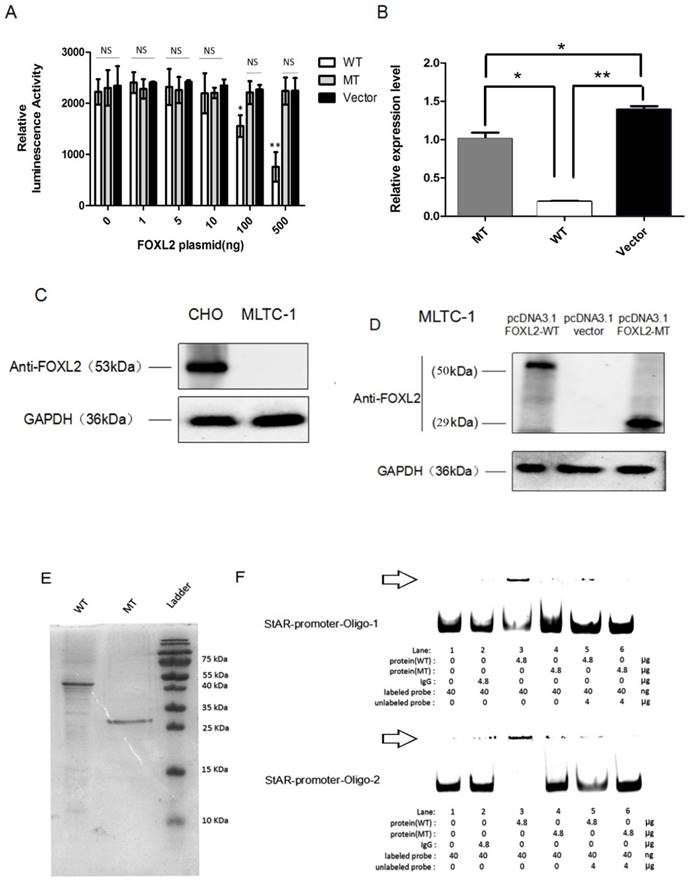
To further verify that decreased transcriptional repression of the StAR gene was due to the indel mutation in FOXL2, we performed real-time PCR analyses to measure the endogenous mRNA expression of the StAR gene following stimulation with both the WT and mutant FOXL2 proteins. As shown in Figure 4B, the MLTC-1 cells transfected with the WT protein showed the lowest endogenous expression of StAR. However, the expression of StAR with the mutant-type FOXL2 protein was similar to that of the empty vector pcDNA 3.1. The real-time PCR results confirmed that the WT FOXL2 protein acts as a repressor of the StAR gene, and the indel mutation resulted in dysregulation of the function of FOXL2 as a regulator of StAR expression. Furthermore, FOXL2 expression was absent in MLTC-1 cells, which was confirmed using Western blotting, as shown in Figure 4C. FOXL2 expression in cells transfected with either pcDNA3.1-FOXL2-WT or pcDNA3.1-FOXL2-MT is shown in Figure 4D.
Mutated FOXL2 fails to bind to the StAR promoter
To confirm whether the indel mutation reduced the binding capacity of FOXL2, recombinantly protein expressed in E. coli was used to perform EMSAs. Two probes for the StAR promoter were used, and the assay showed that the binding activity of the mutant FOXL2 protein to the StAR promoter was substantially reduced. As shown in Figure 4E and 4F, when equal amounts of WT and mutant FOXL2 protein were incubated together, a band shift indicated binding of the WT protein to the StAR promoter, which was evidenced by a prominent high molecular weight band that was observed next to the band, consistent with the labelled probe. In contrast, a faint band was detected when the mutant FOXL2 was incubated with the StAR promoter. In addition, when the probe was incubated without sample or with a non-specific IgG protein, no bands were detected (negative control). In addition, when treated with a 100-fold excess of the unlabelled probe, the binding between the FOXL2 protein and the StAR promoter was partially inhibited. EMSA confirmed that the WT FOXL2 was able to bind to the StAR gene promoter, but this direct interaction was blocked by the indel mutation of FOXL2.
DISCUSSION
FOXL2 is an evolutionarily conserved transcription factor that was first identified as a BPES susceptibility gene. It is a key regulator of sex determination, reproductive system development, metabolism, and oncogenesis and has also been shown to play an important role in the pathogenesis of diverse diseases such as polycystic ovary syndrome (PCOS), keloid and reproductive system tumours [14]. First, this study has shown that FOXL2 knockout mice (homozygous mutation) presented with many anomalies, including high perinatal mortality, low weight, reduced serum levels of IGF1, eyelid malformations and POF. In addition, this study also found that the heterozygous deletion of foxl2 resulted in subfertility [15]. In our opinion, FOXL2 plays a fundamental role in physical growth and female reproductive system development; thus, a one allele mutation could result in decreased functional foxl2 expression, and the mutation of two of the FOXL2 alleles could be lethal. Therefore, typical BPES patients contain the heterozygous mutation.
