Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(6):682-692. doi:10.7150/ijbs.24081 This issue Cite
Research Paper
Berberine suppresses apoptosis and extracellular matrix (ECM) degradation in nucleus pulposus cells and ameliorates disc degeneration in a rodent model
Yu Chen1,2#, Zengming Zheng1,2#, Jianle Wang1, Chengxuan Tang1, Sinan Khor3, Jian Chen1, Xibang Chen1, Zengjie Zhang1, Qian Tang1, Chenggui Wang1, Yiting Lou1, Zhouguang Wang2 ![]() , Jian Xiao1
, Jian Xiao1 ![]() , Xiangyang Wang1
, Xiangyang Wang1 ![]()
1. Department of Orthopaedic Surgery, The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, 325027 Wenzhou, China
2. Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, 325035, Wenzhou, China
3. Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY 10461, U.S.A.
4. Department of Orthopaedic Surgery, Ruian Peoples Hospital & the 3rd Hospital Affiliated to Wenzhou Medical University, 325200 Ruian, China
# These authors contributed equally to this work.
Received 2017-11-29; Accepted 2018-2-13; Published 2018-4-30
Abstract

Intervertebral disc degeneration (IVDD) is a chronic disease with complicated pathology involving nucleus pulposus (NP) cell apoptosis and extracellular matrix (ECM) degradation. Previous studies have shown that moderate autophagy has a protective effect against apoptosis in NP cells. Berberine (BBR) is an alkaloid compound with many beneficial properties including antimicrobial, anti-inflammatory, antioxidative, and anti-apoptotic activity. Recently, it was found to induce autophagy in various tissues as well. Thus, we hypothesized that BBR may exert a therapeutic effect on IVDD through autophagy activation. In this study, we investigated the effects of BBR on IVDD and delineated a potential mechanism. BBR treatment in vitro inhibited the expression of pro-apoptotic proteins induced by tert-butyl hydroperoxide (TBHP), and increased the expression of anti-apoptotic Bcl-2. Furthermore, it prevented ECM degradation by inhibiting the production of matrix-degrading enzymes. Additionally, BBR treatment significantly activated autophagy in NP cells. However, autophagy inhibition markedly suppressed BBR's effects on NP cell apoptosis and ECM degeneration, indicating that autophagy activation with BBR treatment is protective against IVDD. In vivo, BBR treatment increased the expression of LC3 in disc cells and prevented the development of IVDD in a needle puncture-induced rat model. Thus, BBR stimulates autophagy as a protective mechanism against NP cell apoptosis and ECM degeneration, revealing its therapeutic potential in the treatment of IVDD.
Introduction
Low back pain (LBP) induced by intervertebral disc degeneration (IVDD) has been estimated to affect approximately 80% of the adult population at least once in their lives and imposes a heavy social and economic burden on individuals, communities and countries(1). With increased lifespan and slower aging in the general population, the incidence rate of IVDD is predicted to increase in the future. However, there is no effective intervention to prevent IVDD because its pathogenesis involves a variety of pathological pathways and mechanisms that remain unclear. Excessive apoptosis of disc cells has long been considered as a key pathogenic mechanism as well as a potential therapeutic target in IVDD (2).
The intervertebral disc is comprised of a central gelatinous nucleus pulposus (NP) and a tough outer annulus fibrosus (AF), sandwiched between the upper and lower cartilage endplates(3). The NP enables the intervertebral disc to confront diverse mechanical impacts due its highly hydrated structure, which is physiologically important for its function(4). Within the NP, chondrocyte-like NP cells are embedded in a loosely organized matrix composed primarily of type II collagen and Aggrecan, supplemented with other proteoglycans. The extracellular matrix (ECM) is produced by NP cells, which are the main component of the gelatinous tissues of the NP. Excessive apoptosis of NP cells disrupts the normal metabolic activity within the ECM, resulting in an unbalanced synthesis of matrix components and aberrant activity of matrix degradation enzymes such as MMPs and ADAMTS, which eventually leads to IVDD(5).
Autophagy is a lysosome-dependent cellular degradation pathway through which cells degrade altered and dysfunctional organelles, membranes, and cytosolic macromolecules to protect against cellular stress(6). Autophagy is associated with apoptosis in the pathological process of many diseases, including osteoarthritis(7), Alzheimer's disease(8), and diabetic cardiomyopathy(9). Studies from our group and other groups have found evidence that autophagy could be a protective mechanism against excessive apoptosis of NP cells under stress(10).
Berberine (BBR) is an isoquinoline alkaloid isolated from a variety of plants such as rhizoma coptidis, cortex phellodendri and mahonia bealei. BBR is known to exert extensive pharmacological activity as an anti-inflammatory(11), antioxidative (12), and anti-apoptotic(13) compound in various diseases. In several recent studies, BBR was found to have beneficial effects on musculoskeletal disorders such as rheumatoid arthritis due to its anti-inflammatory properties(14). More recently, it was reported that BBR could alleviate high glucose-induced podocyte apoptosis via autophagy(15). In addition, BBR can stimulate autophagy in various tissues including heart(16), lungs(17) and hepar(18), however, this has not been shown in the NP. Considering that oxidative stress, apoptosis and inflammatory activation are all elements of IVDD pathology, we hypothesize that BBR may utilize autophagy to prevent the formation of IVDD.
To study this, we utilized tert-butyl hydroperoxide (TBHP) to induce oxidative stress, which is a common pathological mechanism of apoptosis in NP cells. We investigated the effects of BBR on apoptosis in NP cells under oxidative stress and examined matrix components and matrix metalloproteinases. We further investigated if the protective effects of BBR on stressed NP cells are mediated, at least partially, through regulation of autophagy. Finally, we evaluated the therapeutic potential of BBR in a puncture-induced rat IVDD model.
Materials and methods
Ethics statement
All surgical interventions, treatments and postoperative animal care procedures were performed in strict accordance with the Animal Care and Use Committee of Wenzhou Medical University (wydw 2014-0129).
