Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Neuroplasticity following...
PDE4 and its activity regulation
Effect of PDE4 on cAMP-mediated...
Inhibition of PDE4 promotes...
Inhibition of PDE4 is beneficial...
Inhibition of PDE4 activates...
Inhibition of PDE4 restricts...
Conclusions and future...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1745-1754. doi:10.7150/ijbs.26230 This issue Cite
Review
Targeting phosphodiesterase 4 as a potential therapeutic strategy for enhancing neuroplasticity following ischemic stroke
Haitao Wang1, Uma Gaur2, Jiao Xiao1, Bingtian Xu1, Jiangping Xu1 ![]() , Wenhua Zheng2
, Wenhua Zheng2 ![]()
1. Department of Neuropharmacology and Drug Discovery, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China.
2. Faculty of Health Sciences, University of Macau, Taipa, Macau, China.
Received 2018-3-22; Accepted 2018-6-3; Published 2018-10-3
Abstract

Sensorimotor recovery following ischemic stroke is highly related with structural modification and functional reorganization of residual brain tissues. Manipulations, such as treatment with small molecules, have been shown to enhance the synaptic plasticity and contribute to the recovery. Activation of the cAMP/CREB pathway is one of the pivotal approaches stimulating neuroplasticity. Phosphodiesterase 4 (PDE4) is a major enzyme controlling the hydrolysis of cAMP in the brain. Accumulating evidences have shown that inhibition of PDE4 is beneficial for the functional recovery after cerebral ischemia; i. subtype D of PDE4 (PDE4D) is viewed as a risk factor for ischemic stroke; ii. inhibition of PDE4 enhances neurological behaviors, such as learning and memory, after stroke in rodents; iii.PDE4 inhibition increases dendritic density, synaptic plasticity and neurogenesis; iv. activation of cAMP/CREB signaling by PDE4 inhibition causes an endogenous increase of BDNF, which is a potent modulator of neuroplasticity; v. PDE4 inhibition is believed to restrict neuroinflammation during ischemic stroke. Cumulatively, these findings provide a link between PDE4 inhibition and neuroplasticity after cerebral ischemia. Here, we summarized the possible roles of PDE4 inhibition in the recovery of cerebral stroke with an emphasis on neuroplasticity. We also made some recommendations for future research.
Introduction
Ischemic stroke is usually occurred under the condition of a sudden loss of physiological brain functions due to blockage of a blood vessel in the brain [1]. The incidence of ischemic stroke is much greater than that of hemorrhagic stroke and is one of the leading causes of morbidity and mortality worldwide [2]. Despite years of efforts on the drug development, recombinant tissue plasminogen activator (rt-PA) is the only approved drug by the FDA in United States for the treatment of ischemic stroke. However, rt-PA is also limited by the narrow therapeutic window, and currently there are no evidence to support that rt-PA would help to restore the already lost functions in stroke patients. Under certain conditions, such as in an enriched environment, partial lost neurological functions could be regained in rodents after stroke due to the brain's capacity for neuroplasticity [3, 4]. Similarly, implementation of an enriched environment is also beneficial for the neurological recovery in patients suffering from stroke. The benefits include increased motor, cognitive and sensory functions, enhanced social interaction and decreased degree of boredom [5, 6]. Neuroplasticity refers to the capacity of the brain to rewire or reorganize the structure and functions after an injury [7]. Neurons from the unaffected area and adjacent to the ischemic region are believed to regenerate new neuronal precursor cells and form new neuronal connectivity, which take over part biological functions of damaged neurons [8]. This suggests that some lost functions of the brain due to stroke, such as motor and speech can be restored by certain interventions. Animal and human studies over the past decades have confirmed that the plasticity of the central nervous system (CNS) is the main neurophysiological mechanism of motor function recovery after stroke [8-10]. However, spontaneous neuroplasticity after stroke is limited, post-ischemic interventions which are helpful for the functional recovery after brain lesions are still in great need.
Enhancing neuroplasticity is possibly an efficient way to promote functional recovery following ischemic brain damage. Multiple molecular targets are under consideration in recent years. Phosphodiesterase (PDE) is an enzyme hydrolyzing both cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) in mammalian organs [11, 12], especially in the brain [11]. Among various PDE family proteins, phosphodiesterase 4 (PDE4) specifically hydrolyzes cAMP and inhibition of PDE4 is an ideal strategy for the prevention and treatment of peripheral inflammation and immune diseases, such as pulmonary inflammation and psoriasis [13]. In the CNS, PDE4 plays a key role in both chronic neurodegenerative diseases and acute neurological insults, including stroke [14, 15]. Recently, an increasing number of reports have shown that inhibition of PDE4 is beneficial for neuroplasticity [16-18], making it a promising candidate target that has therapeutic potential for the recovery of stroke. Thus, this review provides a perspective for advancing our understanding of the role of PDE4 in neuroplasticity after stroke, supporting the possibility of exploiting PDE4 inhibitors as effective therapeutics against ischemic stroke.
Neuroplasticity following ischemic stroke
Cerebral vascular diseases, including ischemic stroke, are a serious hazard to human health and life. Neurons are particularly susceptible to hypoxia-ischemia and therefore, insufficient supply of glucose and oxygen leads to comparable neuronal death or apoptosis, which subsequently causes functional disability and mortality [19, 20]. Currently, few drugs or therapeutic interventions are in clinical use that can repair the damaged region of the brain caused by a stroke. Preventing further damage of ischemic tissue in time or preserving as much undamaged neurons as possible is perhaps the most effective way for the recovery of stroke. Excitingly, In the last two decades, scientists have confirmed that nerve tissues, including both brain and spine, are able to rewire or reorganize themselves under certain conditions [21, 22], by a process called neuroplasticity [23, 24]. Neuroplasticity may also occur in various practices in our daily life, for example, when we try to learn a new skill or memorize a new information, persistent functional changes keep happening in our brain [25]. It may also occur under the condition of compensation for neural injury [26].
In the present review, we mainly focused on neuroplasticity following ischemic stroke. Endogenous neuroplasticity and subsequent functional recovery could occur spontaneously following an ischemic stroke [27]. However, spontaneous recovery is usually quite limited and insufficient for the repair of neuronal injury caused by stroke. Patients with stroke are supposed to reach the maximal improvement following stroke after 2-5 months depending on the damaged brain region and severity of their symptoms [6]. Appropriate functional rehabilitation training is proved to accelerate the recovery [28], and it should be implemented at the early stage of stroke [29, 30]. Enhanced functional recovery is usually accompanied by morphological changes in neurons, such as increased dendritic length and spines. Generally, the changes occurred in the cortex organization caused by neuroplasticity include: i. generation of neural progenitor cells and vascular endothelial cells [31]; ii. an increase in the density and strength of synapse [32]; iii. an increase in the number and density of dendrites [33], iv. enhanced expression and function of synaptic receptors [34] and v. the "pruning" of old synapses, and formation of new synapses in peri-infarct cortex [31, 35]. Neurotrophic factors in the brain, such as brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) possesses significant roles in promoting neuroplasticity [36-38]. VEGF promotes neurogenesis and migration of neural stem cells, while BDNF enhances the survival and synaptic development of neuronal cells [38]. As neuroplasticity refers to any long-lasting changes in the brain throughout one's life, the definition is broad and nearly every region of brain shows neuroplasticity under a specific condition [9, 39, 40].