For example, conditional deletion of FOXL2 in mice gives rise to two phenotypes. In the first, granulosa cells are transdifferentiated into testis-specific Sertoli-like cells, and in the second, thecal cells are transformed into Leydig-like cells [16]. These findings provide strong evidence that FOXL2 plays a crucial role in the pathogenesis of PCOS. Additionally, FOXL2 is known to be a main coordinator in GnRH homeostasis and cholesterol metabolism; these hormones directly influence keloid formation [17].
To date, over 110 mutations have been identified in 210 families with BPES worldwide. Intragenic mutations are identified in 70% of BPES cases. Notably, 93% of the in-frame mutations lead to poly-Ala expansions [18]. Before any functional studies were conducted, type I BPES was hypothesised to result from FOXL2 truncation. Poly-Ala expansion account for 30% of the reported mutations in the FOXL2 ORF and often give rise to type II BPES. However, there is not a gold standard to distinguish between the two kinds of BPES. Other studies have also determined that poly-Ala expansion can also give rise to POF [19]. Thus, to put forward a better method of distinguish BPES type according to the mutation of FOXL2 is of promising diagnostic value. Currently, the new classification scheme for BPES is based on whether the FOXL2 mutation will disrupt the protein's function as a transcription factor, causing the loss of its regulatory control over target genes related to ovarian development, such as SIRT-1 or StAR [20].
The major problem for type I BPES female patients, however, is infertility rather than eyelid malformation. In our case, the two affected females (II3/II4) suffered from infertility. While they began menstruating at approximately 14 years old, they each had an irregular menarche cycle, experiencing oligomenorrhea and then amenorrhoea before 38 years of age. In our study, we discovered a novel 77-bp indel mutation located in c.19-c.95 of the FOXL2 gene that gives rise to 2 truncated proteins: FOXL2-MT-1, a 68 amino acid protein due to a pre-stop codon, and FOXL2-MT-2, a 240 amino acid protein that initiates translation at the next ATG. We experimentally confirmed that FOXL2-MT-2 could be translated into a protein and that the subcellular localisation of FOXL2-MT-2 was in both the nucleus and cytoplasm. We next investigated how this type of FOXL2 mutation results in POF. StAR is a crucial gene in ovarian development and has been shown to be closely related to POF. We found that the mutation disrupted the function of FOXL2 as a transcription factor and prevented its binding to the StAR promoter, causing it to fail to down-regulate the expression of StAR. In short, our research identified this novel mutation in FOXL2 and demonstrated its relationship to POF. However, much work still needs to be done. StAR is only one of the target genes of FOXL2, and POF is a complex disease that involves the dysfunction of multiple genes. Is there another gene that is also regulated by FOXL2 and participates in the pathogenesis of POF? Could identifying such a gene assist in treating the side effects of BPES type I? These questions have yet to be answered.
In conclusion, our work provides additional support for previously reported genotype-phenotype correlations and expands the spectrum of known FOXL2 gene mutations (c.19_95del). In type I BPES families, POF is a symptom associated with FOXL2 mutations. We also determined the molecular mechanism through which this mutation disrupts FOXL2-mediated regulation of StAR expression, which results in POF.