Reagents and antibodies
Berberine (PHR1502), tert-butyl hydroperoxide (TBHP) (458139), dimethyl sulphoxide (DMSO) (D2650), 3-methyladenine (3-MA) (M9281) and type II collagenases (1148090) were purchased from Sigma-Aldrich (St Louis, MO, USA). Cell-Counting Kit-8 (CCK-8) (CK04) was purchased from Dojindo (Kumamoto, Japan). Primary antibodies against Collagen II (ab34712), Aggrecan (ab3778), MMP-13 (ab39012), ADAMTS-5 (ab41037), Bax (ab32503), Bcl-2 (ab59348), Cytochrome C (ab90529), P62 (ab56416) and GAPDH (ab8245) were purchased from Abcam (Cambridge, MA, USA). LC3 (12741S), Beclin-1 (3495S), ATG7 (8558S) and Cleaved-caspase3 (9664S) antibodies were acquired from CST (Beverly, MA, USA). Dulbecco's modified Eagle's medium (DMEM)/F12 (11320082), and fetal bovine serum (FBS) (10099141) were purchased from Gibco (Grand Island, NY, USA). FITC-labeled and horseradish peroxidase-labeled secondary antibodies (BS10000, BS13278) were purchased from Bioworld (MN, USA). 4', 6-diamidino-2-phenylindole (DAPI) (C1002) was obtained from Beyotime (Shanghai, China).
Isolation and culture of NP cells
NP cells were isolated and cultured as described previously (19, 20). Thirty young male Sprague-Dawley rats (150-200 g) were euthanized with an overdose of 10 % chloralhydrate. The lumbar vertebrae were removed and disc tissues were collected. NP tissues were isolated and then cut into small pieces, followed by digestion in 0.1% type II collagenase (Gibco) for approximately 4 h at 37°C. Next, the digested tissues were suspended and seeded into tissue culture flasks. NP cells were grown in DMEM/F12 with 10% fetal bovine serum and 1% penicillin/streptomycin antibiotics (Gibco, Invitrogen, Grand Island, NY) and maintained with 5% CO2 at 37 °C. Medium was changed after 7 days incubation when the adherence and migration of NP cells was observed. At 80% confluency, the cells were harvested using 0.25% Trypsin-EDTA (Gibco, Invitrogen). Next, cells were counted and subcultured into 10 cm culture plates at an appropriate density. Cells from passage 1 to 3 were used for all experiments due to no significant changes during cells passaging.
Cell viability assay
Cell viability was determined using a CCK-8 kit. NP cells were seeded onto 96-well plates (5000/well) for 24 h and then treated with different concentrations of BBR with or without TBHP for 24 h. Subsequently, the cells were incubated with CCK-8 solution at 37°C for 4 h. The absorbance of the wells was measured at 450 nm by a micro-plate reader (Leica Microsystems, Germany). BBR used for in vitro experiments was dissolved in DMSO and diluted in phosphate buffered saline (PBS) immediately before use (DMSO < 0.1%). All experiments were performed in triplicate.
Transmission electron microscopy
NP cells were fixed in 2.5% glutaraldehyde overnight, post-fixed in 2% osmium tetroxide for 1 h, and stained with 2% uranyl acetate for another 1 h. Then, samples were dehydrated in ascending acetone solutions, embedded into araldite, and cut into semi-thin sections. Sections were subsequently stained with toluidine blue to locate cells that were finally recorded with a transmission electron microscope (Hitachi, Tokyo, Japan).
Western blotting
Total protein was extracted from the NP cells using ice-cold RIPA and PMSF, and the protein concentration was quantified using the BCA protein assay kit (Beyotime, Shanghai, China). The equivalent of 60 μg protein samples were loaded onto an SDS-PAGE gel and transferred to a PVDF membrane (Bio-Rad, USA). Membranes were blocked with 5% skim milk for 1.5 h at room temperature and subsequently washed three times for 7 min in Tris-buffered saline with Tween-20 (TBST). The membranes were incubated with primary antibodies specific to Collagen II (1:1000), Aggrecan (1:200), MMP-13 (1:1000), ADAMTS-5 (1:1000), Bax (1:1000), Bcl-2 (1:500), Cleaved-caspase3 (1:1000), Cytochrome C (1:500), ATG7 (1:1000), P62 (1:1000), Beclin-1 (1:1000), LC3 (1:1000) and GAPDH (1:5000) in TBST at 4°C overnight. After washing with TBST three times for 7 min, the membranes were incubated with respective secondary antibodies for 1 h at room temperature. Signals were visualized using the ChemiDicTM XRS+ Imaging System (Bio-Rad Laboratories, Hercules, CA, USA), and the band densities were quantified with Image Lab 3.0 software (Bio-Rad).
Immunofluorescence
For LC-3 and Cleaved-caspase3 staining, NP cells were seeded on slices in a six-well plate at a density of 5 × 105 cells/ml and incubated overnight. After treatment, samples were fixed with 4% paraformaldehyde for 15 min followed by permeation using 0.1% Triton X-100 diluted in PBS for 15 min. Cells were blocked with 5% bovine serum albumin (BSA) for 1 h at 37°C, rinsed with PBS, and incubated with primary antibodies diluted in PBS: LC3 (1:200) and Cleaved-caspase3 (1:200) overnight at 4 °C. The following day, glass plates were washed and incubated with Alexa Fluor®488 labeled or Alexa Fluor®594 conjugated second antibodies (1:1000) for 1 h in a 37°C oven and labeled with DAPI for 5 min. Finally, three random microscopic fields per slide were observed under a fluorescence microscope (Olympus Inc., Tokyo, Japan).