As functional recovery mediated by neuroplasticity following stroke is an extremely complicated process, therefore elucidation on the potential mechanisms requires a good framework. Murphy et al., contributed an excellent review on the neuroplasticity after stroke [41]. Activity-dependent rewiring occurs around the damaged region, and this process reconstructs the connectivity between neurons that has been lost because of the sudden shortage of oxygen, and repetitive activation of synapses and neural pathways is supposed to strengthen the synaptic connectivity [41]. Furthermore studies have identified multiple signaling pathways associated with plasticity following stroke [42]. For example, Shh and Notch signaling are involved and thereby influence activity-dependent synaptic plasticity in hippocampus [43]. Phosphatidylinositol-3-kinase/protein kinase B/ Glycogen synthase kinase 3β (PI3K/Akt/GSK3β) signaling pathway mediates axonal regeneration and outgrowth of cortical neurons after stroke [44]. Cyclic adenosine monophosphate/protein kinase A/cAMP response element binding protein /brain-derived neurotrophic factor (cAMP/PKA/CREB/BDNF) pathway regulates neurogenesis, neuronal survival and synaptic plasticity following ischemic stroke [38, 43, 45, 46].
PDE4 and its activity regulation
PDE is a class of enzymes that are critical for the hydrolysis of phosphodiester bonds and thus are important in breaking down cAMP and cyclic guanosine monophosphate (cGMP) [47]. PDE4, 7, and 8 are specific for the degradation of cAMP, PDE5, 6, and 9 specifically degrade cGMP, while PDE1, 2, 3, 10, and 11 have a dual-specificity [48]. PDE12 is a newly discovered enzyme which hydrolyzes 2'-5'-linked oligoadenylate (2-5A) and regulates innate immune response [49]. Among them, PDE4 is a member of the enzyme superfamily which is critical in terminating cAMP signaling in distinct mammalian tissues [50]. PDE4 superfamily consist of 4 subtypes (PDE4A-D), and each subtype contains 3-11 isoforms. For example, there are 3 isoforms for PDE4C (PDE4C1-3), and PDE4D contains 11 isoforms (PDE4D1-11). Hence, there are over 20 isoforms for PDE4s due to alternative mRNA splicing [51, 52]. These isoforms can be grouped into long (PDE4A4/5/8/10/11, PDE4B1/3/4, PDE4C1/2/3, PDE4D3/4/5/7/8/9), short (PDE4B2, PDE4D1, PDE4D2), super short (PDE4A1, PDE4B5, PDE4D6, PDE4D10 and PDE4D11) and dead short (PDE4A7) categories [52, 53]. All PDE4 isoforms contain a conserved catalytic region. Long isoforms have two conserved N-terminal regulatory domains, termed UCR1 (upstream conserved region 1) and UCR2 (upstream conserved region 2), short isoforms only contain UCR2, super short isoforms contain a truncated UCR2 but lack UCR1, dead short isoforms have no enzymatic activity, they lack both UCR1 and UCR2, also the catalytic unit is intact and inactive [54].
The catalytic activity of PDE4 is regulated by post-translational modifications, such as phosphorylation. Except PDE4A, all other PDE4 variants contain an extracellularly regulated protein kinases (ERK) phosphorylation site in the catalytic domain [53, 55]. Interestingly, phosphorylation of PDE4 by ERK changes the hydrolytic activity of PDE4 in a variant-specific fashion. ERK phosphorylation exerts an inhibitory effect on the activity of long form PDE4s and a stimulatory effect on hydrolytic activity of short form PDE4s. While it has no or weaker effect on the super-short form PDE4, there is a possibility that different PDE4 isoforms may have distinct functional roles [53, 55, 56]. In addition, a PKA phosphorylation site locates in the UCR1 domain of long PDE4 isoforms [57]. Binding of PKA to UCR1 domain promotes the phosphorylation of long PDE4 isoforms, which leads to an increase in their activity [58]. Phosphorylation of PDE4 by PKA is involved in the termination of cAMP-mediated signaling timely [59]. The roles of ERK and PKA on the phosphorylation and distinct biological functions of PDE4 indicate that various PDE4 isoforms may perform distinct functional roles in cells through distinct post-translational modifications, especially phosphorylation [58]. Hence, based on the diversity of PDE4 enzymes, it is highly possible that we can design sub-family selective PDE4 inhibitors which might have rather different functional effects.
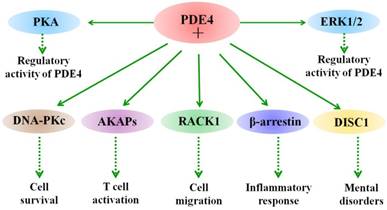
Besides phosphorylation, different PDE4 isoforms show specific biological functions in different cells through interaction with different partner proteins [60-62]. For example, PDE4A binds to A-kinase anchoring proteins (AKAPs) and regulates T cell activation [63]. Binding of PDE4 long isoforms to AKAPs localized in the perinuclear and centrosomal regions restricts the activation of downstream cAMP targets [64]. Hence, interaction of PDE4 and AKAPs terminates the accumulation of local cAMP and the subsequent activation of cAMP/PKA-mediated phosphorylation events. The biological role of their binding in the CNS remains unclear and needs further investigations. Other signaling scaffold proteins, such as β-arrestin 1 and β-arrestin 2 have also been shown to bind to all PDE4 isoforms [65]. This interaction between β-arrestin and PDE4 causes an increase in cAMP degradation [65]. Additionally, the N-terminal of PDE4D5 isoform contains both a β-arrestin-binding site and a binding site for RACK (receptors for activated C-kinase) [66]. Interestingly, PDE45 is able to interact with the signaling scaffold proteins RACK1 and β-arrestin simultaneously [66]. Presumably, their interactions are poised to play a role in regulating cAMP and its downstream signaling in cells. Additional studies are needed to be done in neuronal cells and under pathological conditions, such as ischemic stroke. Figure 1 summarizes the interacting proteins with PDE4. Taken together, these studies provide potential for understanding the role of PDE4 isoforms in molecular pathology as well as in providing novel means of therapeutic intervention.
Interaction of PDE4 with cellular proteins. The activity of PDE4 is regulated by both extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) and protein kinase A (PKA). ERK1/2 exerts an inhibitory effect on the activity of long form PDE4s and a stimulatory effect on hydrolytic activity of short form PDE4s, while phosphorylation of long PDE4 isoforms by PKA leads to an increase in their activity in cells. Under different conditions, PDE4 exerts diverse biological functions through interacting with multiple proteins. For example, PDE4 interacts with DNA-PKs and thus regulates cell death. PDE4 binds to A-kinase-anchoring proteins (AKAPs) and modulates T cell activation. In addition, interaction of PDE4 with disrupted in schizophrenia 1 (DISC1) is involved in the process of mental disorders, such as schizophrenia.