Acknowledgements
This work was supported by the Scientific Research Program of the National Health and Family Planning Commission of China (201402014), the National Natural Science Foundation of China (grant 31470757), the Program for Professors of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning (1410000159), the SMC-ChenXing Yong Scholar Program (2014, Class B), and the Science and Technology Commission of Shanghai (grants 14JC1404100, 14JC1404200, and 14430723100). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
BPES: Blepharophimosis-ptosis-epicanthus inversus syndrome; POF: Premature ovarian failure; FOXL2: Forkhead box L2; PCR: Polymerase chain reaction; StAR: Steroidogenic acute regulatory protein; ROS: Reactive oxygen species; DMEM: Dulbecco's modified Eagle's medium; FOXL2-MT-1: FOXL2-Mutated Type-1; FOXL2-MT-2: FOXL2-Mutated Type-2; FOXL2-WT: FOXL2-wild type; HPFL: horizontal palpebral fissure length; IICD: inner intercanthal distance; IPFH: vertical interpalpebral fissure height; LE: left eye; RE: right eye.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Anttonen M, Pihlajoki M, Andersson N. et al. FOXL2, GATA4, and SMAD3 co-operatively modulate gene expression, cell viability and apoptosis in ovarian granulosa cell tumor cells. PLOS ONE. 2014;9:e85545
2. Alexiadis M, Chu S, Leung D. et al. Transcriptomic analysis of stage 1 versus advanced adult granulosa cell tumors. Oncotarget. 2016;7:14207-19
3. Bertho S, Pasquier J, Pan Q. et al. Foxl2 and its relatives are evolutionary conserved players in gonadal sex differentiation. Sex Dev. 2016;10:111-29
4. De Baere E. [Role of FOXL2 impairment in syndromic and non-syndromic premature ovarian failure (POF)]. Ann Endocrinol (Paris). 2010;71:214. doi: 10.1016/j.ando.2010.03.001
5. Fan JY, Han B, Qiao J. et al. Functional study on a novel missense mutation of the transcription factor FOXL2 causes blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). Mutagenesis. 2011;26:283-9
6. Gulati R, Verdin H, Halanaik D. et al. Co-occurrence of congenital hydronephrosis and FOXL2-associated blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). Eur J Med Genet. 2014;57:576-8
7. Kim JH, Bae J. Differential apoptotic and proliferative activities of wild-type FOXL2 and blepharophimosis-ptosis-epicanthus inversus syndrome (BPES)-associated mutant FOXL2 proteins. J Reprod Dev. 2014;60:14-20
8. Kommoss S, Anglesio MS, Mackenzie R. et al. FOXL2 molecular testing in ovarian neoplasms: diagnostic approach and procedural guidelines. Mod Pathol. 2013;26:860-7
9. Kuo FT, Bentsi-Barnes IK, Barlow GM. et al. Mutant forkhead L2 (FOXL2) proteins associated with premature ovarian failure (POF) dimerize with wild-type FOXL2, leading to altered regulation of genes associated with granulosa cell differentiation. Endocrinology. 2011;152:3917-29
10. Lu W, Zheng X, Liu S. et al. SNP Rs1511412 in FOXL2 gene as a risk factor for keloid by meta analysis. Int J Clin Exp Med. 2015;8:2766-71
11. Men Y, Fan Y, Shen Y. et al. The steroidogenic acute regulatory protein (StAR) is regulated by the H19/let-7 axis. Endocrinology. 2016;158:402-9
12. Moumné L, Dipietromaria A, Batista F. et al. Differential aggregation and functional impairment induced by polyalanine expansions in FOXL2, a transcription factor involved in cranio-facial and ovarian development. Hum Mol Genet. 2008;17:1010-9
13. Fan JY, Zhou YX, Huang XL. et al. The combination of polyalanine expansion mutation and a novel missense substitution in transcription factor FOXL2 leads to different ovarian phenotypes in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) patients. Hum Reprod. 2012;27:3347-57
14. Nakashima M, Chung S, Takahashi A. et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet. 2010;42:768-71
15. Uda M, Ottolenghi C, Crisponi L. et al. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum Mol Genet. 2004;13:1171-81
16. Nallathambi J, Moumné L, De Baere E. et al. A novel Polyalanine expansion in FOXL2: the first evidence for a recessive form of the blepharophimosis syndrome (BPES) associated with ovarian dysfunction. Hum Genet. 2007;121:107-12
17. Settas N, Anapliotou M, Kanavakis E. et al. A novel FOXL2 gene mutation and BMP15 variants in a woman with primary ovarian insufficiency and blepharophimosis-ptosis-epicanthus inversus syndrome. Menopause. 2015;22:1264-8
18. Verdin H, De Baere E. FOXL2 impairment in human disease. Horm Res Paediatr. 2012;77:2-11
19. Wang Y, Yuan K, Li X. et al. Leukemia inhibitory factor stimulates steroidogenesis of rat immature Leydig cells via increasing the expression of steroidogenic acute regulatory protein. Growth Factors. 2016;34:166-76
20. Yang Y, Yang C, Zhu Y. et al. Intragenic and extragenic disruptions of FOXL2 mapped by whole genome low-coverage sequencing in two BPES families with chromosome reciprocal translocation. Genomics. 2014;104:170-6
Author contact
![]() Corresponding authors: He Zhang, Ph.D., Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, 200025, P.R. China, E-mail: zhangheedu.cn. Xianqun Fan, M.D, Ph.D., Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, 200025, P.R. China, E-mail: shfanxqnet
Corresponding authors: He Zhang, Ph.D., Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, 200025, P.R. China, E-mail: zhangheedu.cn. Xianqun Fan, M.D, Ph.D., Ninth People's Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, 200025, P.R. China, E-mail: shfanxqnet