TUNEL assay
DNA fragmentation was detected using an In Situ Cell Death Detection Kit (Roche, South San Francisco, CA, USA). After fixation with 4% paraformaldehyde for 1 h, cells were incubated with 3% H2O2 and 0.1% Triton X-100 for 10 min and washed with PBS three times in every step. In accordance with standard protocols, cells were stained with TUNEL inspection fluid and DAPI under lucifugal conditions. Finally, three fields of each slide were chosen randomly for microscopic observation with a fluorescence microscope (Olympus Inc., Tokyo, Japan).
Surgical procedure
A total of 48 male Sprague Dawley rats, aged 2 months, were used for the in vivo experiments. They were randomly divided into three groups (control group n=16, IVDD group n=16 and BBR group n=16) and anaesthetized by intraperitoneal(i.p.) injection of 10 % chloral hydrate (3.6 ml/kg). The IVDD group and the BBR group underwent the following operation. As described previously(21), the experimental level rat tail disc (Co7/8) was located by digital palpation on the coccygeal vertebrae, which was further confirmed by the trial radiograph. Needles (27G, about 4mm in length) were used to puncture the whole layer of AF though the tail skin perpendicularly. All the needles were rotated 360° and kept in position for 1 min. After surgery, BBR dissolved in 0.9% NaCl solution was administered intragastrically using syringe feeding (150 mg/kg body weight) once per day in the BBR group, while the IVDD group was given an equal amount of saline solution. Daily feeding of solutions started on the day of operation, and continued until the rats were sacrificed. All animals were allowed free movement, and were monitored every day to ensure their well-being.
Magnetic resonance imaging method
Magnetic resonance imaging (MRI) was performed with a 3.0 T clinical magnet (Philips Intera Achieva 3.0MR) on the rats at 0 week, 4 weeks and 8 weeks after operation. Sagittal T2-weighted images were taken to evaluate the signal and structural changes of the discs. T2-weighted sections in the sagittal plane were obtained in the following settings: fast-spin echo sequence with time to repetition (TR) of 5400 ms and time to echo (TE) of 920 ms; 320 (h) 9 256 (v) matrix; field of view of 260; and 4 excitations. The section thickness was 2 mm with a 0-mm gap. The MRIs were evaluated by another blinded orthopedic researcher using the classification of intervertebral disk degeneration as reported by Pfirrmann et al.(22). (1 point = Grade I, 2 points = Grade II, 3 points = Grade III, 4 points = Grade IV).
Histopathologic analysis
After MRI examination, rats were euthanized by i.p. administration of overdose 10% chloral hydrate and the caudal vertebraes including experimental disc segments were removed. Specimens were then fixed in 4% paraformaldehyde for 24 h, decalcified in neutral 10% (v/v) EDTA solution for 1 month, embedded in paraffin, and sectioned (5 µm) along the midsagittal plane. Sections were stained with Safranin-O to assess disc condition. The cellularity and morphology of NP were evaluated in a blinded manner according to a grading scale(21). Histologic score was 5 for normal disc, 6-11 for moderately degenerated disc, and 12-14 for severely degenerated disc.
Immunohistochemical and immunofluorescence for tissues
Sagittal paraffin sections were deparaffinized and rehydrated, and the endogenous peroxidase activity was then blocked with 3% hydrogen peroxide. Subsequently, the sections were incubated with 0.4% pepsin (Gibco, Grand Island, NY, USA) in 5 mM HCl at 37 °C for 20 min for antigen retrieval, and nonspecific binding sites were blocked by 5% BSA for 30 min at room temperature. Sections were then incubated with primary antibody (Cleaved-caspase3, 1:200) overnight at 4 °C. After triple washing in PBS, sections were incubated with an appropriate HRP-conjugated secondary antibody for 1 h at 37°C. The reaction was stopped with 3, 3-diaminobenzidine (DAB). For LC3 staining, sections were incubated with primary antibody (LC3, 1:200) overnight at 4 °C. After washing with PBS, sections were incubated with Alexa Fluor®488 labeled second antibodies (1:1000) for 1 h in a 37°C oven and labeled with DAPI for 5 min. Images were taken using a fluorescence microscope (OlympusInc., Tokyo, Japan).
Statistical analysis
All experiments were performed in triplicate at least. The results are presented as mean ± S.D. Statistical analyses were performed using SPSS statistical software program 20.0. Differences among the experimental groups were identified by one-way analysis of variance (ANOVA) followed by the Tukey's test. Nonparametric data (Pfrrmann grading) were analyzed by the Kruskal-Wallis H test. P values < 0.05 were considered significant.
Results
Effects of BBR on cell viability
The cytotoxicity of BBR on NP cells was determined at various concentrations (1, 5, 10, 25, 50, 100 and 200 μM) for 24 h. As shown in Figure 1A, cell viability was reduced with treatment of BBR above 50 μM at 24 h, indicating no cytotoxicity of BBR to NP cells at concentrations up to 50 μM. Stimulation with TBHP for 24 h remarkably decreased the viability of NP cells in a dose-dependent manner (Figure 1B), while pretreatment with BBR prevented cell death induced by TBHP, especially at 25 μM (Figure 1C). Therefore, 25 μM BBR was used for subsequent experiments.
BBR treatment inhibits NP cell apoptosis and alleviates ECM degeneration induced by TBHP. (A) Cell Counting Kit-8 (CCK-8) results of NP cells treated with different concentrations of BBR for 24 h. (B) Viability of NP cells treated with different concentrations of TBHP for 24 h. (C) CCK-8 results of BBR-pretreated NP cells induced by TBHP. (D-E) Protein levels of Bax, Bcl-2, Cytochrome C and Cleaved-caspase3 of NP cells treated with TBHP and TBHP plus BBR for 24 h. (F-G) Protein content of Aggrecan, CollagenⅡ, MMP-13 and ADAMTS-5 of NP cells as treated above. (H-I) TUNEL assay was performed in NP cells as treated above (original magnification × 200, scale bar: 50 μm). The data in the figures represent the averages ± S.D. Significant differences between the treatment and control groups are indicated as **P<0.01,*P<0.05, n = 3.