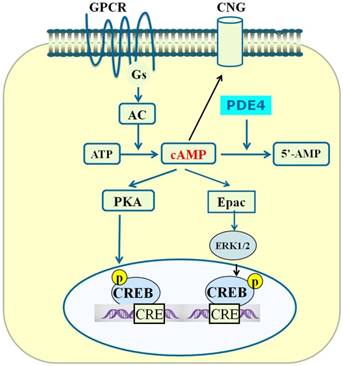
Effect of PDE4 on cAMP-mediated signaling
cAMP is a second messenger which is involved in multiple biological processes, such as release of hormones and signal transduction mediated by neurotransmitters [67]. In ischemic brain, the level of cAMP is significantly lower than that in the contralateral side as early as 3 hours after the onset of ischemia and remains below the contralateral side [68]. ATP is the source of cAMP and adenylyl cyclase (AC) which is responsible for the conversion of ATP to cAMP. The intracellular concentration of cAMP is regulated by the balance between AC and PDE, and PDE4 is the major enzyme degrading cAMP due to its high affinity for cAMP [54].
In the CNS, binding of neurotransmitters to their receptors activates the α subunit of the Gs protein, which leads to consequent activation of AC [69]. cAMP is generated as a consequence of AC activation, and increased cAMP subsequently activates several downstream effectors, such as PKA. PDE4 is used to catalyze the abundant cAMP, so that the signal transduction is terminated in time [53].
PKA is one of the downstream effectors mediating cAMP signaling. cAMP activates PKA and promotes the phosphorylation of multiple downstream substrates, including CREB, cAMP responsive modulator (CREM), and ATF1 [70]. In the nuclear, phosphorylated CREB binds to other co-activator proteins, such as CREB binding protein (CBP) and p300. Activated CREB binds to CRE in the promoter region of target genes and regulates the transcription of these genes, including BDNF [71].
Another important effector for cAMP is Epac (exchange protein activated by cAMP). Epac has two isoforms, namely Epac1 and Epac2, and the latter has a higher affinity to cAMP than Epac1 [72]. Epac functions as a guanine nucleotide exchange factor for the GTPases [73]. Through acting on GTPases, Epac is involved in multiple cellular processes such as cell adhesion and neurotransmitter release [73]. Epac also modulates cAMP-mediated signaling synergistically with PKA [74, 75].
Finally, cAMP binds to and regulates the biological functions of cyclic-nucleotide-gated (CNG) ion channels [76], which have been identified in a number of other tissues, including the brain [77]. CNG channels are nonselective cation channels. Currently, the biological functions of CNG channels are highly related to signal transduction in sensory neurons (such as photoreceptors in the eyes) [78]. while, their roles in synaptic plasticity in the brain is not very well understood.
We have summarized three main signaling effectors of cAMP above. Summary of the PDE4-cAMP-mediated signaling pathways is shown in Figure 2. As PDE4 is an enzyme that degrades cAMP inside cells, the attenuation of cAMP signaling could be achieved through enhancing the activity of PDE4, which is able to hydrolyze cAMP to 5'-AMP [79]. Since PDE is the sole route for the degradation of cAMP and PDE4 is the main enzyme acting on cAMP in cells [80], PDE4 plays a key role in controlling the concentration of cAMP and cAMP-mediated signaling. In the CNS, PDE4A, PDE4B, and PDE4D are widely expressed in neuronal cells, whereas PDE4C is expressed primarily in peripheral tissues [53]. Structure- and scaffold-based design of relatively selective inhibitors of one or two PDE4 subtypes have been proposed to be promising for the treatment of neurological disorders.
PDE4-cAMP-mediated signaling pathways in neuronal cells. Activation of GPCR activates Gsα subunit and adenylyl cyclase (AC), which subsequently catalyzes the conversion of ATP to cAMP. Increase in the level of local intracellular cAMP leads to activation of protein kinase A (PKA), exchange proteins activated by cAMP (Epac) and cyclic nucleotide-gated (CNG). Activated PKA phosphorylates a number of other proteins including cAMP-responsive element-binding protein (CREB). Phosphorylated CREB binds to the cAMP-responsive element (CRE) and thereby modulates gene transcription. Activated Epac promotes the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2). Inhibition of PDE4 triggers an increase in cAMP which in turn exerts its downstream effects via activation of PKA/CREB and Epac/ERK signaling pathways.
Inhibition of PDE4 promotes neurogenesis and neuroplasticity
Neuroplasticity or remodeling is a critical process that underlies brain functions during normal and pathological conditions [81, 82]. Brain makes appropriate responses in order to adapt to the changed environment [83]. PDE4 inhibitors has caused extensive concern as they are expected to modulate a variety of cellular functions, including neuroplasticity and neuroprotection [84]. All the three PDE4 isoforms (PDEA, B, D) expressed in the brain show inhibitory effects during neurogenesis and neuroplasticity. In a sleep deprivation animal model, sleep deprivation dramatically decreases number and dendrite length of neurons in hippocampal CA1 region, while mutation of PDE4A5 reversed the morphological alterations and memory impairments caused by sleep deprivation [85]. Further studies showed that inhibition of PDE4A5 mediated changes in dendritic structure through degrading cAMP [85]. Similar to PDE4A, PDE4B is widely distributed throughout the brain in humans and rodents with prominent expression in cortex, hippocampus and amygdala [86]. Deficiency of PDE4B in mice resulted in a significant increase in the proliferation of neuronal cells in the hippocampal dentate gyrus [87]. PDE4B knockout or mutant mice also displayed markedly enhanced basal postsynaptic responses to stimulation, neurogenesis and ability of learning and memory as compared to wild-type littermates [88, 89]. PDE4D is important in mediating antidepressant activity and memory [90]. Treatment with the pan-PDE4 inhibitor rolipram enhances the local level of cAMP [91], and increases the production of neural progenitor cells in the dentate gyrus [91]. These results were supported by the findings that PDE4D deficiency leads to increased BrdU-positive cells in the dentate gyrus and memory enhancement [90]. Collectively, current investigations demonstrate that inhibition of PDE4D activates cAMP/PKA/CREB signaling in the brain and enhances neurogenesis, dendritic density and neuroplasticity.
Inhibition of PDE4 is beneficial for the recovery from ischemic stroke
In response to various stimuli, PDE4 mediates cAMP signaling and is associated with both normal physiological functions and pathological conditions. Specifically, the biological role of the PDE4 enzyme family relates to individual isoforms [15]. PDE4A is mainly associated with depression [15], PDE4B is linked to the pathology of schizophrenia and PDE4D is highly related with ischemic stroke [15]. Actually, PDE4D has been viewed as a risk factor of ischemic stroke, especially carotid and cardiogenic stroke [92]. Microsatellite allelic association showed that mutations of microsatellite markers, especially AC008818-1 is associated with cardiogenic stroke, and interestingly, this marker is located within PDE4D [92]. Further studies on the single-nucleotide polymorphism (SNP) showed that SNP41, SNP45, SNP56, SNP87 and SNP89 are in connection with an increased incidence of ischemic stroke in Icelandic patients, and all these SNPs are located in intron regions in the 5'-end of the PDE4D gene [15, 92]. Therefore knocking down or inhibiting the expression of one or more PDE4D isoforms specifically might decrease the risk of ischemic stroke.