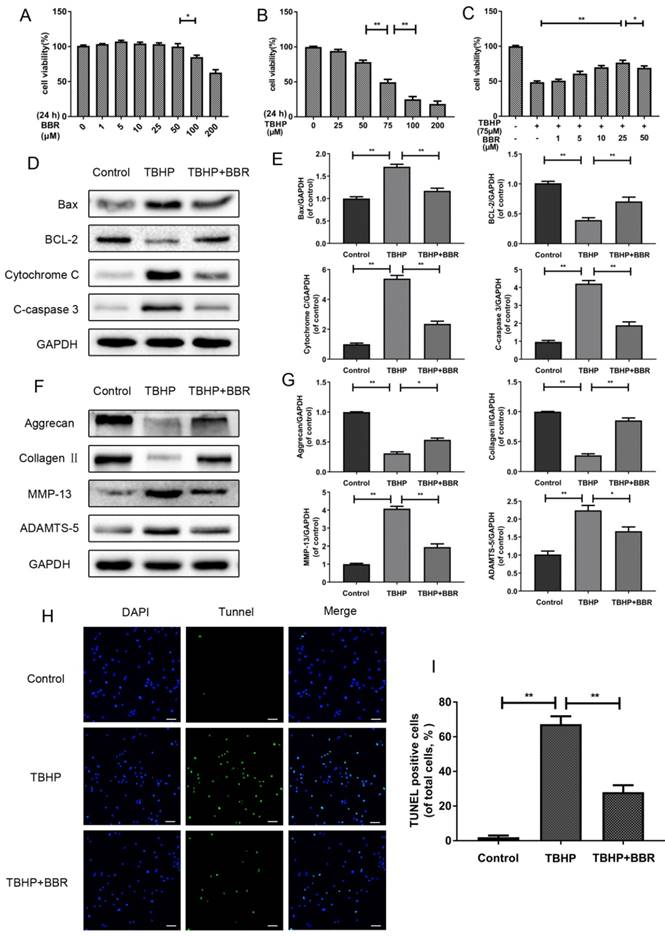
BBR treatment protects against TBHP-induced apoptosis in NP cells
In order to assess the effects of BBR on TBHP-induced apoptosis, NP cells were pretreated with 25 μM BBR for 2 h before subsequent TBHP stimulation for 24 h. TBHP (100 μM) markedly increased the expression of pro-apoptotic proteins (Cleaved-caspase3, Bax and Cytochrome C) and decreased the expression of anti-apoptotic Bcl-2 (Figure 1D-E). However, pretreatment with BBR significantly reversed these alterations, which was consistent with the TUNEL results (Figure 1H-I).
Effects of BBR on ECM synthesis and degradation in TBHP-induced rat NP cells
To evaluate the effects of BBR on the ECM, we detected the protein levels of matrix components (Collagen Ⅱ and Aggrecan) and matrix degrading enzymes (MMP-13 and ADAMTS-5) after TBHP stimulation with or without BBR pretreatment. As shown in Figure 1F-G, TBHP significantly inhibited Collagen II and Aggrecan synthesis, but increased the expression of MMP-13 and ADAMTS-5. Importantly, BBR administration reduced the loss of Collagen II and Aggrecan, and suppressed matrix degrading enzyme expression.
BBR promotes autophagy in NP cells
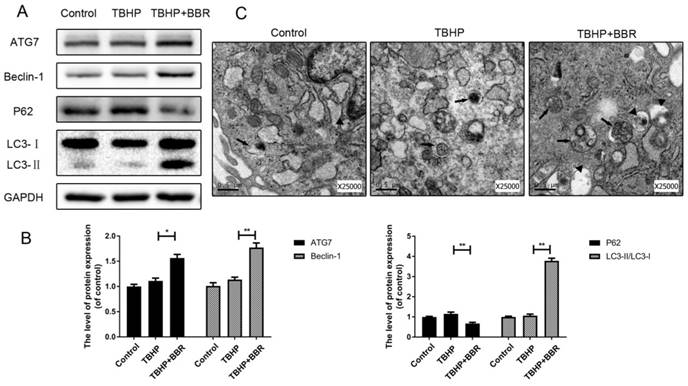
To assess the effects of BBR on autophagy, we detected autophagy markers including ATG7, Beclin-1, P62, and LC3-II/LC3-I ratio. Pretreatment with BBR followed by TBHP increased the LC3-II/LC3-I ratio and expression of ATG7 and Beclin-1 in NP cells, but decreased the expression of P62 (Figure 2A-B). Autophagosomes and autophagolysosomes were observed by transmission electron microscopy, which is a standard method to detect autophagy formation. BBR-pretreated cells contained increased autophagosomes and autophagolysosomes in their cytoplasm with TBHP stimulation in comparison to untreated cells (Figure 2C).
BBR treatment promotes autophagy in NP cells. (A-B) Protein levels of ATG7, Beclin-1, P62 and LC3 of NP cells as treated above. (C) Autophagosomes and autophagolysosomes in NP cells were observed by transmission electron microscopy (×25000). (Black arrow: autophagosome; black triangle: autophagolysosome). The data in the figures represent the averages ± S.D. Significant differences between the treatment and control groups are indicated as **P<0.01, *P<0.05, n = 3.
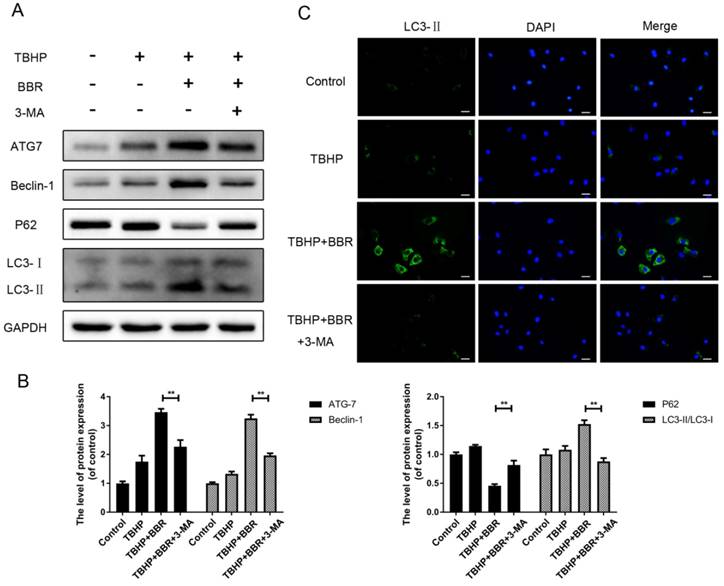
Activation of autophagy is important for BBR's protective effects in NP cells
To investigate the role of autophagy in BBR's beneficial effects on NP cells, cells were pretreated with the autophagy inhibitor 3-MA before BBR administration. Compared with the BBR group, 3-MA pretreatment significantly decreased the amount of ATG7 and Beclin-1, as well as the LC3-II/LC3-I ratio, and concordantly increased P62 levels (Figure 3A-B). By immunofluorescence, LC3 puncta in the cytoplasm of NP cells were up-regulated with BBR administration, whereas pretreatment with 3-MA markedly reversed this alteration (Figure 3C).
Pretreatment with 3-MA inhibits BBR-induced autophagy in NP cells. (A-B) NP cells were pretreated with 3-MA and followed with BBR and TBHP stimulation as described above, then the protein expression of ATG7, Beclin-1, P62 and LC3 was detected. (C)The representative LC3-positive autophagic vesicles were detected by immunofluorescence staining combined with DAPI staining for nuclei (scale bar:25 μm). The data in the figures represent the averages ± S.D. Significant differences between the treatment and control groups are indicated as **P<0.01, *P<0.05, n = 3.
To further link autophagy to BBR's effects, cells were pretreated with 3-MA, which markedly attenuated BBR's effects on apoptotic protein expression (Figure 4A-B). By immunofluorescence, Cleaved- caspase3 was significantly increased in both the TBHP group and 3-MA group, compared to the BBR group (Figure 4C).
BBR's effects on apoptosis and ECM degradation are reversed by 3-MA. (A-B) Protein expression of Bax, Bcl-2, Cytochrome C and Cleaved-caspase3 of NP cells which pretreated with 3-MA and followed with BBR and TBHP stimulation as described above. (C) The representative Cleaved-caspase3 were detected by the immunofluorescence combined with DAPI staining fornuclei (scale bar: 25 μm). (D-E) Protein content of Aggrecan, CollagenⅡ, MMP-13 and ADAMTS-5 of NP cells as treated above. The data in the figures represent the averages ± S.D. Significant differences between the treatment and control groups are indicated as **P<0.01, *P<0.05, n = 3.
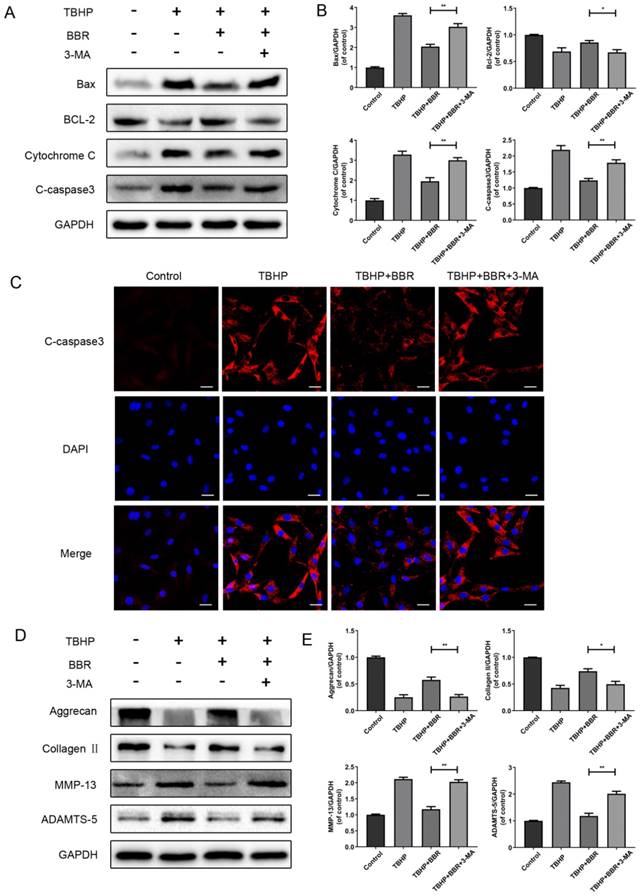
We then assessed if autophagy affects ECM synthesis and degradation of NP cells. As shown in Figures 4D-E, BBR's effects on ECM synthesis and degradation marker proteins were markedly decreased with 3-MA treatment. Taken together, these results indicate that autophagy is involved in the beneficial effects of BBR on NP cells.
BBR ameliorates disc degeneration in rats in vivo.