On the other hand, PDE4 affects the sensitivity or vulnerability of neuronal tissues to cerebral ischemic lesion. As we know, inflammation is a destructive factor limiting the functional recovery of ischemic brain at the early stage [93]. Ischemia causes an elevated expression of cytokines and increased activity of inducible nitric oxide synthases (iNOS), which subsequently enhances the production of free radicals [94]. Overproduction of free radicals following cerebral ischemia destroys neuronal cell membrane and causes the activation of apoptotic signaling in the brain, whereas inhibition of NO overproduction by L-NAME reduced infarct volume and histopathological changes of ischemic striatum [95]. Notably, enhanced intracellular cAMP attenuates the production of iNOS induced by other cytokines [96]. PDE4 inhibitors also suppressed the level of iNOS through elevating the concentration of cAMP both in vitro and in vivo [97]. These observations were also supported by the findings that treatment with the PDE4 inhibitor roflumilast ameliorated NO-induced apoptosis and myocardial ischemia/reperfusion injury via both cAMP/PKA/CREB and Epac/Akt-dependent pathway [98]. PDE7 is another enzyme responsible for the hydrolysis of cAMP. Studies showed that inhibition of PDE7 exhibited neuroprotective effects in both cellular cultures and in a permanent middle cerebral artery occlusion stroke animal model [99]. This study also suggested the importance of cAMP in the regulation of cellular survival.
Based on the above findings, it is safe to predict an association of PDE4 mutations and risk of ischemic stroke. Enhanced activity of PDE4 after ischemic stroke plays a devastating role in mediating neuronal cell death and PDE4 inhibitors may be useful drugs which might work through their specific regulation of the intracellular cAMP level. Treatment with rolipram, a classical PDE4 inhibitor, reduces infarct volume in both the CA1 region of the hippocampus and the striatum in rats and gerbil [100, 101]. These findings are consistent with the observations that Rolipram and HT-0712, a novel PDE4 inhibitor, significantly enhanced motor recovery, sensorimotor function and cortical reorganization after ischemic insult [102, 103]. Our previous studies also showed that treatment with Rolipram inhibited PDE4 activity in the hippocampus and prevented cerebral ischemia-induced memory deficits [104]. These results indicate that PDE4 may play a pivotal role in cAMP signaling-mediated neurological deficits induced by ischemic stroke and PDE4 inhibitors could be the promising candidate drugs that may have therapeutic benefit in patients suffering from stroke.
Inhibition of PDE4 activates cAMP/PKA/CREB/BDNF signaling pathway and enhances neuroplasticity following stroke
Though PDE4 inhibition is a potential therapeutic strategy for the treatment of stroke, the underlying mechanisms are not fully explored. Increasing studies indicate that PDE4 inhibition enhances neuroplasticity in various models [17, 105, 106]. Considering that neuroplasticity is a pivotal strategy to promote functional recovery after stroke, the role of PDE4 inhibition on neuroplasticity and its underlying mechanisms have aroused widespread concern.
Firstly, PDE4-cAMP regulates BDNF transcription by a PKA/CREB-dependent mechanism. In mature neurons, BDNF plays a positive role in neuroplasticity [107-109]. Increased expression of BDNF facilitates neuroplasticity and promotes the regain of motor and sensory abilities after stroke [110]. In immature neurons, BDNF participates in the neurite outgrowth and maturation of progenitor cells [111]. Under the condition of ischemic stroke, larger stroke infarct volumes are directly associated with the lower levels of BDNF in serum [112]. Interestingly, low level of BDNF in the serum is positively associated with poor long-term functional recovery in both rodents and patients [113, 114]. Consistently, cerebral ischemia led to increases in activity of PDE4 [104] and inhibition of PDE4 by rolipram activated the cAMP/CREB pathway to facilitate functional recovery following ischemic stroke [115]. Motor recovery following ischemic stroke is assigned to both structural and functional reorganization of residual neural tissues [116]. Inhibition of PDE4 by rolipram and HT-0712 activates cAMP/CREB signaling, enhances cognitive function, and restores motor skill after cerebral ischemia [102]. Notably, PDE4 inhibitors facilitate behavioral recovery and cortical reorganization to a significantly greater level when comparing to the rehabilitation alone [102]. Neuroplasticity after stroke could also be strengthened through running exercise and enriched environment, where enriched environment has also been shown to enhance the phosphorylation of CREB in the adult hippocampus [117]. Phosphorylated CREB binds the canonical cAMP response element (CRE), 5'-TGACGTCA-3' in the promoter region of BDNF [118]. Binding of CREB to the motif promotes the transcription of BDNF [118]. Our previous study showed that FFPM, a PDE4 inhibitor ameliorated the cognitive deficits in APP/PS1 transgenic mice and stimulated the expression of BDNF through cAMP/PKA/CREB pathway [119]. Hence, pharmacological activation of cAMP-CREB signaling after ischemia by administration of the PDE inhibitor enhances the expression of BDNF, which is supposed to mediate morphological and functional changes in neurons after stroke.
Inhibition of PDE4 restricts neuroinflammation in the brain and promotes neuroplasticity after stroke
Evidence suggests that the inflammatory response may exacerbate brain injury or contribute to the brain recovery in the different stage of stroke [120]. Studies are still needed to understand the dynamic balance between the protective and detrimental effects of pro-inflammatory factors in different stages of ischemic stroke [120]. It has been widely accepted that at relatively low “physiological” levels, pro-inflammatory factors and other immune mediators are beneficial for maintenance of neuroplasticity [121]. In this case, binding of cytokines to their receptors activates mitogen activated protein kinases (MAPKs) and nuclear factor-kappa B (NF-κB), which influence the expression of neuronal genes and promote neuroplasticity [122]. On the other hand, after neurological injuries such as ischemic stroke, cytokines and other immune mediators are induced and over-expressed, which show negative roles in neuroplasticity [121]. Furthermore, among various cytokines, IL-1 seems to mediate neurotoxic effect during traumatic brain injury, whereas IL-1ra, IL-10 and TGF-β play a neuroprotective effect. Notably, IL-6 and TNF-α may have a dual role where they may produce a neuroprotective or neurotoxic effect, probably depending on the stage and the extent up to which they are induced [123].
Pro-inflammatory factors are produced in phases in experimental and human stroke [124]. IL-1 is proved to be upregulated early in peri-infarct microglia [124]. A meta-analysis suggested that IL-1α (-889C/T) polymorphism is associated with stroke risk [125]. This observation was further confirmed by the finding that IL-1 deficient mice exhibited dramatically reduced ischemic infarct volumes after ischemic brain damage [126]. The role of IL-1 in reducing neurogenesis and neuroplasticity has been repeatedly shown in different animal models [127]. Treatment with IL-1 inducers resulted in significant inhibition of hippocampal neurogenesis in rats [128, 129].
Inhibition of PDE4 is effective to suppress the production of IL-1. Our previous studies have shown that inhibition or silencing of PDE4 attenuated the level of IL-1β in β amyloid-injected mice [50], APP/PS1 transgenic mice [119] and lipopolysaccharide-treated mice [14, 130]. Consistently, another independent study showed that the SNPs in the PDE4D (83T/C) and IL-1 (-889C/T) were related with high risk of the occurrence of ischemic stroke [131]. We propose that inhibition of PDE4 suppresses the expression of IL-1, especially at the early stage of stroke and is beneficial for the functional recovery and this effect is highly related to enhanced neurogenesis and neuroplasticity.