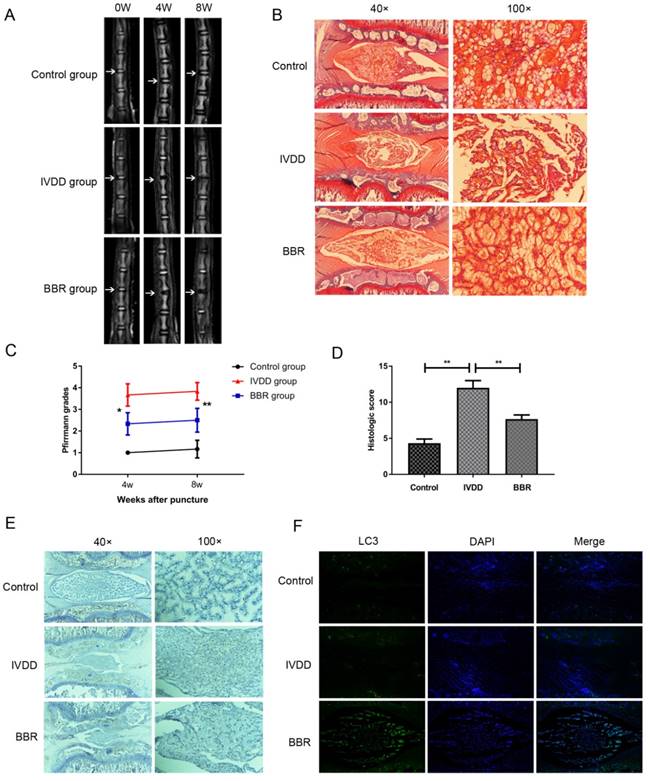
To assess the therapeutic effect of BBR on a rat IVDD model, MRI and Pfirrmann MRI grade scores were obtained at 4 and 8 weeks after puncture. The BBR group showed stronger T2-weighted signal intensities than the IVDD group both at 4 weeks and 8 weeks (Figure 5A). Pfirrmann MRI grade scores, which indicate the levels of disc degeneration, were markedly lower in BBR-treated rats than those in the IVDD group at 4 weeks and 8 weeks (Figure 5C). The beneficial effects of BBR were further confirmed by Safranin O staining in the degenerative discs at 4 and 8 weeks (data not shown). As shown in Figure 5B, the cells in control discs were mostly stellar-shaped and equably embedded in abundant extracellular matrix. Compared to controls, cells in the IVDD group migrated into clusters, separated by wispy proteoglycan matrix, indicating critical degeneration of the disc. However, BBR treatment alleviated the degeneration of proteoglycan matrix and maintained NP cell morphology. Moreover, the histologic score of the BBR group was significantly lower than that of the IVDD group (Figure 5D). Finally, BBR treatment increased LC3 expression and decreased the amount of Cleaved-caspase3 in the degenerative rat discs (Figures 5E-F), which further confirmed the anti-apoptotic effect of BBR in vivo.
BBR ameliorates IVDD development in a needle puncture-induced rat model in vivo. (A, C) T2-weighted MRI of rat tails from different experimental groups at 4 and 8 weeks post surgery (white arrows) with the corresponding Pfirrmann MRI grade scores. (B, D) Representative Safrain O staining of disc samples from different experimental groups at 4 weeks post surgery (original magnification×40 or ×100) with the corresponding histological grades. (E) Immunohistochemical staining of Cleaved-caspase3 expression in the disc samples at 4 weeks post-surgery (original magnification×40 or ×100). (F) Immunofluorescence staining of LC3 expression combined with DAPI staining for nuclei in the disc samples at 4 weeks post-surgery (original magnification×80). The data in the figures represent the averages ± S.D. Significant differences between the treatment and control groups are indicated as **P<0.01, *P<0.05, n = 8.
Discussion
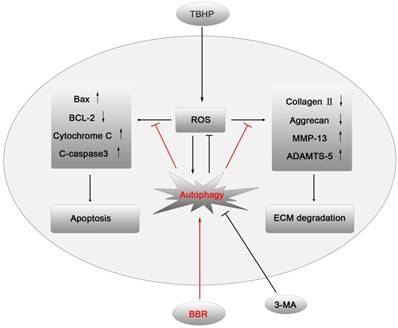
IVDD is a complex process involving a variety of pathophysiological mechanisms, including excessive apoptosis of disc cells(23). Although several drugs have been reported that may have anti-apoptotic effects on disc cells, few of them have been applied in clinical practice due to excessive side effects. BBR, an isoquinoline alkaloid extracted from Coptidis rhizome and Cortex phellodendri, has been shown to have potent anti-apoptotic activity with minor side effects(24). This study demonstrated for the first time that BBR can inhibit oxidative stress-induced NP cell apoptosis via autophagy activation, which subsequently attenuates the ECM degradation of NP cells under stress (Figures 6). These results were corroborated by in vivo results, suggesting that BBR may play a protective role in IVDD in a puncture-induced rodent model.
A model illustrating the protective effects of BBR on NP cells. TBHP increased the level of ROS in NP cells, which subsequently up-regulated the expression of pro-apoptotic proteins and matrix-degrading enzymes, leading to apoptosis and ECM degradation of NP cells. BBR treatment attenuated NP cell apoptosis and mitigated the ECM degeneration via autophagy activation, which was inhibited by 3-MA.
As a potent pro-apoptotic factor for human NP cells, reactive oxygen species (ROS) are a vital mediator of the occurrence and progression of IVDD(25). Pathogenic factors affecting IVDD include inflammatory cytokines, mechanical loading, and hyperglycemia, which can induce apoptosis through ROS(26-28). Thus, hydroperoxide is widely used to induce apoptosis in vitro. In this study, the administration of TBHP markedly increased the expression of pro-apoptotic proteins and matrix degradation enzymes, and dramatically decreased the viability of NP cells, as well as ECM components, which is one of the most prominent features of IVDD.
Autophagy is an essentially intracellular catabolic process in which damaged organelles and cytosolic macromolecules can be degraded and recycled in response to increased metabolic demands or stresses(29). In recent years, there is abundant evidence showing that moderate autophagy is involved in the maintenance of cellular homeostasis in discs(30). As the largest avascular organ in the body, the intervertebral disc is a hypoxic niche with NP cells residing there. It was recently reported that NP cells adapt to the hypoxic microenvironment through autophagy activation, indicating that autophagy is essential for their normal metabolism(31). Interestingly, it appears contradictory that either higher or lower levels of autophagy were observed in degenerative disc cells. According to previous reports, autophagy is significantly increased both in degenerative NP and AF cells(32, 33). However, a recent study found fewer autophagosomes and decreased expression of autophagy-related genes in human degenerated discs compared to normal discs(34). To resolve this apparent contradiction, we believe that moderate autophagy could be enhanced in disc cells as a protective mechanism in the early stages of IVDD. However, aggravating disc degeneration potentiates autophagy dysfunction, leading to irreversible destruction of intervertebral discs. In this study, we detected autophagy markers, which have crucial functions in autophagy. Furthermore, we observed autophagosomes and autophagolysosomes by transmission electron microscopy, which is a standard method to check for autophagy activation. Our results showed for the first time that BBR could activate autophagy in NP cells.