Conclusions and future perspectives
PDE4 is highly expressed in the frontal cortex, hippocampus and cerebellum, which are important brain regions in modulating sensory activity, recognition, and motor activity [132]. Recent studies have made great progress in understanding the roles of PDE4 in the CNS both under physiological and pathological conditions [11]. Emerging evidence has indicated that inhibition of PDE4 is beneficial for the outcome of stroke [100-104]. Figure 3 shows the potential mechanism underlying the role of PDE4 inhibitors in neuroplasticity after stroke. However, complete understanding of PDE4 function in the pathology of stroke is still lacking. Especially, the role of PDE4 in the process of neuroplasticity following ischemic stroke requires further investigation.
The potential mechanism underlying the role of PDE4 inhibitors in neuroplasticity after stroke. Under the condition of ischemic stroke, the expression and activity of PDE4 are extensively increased in neural cells, which subsequently lead to decreased level of cAMP and the inhibition of PKA/CREB and Epac/ERK1/2 signaling pathways. cAMP/PKA/CREB pathway plays a critical role in promoting the expression of BDNF, which is a potent factor triggering neuroplasticity. Activation of Epac/ERK1/2 and cAMP/PKA/CREB pathways inhibits the expression of pro-inflammtory factors, such as IL-1β, which are viewed as negative factors delaying the process of neuroplasticity, especially at the early stage of stroke. PDE4 inhibition is supposed to enhance neuroplasticity through increasing the levels of cAMP and BDNF, and decreasing the expression of pro-inflammatory factors.
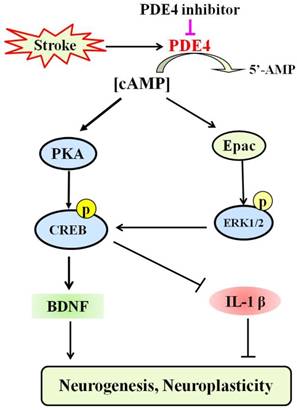
Firstly, a larger number of studies focusing on the PDE4 gene polymorphism, SNP mutations and cerebral infarction risk are needed to determine the role of PDE4 in ischemic stroke. PDE4D gene is associated with an enhanced risk of ischemic stroke [133], while it is still unknown whether other PDE4 isoforms and variants are also the risk factors for ischemic stroke. On the other hand, roles of PDE4D in stroke are only beginning to be unveiled and the possible mechanisms are still needed to be investigated in detail. Specifically, the expression levels and activity of PDE4D are necessary to be fully investigated during ischemic stroke, in particular, the role of PDE4D in neuroplasticity following stroke. To achieve this purpose, further investigation is necessary to explore the functional role of PDE4D, the mutations and effect of selective inhibitors or activators.
Secondly, PDE4-cAMP signaling pathway has been shown to play important roles in ischemic stroke and neuroplasticity. Therefore, targeting PDE4 would be a promising strategy in developing therapeutic agents for the treatment of stroke. However, most of the currently available PDE4 inhibitors produce serious side effects, such as nausea and vomiting [134]. So it is necessary to develop novel and selective inhibitors that affect individual subtypes and isoforms of PDE4 selectively and have little emetic potential. The good news is that selective PDE4 inhibitors without the dose-limiting side effects such as nausea and vomiting have been reported [135, 136]. We also reported some PDE4 inhibitors which show efficiency in the brain at non-emetic doses [119, 137]. Their effect for the treatment of stroke and their role in neuroplasticity after ischemia are being studied. Such information and tools will provide a better understanding of the development of drugs to treat ischemic stroke.
Finally, it is proposed that there are three pivotal processes implicated in neurorepair after stroke: angiogenesis, neurogenesis and synaptic plasticity [138]. Under the condition of stroke, vascular remodeling is significantly increased around the areas of newly-born neurons [138, 139]. The presence of vascular endothelial cells promotes neurogenesis and the survival of newly formed neurons through secreting growth factors and chemokines, such as erythropoietin (EPO) [139]. Interestingly, human vascular endothelial cells express high level of PDE4 [140], inhibition of PDE4 by rolipram effectively attenuates angiogenesis and the expression of VEGF in a chronic subcutaneous inflammatory animal model [141]. This result was further verified by the finding that upregulation of PDE4 in human umbilical vein endothelial cells causes increased hydrolysis of cAMP and enhanced level of angiogenesis [142]. Currently, the roles of PDE4 on angiogenesis and the expression of related factors (such as EPO and VEGF) are largely unknown in the brain following stroke. As angiogenesis is pivotal for neurogenesis and neuroplasticity after stroke, it is worthwhile to systematically study the involvement of PDE4 in the production, proliferation and the biological functions of vascular endothelial cells during the process of cerebral ischemia.
Acknowledgements
This research was supported by National Natural Science Foundation of China (No. 81301099, No. 31371088), Science and Technology Planning Project of Guangdong Province (No. 2011B050200005), and MYRG2016-00052-FHS from University of Macau, and the Science and Technology Development Fund (FDCT) of Macao (FDCT 021/2015/A1).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sporns PB, Hanning U, Schwindt W. et al. Ischemic Stroke: What Does the Histological Composition Tell Us About the Origin of the Thrombus? Stroke. 2017;48:2206-10
2. Wendelboe AM, Raskob GE. Global Burden of Thrombosis: Epidemiologic Aspects. Circ Res. 2016;118:1340-7
3. Mering S, Jolkkonen J. Proper housing conditions in experimental stroke studies-special emphasis on environmental enrichment. Front Neurosci. 2015;9:106
4. Wang Y, Bontempi B, Hong SM. et al. A comprehensive analysis of gait impairment after experimental stroke and the therapeutic effect of environmental enrichment in rats. J Cereb Blood Flow Metab. 2008;28:1936-50
5. White JH, Bartley E, Janssen H. et al. Exploring stroke survivor experience of participation in an enriched environment: a qualitative study. Disabil Rehabil. 2015;37:593-600
6. Jorgensen HS, Nakayama H, Raaschou HO. et al. Stroke. Neurologic and functional recovery the Copenhagen Stroke Study. Phys Med Rehabil Clin N Am. 1999;10:887-906
7. Small SL, Buccino G, Solodkin A. Brain repair after stroke-a novel neurological model. Nat Rev Neurol. 2013;9:698-707
8. Hermann DM, Chopp M. Promoting brain remodelling and plasticity for stroke recovery: therapeutic promise and potential pitfalls of clinical translation. Lancet Neurol. 2012;11:369-80
9. Qu HL, Zhao M, Zhao SS. et al. Forced limb-use enhanced neurogenesis and behavioral recovery after stroke in the aged rats. Neuroscience. 2015;286:316-24
10. Alia C, Spalletti C, Lai S. et al. Neuroplastic Changes Following Brain Ischemia and their Contribution to Stroke Recovery: Novel Approaches in Neurorehabilitation. Front Cell Neurosci. 2017;11:76
11. Heckman PR, Blokland A, Ramaekers J. et al. PDE and cognitive processing: beyond the memory domain. Neurobiol Learn Mem. 2015;119:108-22
12. Garcia-Barroso C, Ugarte A, Martinez M. et al. Phosphodiesterase inhibition in cognitive decline. J Alzheimers Dis. 2014;42(Suppl 4):S561-73
13. Parikh N, Chakraborti AK. Phosphodiesterase 4 (PDE4) Inhibitors in the Treatment of COPD: Promising Drug Candidates and Future Directions. Curr Med Chem. 2016;23:129-41
14. Zou ZQ, Chen JJ, Feng HF. et al. Novel Phosphodiesterase 4 Inhibitor FCPR03 Alleviates Lipopolysaccharide-Induced Neuroinflammation by Regulation of the cAMP/PKA/CREB Signaling Pathway and NF-kappaB Inhibition. J Pharmacol Exp Ther. 2017;362:67-77
15. Jorgensen C, Yasmeen S, Iversen HK. et al. Phosphodiesterase4D (PDE4D)-A risk factor for atrial fibrillation and stroke? J Neurol Sci. 2015;359:266-74
16. Vogel EW 3rd, Morales FN, Meaney DF. et al. Phosphodiesterase-4 inhibition restored hippocampal long term potentiation after primary blast. Exp Neurol. 2017;293:91-100
17. Havekes R, Park AJ, Tolentino RE. et al. Compartmentalized PDE4A5 Signaling Impairs Hippocampal Synaptic Plasticity and Long-Term Memory. J Neurosci. 2016;36:8936-46
18. Wang ZZ, Yang WX, Zhang Y. et al. Phosphodiesterase-4D Knock-down in the Prefrontal Cortex Alleviates Chronic Unpredictable Stress-Induced Depressive-Like Behaviors and Memory Deficits in Mice. Sci Rep. 2015;5:11332
19. Kann O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol Dis. 2016;90:75-85
20. Miljkovic-Lolic M, Silbergleit R, Fiskum G. et al. Neuroprotective effects of hyperbaric oxygen treatment in experimental focal cerebral ischemia are associated with reduced brain leukocyte myeloperoxidase activity. Brain Res. 2003;971:90-4
21. Hedrick NG, Yasuda R. Regulation of Rho GTPase proteins during spine structural plasticity for the control of local dendritic plasticity. Curr Opin Neurobiol. 2017;45:193-201
22. Phillips C. Lifestyle Modulators of Neuroplasticity: How Physical Activity, Mental Engagement, and Diet Promote Cognitive Health during Aging. Neural Plast. 2017;2017:3589271
23. Duffau H. Brain plasticity: from pathophysiological mechanisms to therapeutic applications. J Clin Neurosci. 2006;13:885-97
24. Buffington SA, Huang W, Costa-Mattioli M. Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37:17-38
25. Raymaekers SR, Darras VM. Thyroid hormones and learning-associated neuroplasticity. Gen Comp Endocrinol. 2017;247:26-33
26. Wang Y, Li WY, Li ZG. et al. Transcriptional and Epigenetic Regulation in Injury-Mediated Neuronal Dendritic Plasticity. Neurosci Bull. 2017;33:85-94
27. Krakauer JW. Arm function after stroke: from physiology to recovery. Semin Neurol. 2005;25:384-95
28. Hatem SM, Saussez G, Della Faille M. et al. Rehabilitation of Motor Function after Stroke: A Multiple Systematic Review Focused on Techniques to Stimulate Upper Extremity Recovery. Front Hum Neurosci. 2016;10:442
29. Constans A, Pin-Barre C, Temprado JJ. et al. Influence of Aerobic Training and Combinations of Interventions on Cognition and Neuroplasticity after Stroke. Front Aging Neurosci. 2016;8:164
30. Krakauer JW, Carmichael ST, Corbett D. et al. Getting neurorehabilitation right: what can be learned from animal models? Neurorehabil Neural Repair. 2012;26:923-31
31. Felling RJ, Song H. Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp Neurol. 2015;268:37-45
32. Kim SY, Allred RP, Adkins DL. et al. Experience with the "good" limb induces aberrant synaptic plasticity in the perilesion cortex after stroke. J Neurosci. 2015;35:8604-10
33. Johnston DG, Denizet M, Mostany R. et al. Chronic in vivo imaging shows no evidence of dendritic plasticity or functional remapping in the contralesional cortex after stroke. Cereb Cortex. 2013;23:751-62
34. Huang L, Li Q, Wen R. et al. Rho-kinase inhibitor prevents acute injury against transient focal cerebral ischemia by enhancing the expression and function of GABA receptors in rats. Eur J Pharmacol. 2017;797:134-42
35. Mostany R, Portera-Cailliau C. Absence of large-scale dendritic plasticity of layer 5 pyramidal neurons in peri-infarct cortex. J Neurosci. 2011;31:1734-8
36. Zepeda R, Contreras V, Pissani C. et al. Venlafaxine treatment after endothelin-1-induced cortical stroke modulates growth factor expression and reduces tissue damage in rats. Neuropharmacology. 2016;107:131-45
37. Ishrat T, Pillai B, Soliman S. et al. Low-dose candesartan enhances molecular mediators of neuroplasticity and subsequent functional recovery after ischemic stroke in rats. Mol Neurobiol. 2015;51:1542-53
38. Chen J, Zhang C, Jiang H. et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281-90
39. Chao CC, Karabanov AN, Paine R. et al. Induction of motor associative plasticity in the posterior parietal cortex-primary motor network. Cereb Cortex. 2015;25:365-73
40. Nodera H, Manto M. Cerebellum tunes the excitability of the motor system: evidence from peripheral motor axons. Cerebellum. 2014;13:663-5
41. Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861-72
42. Chen J, Zacharek A, Cui X. et al. Treatment of stroke with a synthetic liver X receptor agonist, TO901317, promotes synaptic plasticity and axonal regeneration in mice. J Cereb Blood Flow Metab. 2010;30:102-9
43. Yao PJ, Petralia RS, Mattson MP. Sonic Hedgehog Signaling and Hippocampal Neuroplasticity. Trends Neurosci. 2016;39:840-50
44. Ueno Y, Chopp M, Zhang L. et al. Axonal outgrowth and dendritic plasticity in the cortical peri-infarct area after experimental stroke. Stroke. 2012;43:2221-8
45. Fouda AY, Alhusban A, Ishrat T. et al. Brain-Derived Neurotrophic Factor Knockdown Blocks the Angiogenic and Protective Effects of Angiotensin Modulation After Experimental Stroke. Mol Neurobiol. 2017;54:661-70
46. Qu H, Zhao M, Zhao S. et al. Forced limb-use enhances brain plasticity through the cAMP/PKA/CREB signal transduction pathway after stroke in adult rats. Restor Neurol Neurosci. 2014;32:597-609
47. Nthenge-Ngumbau DN, Mohanakumar KP. Can Cyclic Nucleotide Phosphodiesterase Inhibitors Be Drugs for Parkinson's Disease?. Mol Neurobiol. 2017
48. Blokland A, Menniti FS, Prickaerts J. PDE inhibition and cognition enhancement. Expert Opin Ther Pat. 2012;22:349-54
49. Kohno T, Yamaguchi H, Hakoshima T. Crystallization and preliminary X-ray crystallographic analysis of human phosphodiesterase 12. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:520-2
50. Zhang C, Cheng Y, Wang H. et al. RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-beta42-induced memory deficits in mice. J Alzheimers Dis. 2014;38:269-80
51. Richter W, Menniti FS, Zhang HT. et al. PDE4 as a target for cognition enhancement. Expert Opin Ther Targets. 2013;17:1011-27