Apoptosis has been found to be essential in the development of IVDD, which can be activated by various stimuli via different signal transduction pathways such as the death receptor, the mitochondrial pathway, and the endoplasmic reticulum(2, 23). In IVDD, oxidative stress caused by various pathogenic factors mediates mitochondrial pathway apoptosis, decreases Bcl-2 expression, and increases the expression of pro-apoptotic Bcl-2 family members such as Bax and Bak, which leads to the release of Cytochrome C(35, 36). Subsequently, caspase3 is activated as the final executor of apoptosis(37). Our study found that BBR administration attenuated these changes in apoptotic proteins under oxidative stress, suggesting that BBR is protective against apoptosis in NP cells.
The ECM produced by NP cells performs a vital role in the maintenance of disc physiology, and ECM degradation is a major feature of IVDD(38). MMPs and ADAMTS are catabolic enzymes that degrade the ECM in IVDD(39). Our data indicated that BBR could effectively inhibit ECM degradation. To further confirm that BBR stimulates autophagy, autophagy inhibition using 3-MA was capable of reversing BBR's beneficial effects.
Finally, needle puncture-induced degenerative rat discs were evaluated 4 and 8 weeks after successful modeling. We found severely degenerated intervertebral discs in the IVDD group, which was decreased by BBR treatment, indicating that BBR could ameliorate disc degeneration in vivo. Furthermore, BBR treatment promoted autophagy and suppressed the expression of apoptotic proteins in the degenerative rat discs, which further confirmed the results from our in vitro experiments. In addition, no differences in terms of survival rate, weight, and mobility were observed between the BBR group and IVDD group, suggesting that the dosage of BBR used in rats was safe.
However, this study has some limitations. First, the underlying mechanism of BBR-induced autophagy including the upstream signaling pathway involved has not been fully elucidated. Other apoptotic pathways involved in IVDD, such as endoplasmic reticulum stress, were not investigated in the current study and require further research. Moreover, linking the protective effects of BBR against ECM degeneration to its anti-inflammatory properties remains unclear. In addition, the optimal concentration of BBR for animal use needs further confirmation.
In conclusion, the current study demonstrates that BBR treatment attenuates NP cell apoptosis and mitigates the ECM degeneration via autophagy activation, presenting the possibility for an ideal therapeutic agent for IVDD treatment in the future.
Acknowledgements
This work is supported by grant from National Nature Foundation of China (Grant no.81371988, 81722028). Science and technology project of Zhejiang Province (LY17H090017 to Z.W.). Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (to J.X.), and the Wenzhou Science and Technology Foundation (Y20140681).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Takahashi K, Aoki Y, Ohtori S. Resolving discogenic pain. Eur Spine J. 2008;17(Suppl 4):428-31
2. Zhao CQ, Jiang LS, Dai LY. Programmed cell death in intervertebral disc degeneration. Apoptosis. 2006;11(12):2079-88
3. Colombier P, Clouet J, Hamel O, Lescaudron L, Guicheux J. The lumbar intervertebral disc: from embryonic development to degeneration. Joint Bone Spine. 2014;81(2):125-9
4. Setton LA, Chen J. Cell mechanics and mechanobiology in the intervertebral disc. Spine (Phila Pa 1976). 2004;29(23):2710-23
5. Walker MH, Anderson DG. Molecular basis of intervertebral disc degeneration. Spine J. 2004;4(6 Suppl):158S-66S
6. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861-73
7. Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy Is a Protective Mechanism in Normal Cartilage, and Its Aging-Related Loss Is Linked With Cell Death and Osteoarthritis. Arthritis Rheum-Us. 2010;62(3):791-801
8. Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H. et al. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106-107:33-54
9. Zhang M, Lin J, Wang S, Cheng Z, Hu J, Wang T. et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res. 2017:63 (2)
10. Miyazaki S, Kakutani K, Yurube T, Maeno K, Takada T, Zhang Z. et al. Recombinant human SIRT1 protects against nutrient deprivation-induced mitochondrial apoptosis through autophagy induction in human intervertebral disc nucleus pulposus cells. Arthritis Res Ther. 2015;17:253
11. Wang Z, Chen Z, Yang S, Wang Y, Huang Z, Gao J. et al. Berberine ameliorates collagen-induced arthritis in rats associated with anti-inflammatory and anti-angiogenic effects. Inflammation. 2014;37(5):1789-98
12. Li Z, Geng YN, Jiang JD, Kong WJ. Antioxidant and anti-inflammatory activities of berberine in the treatment of diabetes mellitus. Evid Based Complement Alternat Med. 2014;2014:289264
13. Liang Y, Huang M, Jiang X, Liu Q, Chang X, Guo Y. The neuroprotective effects of Berberine against amyloid beta-protein-induced apoptosis in primary cultured hippocampal neurons via mitochondria-related caspase pathway. Neurosci Lett. 2017;655:46-53
14. Wang X, He X, Zhang CF, Guo CR, Wang CZ, Yuan CS. Anti-arthritic effect of berberine on adjuvant-induced rheumatoid arthritis in rats. Biomed Pharmacother. 2017;89:887-93