52. O'Donnell JM, Zhang HT. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4). Trends Pharmacol Sci. 2004;25:158-63
53. Zhang HT. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr Pharm Des. 2009;15:1688-98
54. Houslay MD, Adams DR. Putting the lid on phosphodiesterase 4. Nat Biotechnol. 2010;28:38-40
55. Baillie GS, MacKenzie SJ, McPhee I. et al. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br J Pharmacol. 2000;131:811-9
56. Hoffmann R, Baillie GS, MacKenzie SJ. et al. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18:893-903
57. Collins DM, Murdoch H, Dunlop AJ. et al. Ndel1 alters its conformation by sequestering cAMP-specific phosphodiesterase-4D3 (PDE4D3) in a manner that is dynamically regulated through Protein Kinase A (PKA). Cell Signal. 2008;20:2356-69
58. MacKenzie SJ, Baillie GS, McPhee I. et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br J Pharmacol. 2002;136:421-33
59. Byrne AM, Elliott C, Hoffmann R. et al. The activity of cAMP-phosphodiesterase 4D7 (PDE4D7) is regulated by protein kinase A-dependent phosphorylation within its unique N-terminus. FEBS Lett. 2015;589:750-5
60. Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91-100
61. Contreras S, Milara J, Morcillo E. et al. Selective Inhibition of Phosphodiesterases 4A, B, C and D Isoforms in Chronic Respiratory Diseases: Current and Future Evidences. Curr Pharm Des. 2017;23:2073-83
62. Eskandari N, Tashrifi F, Bastan R. et al. Cyclic nucleotide phosphodiesterase isoforms in human basophils and mast cells. Int J Immunopathol Pharmacol. 2016;29:654-65
63. Asirvatham AL, Galligan SG, Schillace RV. et al. A-kinase anchoring proteins interact with phosphodiesterases in T lymphocyte cell lines. J Immunol. 2004;173:4806-14
64. Willoughby D, Wong W, Schaack J. et al. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 2006;25:2051-61
65. Perry SJ, Baillie GS, Kohout TA. et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298:834-6
66. Bolger GB, Baillie GS, Li X. et al. Scanning peptide array analyses identify overlapping binding sites for the signalling scaffold proteins, beta-arrestin and RACK1, in cAMP-specific phosphodiesterase PDE4D5. Biochem J. 2006;398:23-36
67. Gnegy ME. Calmodulin in neurotransmitter and hormone action. Annu Rev Pharmacol Toxicol. 1993;33:45-70
68. Flamm ES, Schiffer J, Viau AT. et al. Alterations of cyclic AMP in cerebral ischemia. Stroke. 1978;9:400-2
69. Torres-Quesada O, Mayrhofer JE, Stefan E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017;37:1-11
70. Lim J, Yang C, Hong SJ. et al. Regulation of tyrosine hydroxylase gene transcription by the cAMP-signaling pathway: involvement of multiple transcription factors. Mol Cell Biochem. 2000;212:51-60
71. Wan Q, Ma X, Zhang ZJ. et al. Ginsenoside Reduces Cognitive Impairment During Chronic Cerebral Hypoperfusion Through Brain-Derived Neurotrophic Factor Regulated by Epigenetic Modulation. Mol Neurobiol. 2017;54:2889-900
72. Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670-709
73. Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680-6
74. Hochbaum D, Hong K, Barila G. et al. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J Biol Chem. 2008;283:4464-8
75. Huston E, Lynch MJ, Mohamed A. et al. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:12791-6
76. Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012:4
77. Ku SM, Han MH. HCN Channel Targets for Novel Antidepressant Treatment. Neurotherapeutics. 2017;14:698-715
78. Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769-824
79. Sarfati M, Mateo V, Baudet S. et al. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood. 2003;101:265-9
80. Heystek HC, Thierry AC, Soulard P. et al. Phosphodiesterase 4 inhibitors reduce human dendritic cell inflammatory cytokine production and Th1-polarizing capacity. Int Immunol. 2003;15:827-35
81. Nie J, Yang X. Modulation of Synaptic Plasticity by Exercise Training as a Basis for Ischemic Stroke Rehabilitation. Cell Mol Neurobiol. 2017;37:5-16
82. Caleo M. Rehabilitation and plasticity following stroke: Insights from rodent models. Neuroscience. 2015;311:180-94
83. Reines A, Cereseto M, Ferrero A. et al. Maintenance treatment with fluoxetine is necessary to sustain normal levels of synaptic markers in an experimental model of depression: correlation with behavioral response. Neuropsychopharmacology. 2008;33:1896-908
84. Heckman PR, Wouters C, Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer's disease: a translational overview. Curr Pharm Des. 2015;21:317-31
85. Havekes R, Park AJ, Tudor JC. et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife. 2016:5
86. Reyes-Irisarri E, Perez-Torres S, Miro X. et al. Differential distribution of PDE4B splice variant mRNAs in rat brain and the effects of systemic administration of LPS in their expression. Synapse. 2008;62:74-9
87. Zhang HT, Huang Y, Masood A. et al. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology. 2008;33:1611-23
88. Rutten K, Wallace TL, Works M. et al. Enhanced long-term depression and impaired reversal learning in phosphodiesterase 4B-knockout (PDE4B-/-) mice. Neuropharmacology. 2011;61:138-47
89. McGirr A, Lipina TV, Mun HS. et al. Specific Inhibition of Phosphodiesterase-4B Results in Anxiolysis and Facilitates Memory Acquisition. Neuropsychopharmacology. 2016;41:1080-92
90. Li YF, Cheng YF, Huang Y. et al. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci. 2011;31:172-83
91. Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol. 2007;18:391-418
92. Gretarsdottir S, Thorleifsson G, Reynisdottir ST. et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003;35:131-8
93. Hagberg H, Mallard C, Ferriero DM. et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol. 2015;11:192-208
94. Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97
95. Mohammadi MT. Overproduction of nitric oxide intensifies brain infarction and cerebrovascular damage through reduction of claudin-5 and ZO-1 expression in striatum of ischemic brain. Pathol Res Pract. 2016;212:959-64
96. Zhang B, Nweze I, Lakshmanan J. et al. Activation of a cyclic amp-guanine exchange factor in hepatocytes decreases nitric oxide synthase expression. Shock. 2013;39:70-6
97. Markovic M, Miljkovic D, Trajkovic V. Regulation of inducible nitric oxide synthase by cAMP-elevating phospho-diesterase inhibitors. Curr Drug Targets Inflamm Allergy. 2003;2:63-79
98. Kwak HJ, Park KM, Choi HE. et al. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008;20:803-14
99. Redondo M, Zarruk JG, Ceballos P. et al. Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur J Med Chem. 2012;47:175-85
100. Block F, Tondar A, Schmidt W. et al. Delayed treatment with rolipram protects against neuronal damage following global ischemia in rats. Neuroreport. 1997;8:3829-32
101. Kato H, Araki T, Itoyama Y. et al. Rolipram, a cyclic AMP-selective phosphodiesterase inhibitor, reduces neuronal damage following cerebral ischemia in the gerbil. Eur J Pharmacol. 1995;272:107-10
102. MacDonald E, Van der Lee H, Pocock D. et al. A novel phosphodiesterase type 4 inhibitor, HT-0712, enhances rehabilitation-dependent motor recovery and cortical reorganization after focal cortical ischemia. Neurorehabil Neural Repair. 2007;21:486-96
103. Hatinen S, Sairanen M, Sirvio J. et al. Improved sensorimotor function by rolipram following focal cerebral ischemia in rats. Restor Neurol Neurosci. 2008;26:493-9
104. Li LX, Cheng YF, Lin HB. et al. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab Brain Dis. 2011;26:37-47
105. Wiescholleck V, Manahan-Vaughan D. PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl Psychiatry. 2012;2:e89
106. Zhong P, Wang W, Yu F. et al. Phosphodiesterase 4 inhibition impairs cocaine-induced inhibitory synaptic plasticity and conditioned place preference. Neuropsychopharmacology. 2012;37:2377-87
107. Di Lazzaro V, Pellegrino G, Di Pino G. et al. Val66Met BDNF gene polymorphism influences human motor cortex plasticity in acute stroke. Brain Stimul. 2015;8:92-6
108. Rothman SM, Griffioen KJ, Wan R. et al. Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Ann N Y Acad Sci. 2012;1264:49-63
109. Kotlega D, Peda B, Zembron-Lacny A. et al. The role of brain-derived neurotrophic factor and its single nucleotide polymorphisms in stroke patients. Neurol Neurochir Pol. 2017;51:240-6
110. Mang CS, Campbell KL, Ross CJ. et al. Promoting neuroplasticity for motor rehabilitation after stroke: considering the effects of aerobic exercise and genetic variation on brain-derived neurotrophic factor. Phys Ther. 2013;93:1707-16
111. Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81-9
112. Qiao HJ, Li ZZ, Wang LM. et al. Association of lower serum Brain-derived neurotrophic factor levels with larger infarct volumes in acute ischemic stroke. J Neuroimmunol. 2017;307:69-73
113. Stanne TM, Aberg ND, Nilsson S. et al. Low Circulating Acute Brain-Derived Neurotrophic Factor Levels Are Associated With Poor Long-Term Functional Outcome After Ischemic Stroke. Stroke. 2016;47:1943-5
114. Xu HB, Xu YH, He Y. et al. Decreased Serum Brain-Derived Neurotrophic Factor May Indicate the Development of Poststroke Depression in Patients with Acute Ischemic Stroke: A Meta-Analysis. J Stroke Cerebrovasc Dis. 2018;27:709-15
115. Hu S, Cao Q, Xu P. et al. Rolipram stimulates angiogenesis and attenuates neuronal apoptosis through the cAMP/cAMP-responsive element binding protein pathway following ischemic stroke in rats. Exp Ther Med. 2016;11:1005-10
116. McClung CA, Nestler EJ. Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology. 2008;33:3-17
117. Young D, Lawlor PA, Leone P. et al. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448-53
118. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141-51
119. Guo H, Cheng Y, Wang C. et al. FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology. 2017;116:260-9
120. Jin R, Liu L, Zhang S. et al. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res. 2013;6:834-51
121. Kriz J, Lalancette-Hebert M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009;117:497-509
122. Di Filippo M, Sarchielli P, Picconi B. et al. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci. 2008;29:402-12
123. Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734-44
124. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677-98
125. Zou L, Zhao H, Gong X. et al. The association between three promoter polymorphisms of IL-1 and stroke: a meta-analysis. Gene. 2015;567:36-44
126. Boutin H, LeFeuvre RA, Horai R. et al. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21:5528-34
127. Zunszain PA, Anacker C, Cattaneo A. et al. Interleukin-1beta: a new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology. 2012;37:939-49
128. Ekdahl CT, Claasen JH, Bonde S. et al. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632-7
129. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760-5
130. You T, Cheng Y, Zhong J. et al. Roflupram, a Phosphodiesterase 4 Inhibitior, Suppresses Inflammasome Activation through Autophagy in Microglial Cells. ACS Chem Neurosci. 2017
131. Li N, He Z, Xu J. et al. Association of PDE4D and IL-1 gene polymorphism with ischemic stroke in a Han Chinese population. Brain Res Bull. 2010;81:38-42
132. Xu Y, Zhang HT, O'Donnell JM. Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders. Handb Exp Pharmacol. 2011:447-85
133. Wu WL, Feng XW, Qiu CF. et al. A meta-analysis of PDE-gene polymorphism and cerebral infarction risk. Exp Ther Med. 2017;13:2905-11
134. Rock EM, Benzaquen J, Limebeer CL. et al. Potential of the rat model of conditioned gaping to detect nausea produced by rolipram, a phosphodiesterase-4 (PDE4) inhibitor. Pharmacol Biochem Behav. 2009;91:537-41
135. Rutter AR, Poffe A, Cavallini P. et al. GSK356278, a potent, selective, brain-penetrant phosphodiesterase 4 inhibitor that demonstrates anxiolytic and cognition-enhancing effects without inducing side effects in preclinical species. J Pharmacol Exp Ther. 2014;350:153-63
136. Burgin AB, Magnusson OT, Singh J. et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63-70
137. Zhou ZZ, Cheng YF, Zou ZQ. et al. Discovery of N-Alkyl Catecholamides as Selective Phosphodiesterase-4 Inhibitors with Anti-neuroinflammation Potential Exhibiting Antidepressant-like Effects at Non-emetic Doses. ACS Chem Neurosci. 2017;8:135-46
138. Font MA, Arboix A, Krupinski J. Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr Cardiol Rev. 2010;6:238-44
139. Tsai PT, Ohab JJ, Kertesz N. et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci. 2006;26:1269-74
140. Netherton SJ, Maurice DH. Vascular endothelial cell cyclic nucleotide phosphodiesterases and regulated cell migration: implications in angiogenesis. Mol Pharmacol. 2005;67:263-72
141. Mendes JB, Rocha MA, Araujo FA. et al. Differential effects of rolipram on chronic subcutaneous inflammatory angiogenesis and on peritoneal adhesion in mice. Microvasc Res. 2009;78:265-71
142. Muzaffar S, Jeremy JY, Angelini GD. et al. NADPH oxidase 4 mediates upregulation of type 4 phosphodiesterases in human endothelial cells. J Cell Physiol. 2012;227:1941-50
Author contact
![]() Corresponding authors: Prof. Jiangping Xu, MD, PhD, Department of Neuropharmacology and Drug Discovery, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China. Email: jpxedu.cn and Prof. Wenhua Zheng, MD, PhD, Faculty of Health Sciences, University of Macau, Room 4021, Building E12, Taipa, Macau. E-mail address: wenhuazhengmo
Corresponding authors: Prof. Jiangping Xu, MD, PhD, Department of Neuropharmacology and Drug Discovery, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China. Email: jpxedu.cn and Prof. Wenhua Zheng, MD, PhD, Faculty of Health Sciences, University of Macau, Room 4021, Building E12, Taipa, Macau. E-mail address: wenhuazhengmo