15. Jin Y, Liu S, Ma Q, Xiao D, Chen L. Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur J Pharmacol. 2016
16. Coelho AR, Martins TR, Couto R, Deus C, Pereira CV, Simões RF. et al. Berberine-induced Cardioprotection and Sirt3 Modulation in Doxorubicin-treated H9c2 Cardiomyoblasts. Biochim Biophys Acta. 2017
17. Chitra P, Saiprasad G, Manikandan R, Sudhandiran G. Berberine inhibits Smad and non-Smad signaling cascades and enhances autophagy against pulmonary fibrosis. J Mol Med (Berl). 2015;93(9):1015-31
18. He Q, Mei D, Sha S, Fan S, Wang L, Dong M. ERK-dependent mTOR pathway is involved in berberine-induced autophagy in hepatic steatosis. J Mol Endocrinol. 2017;59(4):X1
19. Chen D, Xia D, Pan Z, Xu D, Zhou Y, Wu Y. et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016;7(10):e2441
20. Xu D, Jin H, Wen J, Chen J, Chen D, Cai N. et al. Hydrogen sulfide protects against endoplasmic reticulum stress and mitochondrial injury in nucleus pulposus cells and ameliorates intervertebral disc degeneration. Pharmacol Res. 2017;117:357-69
21. Han B, Zhu K, Li FC, Xiao YX, Feng J, Shi ZL. et al. A simple disc degeneration model induced by percutaneous needle puncture in the rat tail. Spine (Phila Pa 1976). 2008;33(18):1925-34
22. Pfirrmann CW, Metzdorf A, Zanetti M, Hodler J, Boos N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine (Phila Pa 1976). 2001;26(17):1873-8
23. Ding F, Shao ZW, Xiong LM. Cell death in intervertebral disc degeneration. Apoptosis. 2013;18(7):777-85
24. Chen K, Li G, Geng F, Zhang Z, Li J, Yang M. et al. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-Akt signaling in diabetic rats. Apoptosis. 2014;19(6):946-57
25. Feng C, Yang M, Lan M, Liu C, Zhang Y, Huang B. et al. ROS: Crucial Intermediators in the Pathogenesis of Intervertebral Disc Degeneration. Oxid Med Cell Longev. 2017;2017:5601593
26. Yang L, Zhu L, Dong W, Cao Y, Lin L, Rong Z. et al. Reactive oxygen species-mediated mitochondrial dysfunction plays a critical role in high glucose-induced nucleus pulposus cell injury. Int Orthop. 2013
27. Ding F, Shao ZW, Yang SH, Wu Q, Gao F, Xiong LM. Role of mitochondrial pathway in compression-induced apoptosis of nucleus pulposus cells. Apoptosis. 2012;17(6):579-90
28. Roberts S, Evans H, Trivedi J, Menage J. Histology and pathology of the human intervertebral disc. J Bone Joint Surg Am. 2006;88(Suppl 2):10-4
29. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280-93
30. Zhang SJ, Yang W, Wang C, He WS, Deng HY, Yan YG. et al. Autophagy: A double-edged sword in intervertebral disc degeneration. Clin Chim Acta. 2016
31. Choi H, Merceron C, Mangiavini L, Seifert EL, Schipani E, Shapiro IM. et al. Hypoxia promotes noncanonical autophagy in nucleus pulposus cells independent of MTOR and HIF1A signaling. Autophagy. 2016;12(9):1631-46
32. Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu W. et al. Age-related increases of macroautophagy and chaperone-mediated autophagy in rat nucleus pulposus. Connect Tissue Res. 2011;52(6):472-8
33. Shen C, Yan J, Jiang LS, Dai LY. Autophagy in rat annulus fibrosus cells: evidence and possible implications. Arthritis Res Ther. 2011;13(4):R132
34. Jiang W, Zhang X, Hao J, Shen J, Fang J, Dong W. et al. SIRT1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Sci Rep. 2014;4:7456
35. Nomura M, Shimizu S, Ito T, Narita M, Matsuda H, Tsujimoto Y. Apoptotic cytosol facilitates Bax translocation to mitochondria that involves cytosolic factor regulated by Bcl-2. Cancer Res. 1999;59(21):5542-8
36. Peña-Blanco A, García-Sáez AJ. Bax, Bak and beyond: mitochondrial performance in apoptosis. Febs j. 2017
37. Min JJ, Huo XL, Xiang LY, Qin YQ, Chai KQ, Wu B. et al. Protective effect of Dl-3n-butylphthalide on learning and memory impairment induced by chronic intermittent hypoxia-hypercapnia exposure. Sci Rep. 2014;4:5555
38. Wang XH, Zhu L, Hong X, Wang YT, Wang F, Bao JP. et al. Resveratrol attenuated TNF-α-induced MMP-3 expression in human nucleus pulposus cells by activating autophagy via AMPK/SIRT1 signaling pathway. Exp Biol Med (Maywood). 2016
39. Li K, Li Y, Xu B, Mao L, Zhao J. Sesamin inhibits lipopolysaccharide induced inflammation and extracellular matrix catabolism in rat intervertebral disc. Connect Tissue Res. 2016
Author contact
![]() Corresponding authors: Jian Xiao, Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, 325035, Wenzhou, China. E-mail: xfxj2000com and Zhouguang Wang, Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, 325035, Wenzhou, China. Tel.:+86-577-85773087; E-mail: wzhouguangcom and Xiangyang Wang, Department of Orthopaedic Surgery, The Second Affiliated Hospital of Wenzhou Medical University, Xueyuan Xi Road, Wenzhou, Zhejiang, 325027, China. Tel.: 86-577-88829799; Email: xiangyangwangcom
Corresponding authors: Jian Xiao, Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, 325035, Wenzhou, China. E-mail: xfxj2000com and Zhouguang Wang, Molecular Pharmacology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, 325035, Wenzhou, China. Tel.:+86-577-85773087; E-mail: wzhouguangcom and Xiangyang Wang, Department of Orthopaedic Surgery, The Second Affiliated Hospital of Wenzhou Medical University, Xueyuan Xi Road, Wenzhou, Zhejiang, 325027, China. Tel.: 86-577-88829799; Email: xiangyangwangcom