Impact Factor ISSN: 1449-2288
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(12):1755-1768. doi:10.7150/ijbs.28142 This issue Cite
Research Paper
Inhibition of Estrogen Signaling Reduces the Incidence of BRCA1-associated Mammary Tumor Formation
Hye Jung Baek1*, Sun Eui Kim1*, Eun Kyung Choi1, Jong Kwang Kim1, Dong Hoon Shin1, Eun Jung Park1, Tae Hyun Kim1, Joo-Young Kim1, Kwang Gi Kim2, Chu-Xia Deng3, Sang Soo Kim1 ![]()
1. Research Institute, National Cancer Center, Goyang, 10408, Korea,
2. Department of Biomedical Engineering, Gachon University College of Medicine, Incheon, 21565, Korea,
3. Cancer Centre, Faculty of Health Sciences, University of Macau, Macau SAR 999078, China.
*These authors contribute equally.
Received 2018-6-26; Accepted 2018-8-30; Published 2018-10-3
Abstract

BRCA1-deficient breast cancer is a very well-known hereditary cancer. However, except for resection of normal mammary glands and ovaries, there is no acceptable measure for proactively preventing tumor development. Importantly, inherited BRCA1 mutations are closely associated with tumors in hormone-responsive tissues. Here, we examined the effects of estrogen on the accumulation of genetic instabilities upon loss of BRCA1, and assessed the contribution of estrogen signaling to the incidence and progression of Brca1-mutated mammary tumors. Our in vitro studies showed that treatment of BRCA1-depleted breast cancer cells with estrogen induced proliferation. Additionally, estrogen reduced the ability of these BRCA1-knockdown cells to sense radiation-induced DNA damage and also facilitated G1/S progression. Moreover, long-term treatment of Brca1-mutant (Brca1co/coMMTV-Cre) mice with the selective estrogen receptor (ER)-α degrader, fulvestrant, decreased the tumor formation rate from 64% to 36%, and also significantly reduced mammary gland density in non-tumor-bearing mice. However, in vivo experiments showed that fulvestrant treatment did not alter the progression of ER-positive Brca1-mutant tumors, which were frequently identified in the aged population and showed less aggressive tendencies. These findings enhance our understanding of how ER-α signaling contributes to BRCA1-deficient mammary tumors and provide evidence suggesting that targeted inhibition of ER-α signaling may be useful for the prevention of BRCA1-mutated breast cancer.
Keywords: BRCA1, estrogen, fulvestrant, cancer prevention
Introduction
The BRCA1 (breast cancer type 1 susceptibility protein) protein is a tumor suppressor that makes a critical contribution to the maintenance of genomic integrity by integrating important cellular processes that regulate genetic stability, such as DNA-damage repair, cell-cycle control, centrosome duplication, and apoptosis [1]. Germline mutations in BRCA1 are responsible for a considerable portion of hereditary breast and ovarian cancers [2]. Women with germline mutations in BRCA1 have a 57% (95% confidence interval [CI], 47%-66%) risk of developing breast cancer and a 40% (95% CI, 35%-46%) risk of developing ovarian cancer by the age of 70 [3]. Gene and protein expression profiling have revealed that cancers arising owing to BRCA1 mutations show triple-negative and basal-like properties, tend to be aggressive, and are typically associated with a poor prognosis [4].
The National Comprehensive Cancer Network (NCCN) recommends that BRCA1-mutation-positive women undergo periodic breast screening and consider mastectomy and salpingo-oophorectomy to reduce their cancer risk (NCCN guideline Ver. 2.2017). Unfortunately, we currently lack the means to actively prevent the initiation and progression of tumors in these women when they do occur, apart from resection of tumors followed by adjuvant chemotherapy. However, resection and chemotherapy may not be an effective option in all patients. Thus, despite considerable research efforts to prevent tumor formation and develop suitable therapies, there remains an urgent need to develop means for improving the prevention and treatment of BRCA1-associated breast cancer.
In approaching the control of BRCA1-associated tumorigenesis, two long-standing questions need to be answered: Why do most tumors caused by a loss of BRCA1 arise in hormone-responsive tissues (e.g., breast and ovary)? And how do BRCA1-mutant cells survive until the acquisition of self-supporting properties that protect them from senescence and apoptosis [5, 6]? One possible answer to this question is estrogen signaling, and attempts have been made to control BRCA1-deficient mammary tumors by inhibiting this signaling axis. Interestingly, two previous in vivo studies using the same mouse model (Brca1co/coMMTV-Cre Tp53+/-) have reached different conclusions. Oophorectomy of these Brca1-mutant mice reducing endogenous estrogen levels, resulting in a much lower tumor incidence than observed in controls from 4 months after surgery [7]. In contrast, treatment of Brca1-mutant mice with tamoxifen was shown to produce no chemopreventive effects against mammary cancer development [8]. A recent study showed that responsiveness to the anti-estrogen tamoxifen, which acts by blocking estrogen binding to the estrogen receptor (ER), is determined by the proto-oncogene ErbB2/Her-2 and other factors [9]. These results suggest that treatment with tamoxifen is inadequate for tumor prevention in normal mammary glands, but is suitable for tumor progression of ER(+) and/or Her-2(+) breast cancers.
Here, to examine the contribution of estrogen signaling to BRCA1-associated breast cancer, we tested fulvestrant (brand name, Faslodex) in Brca1-mutant mice. Fulvestrant, a different type of ER antagonist that lacks agonist activity [10] [11], binds to the ER, blocking its function and causing its rapid degradation [12]. Although fulvestrant has a distinct mode of action, its effectiveness is comparable to that of tamoxifen and an aromatase inhibitor in advanced breast cancer, and reduces the risk of cross-resistance to other endocrine agents [13]. In the present study, we suppressed estrogen signaling with fulvestrant, and examined tumor initiation and progression in Brca1co/coMMTV-cre mice, which simulate human BRCA1-mutated mammary tumors.
Methods
Cell culture
MCF7 cells (wild-type BRCA1) and BRCA1-mutant HCC1937 cells were obtained from the American Type Culture Collection. The authenticity of human cell lines was confirmed by short tandem repeat (STR) analysis performed by the Omics Core of the National Cancer Center. For growth assays, cells were plated at 2 × 104 cells per well in 4-well plates (in quadruplicate), and treated as indicated, after which cell viability was determined using an MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay kit (Sigma) according to the manufacturer's instructions. Expression of BRCA1 in MCF7 cells was knocked down by transfecting cells with a pool of three BRCA1-targeting small interfering RNAs (siRNAs; Santa Cruz) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol; scrambled siRNA (Dharmacon) was used as a negative control. For experiments designed to monitor the effects of estrogen, cells were maintained in hormone-depleted, phenol red-free Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% dextran/charcoal-treated fetal bovine serum (FBS; Gemini).
Animal experiments
Conditional Brca1-knockout and MMTV-Cre transgenic mice were provided by the National Cancer Institute mouse repository. Female Brca1-mutant mice were generated by crossing Brca1 conditional-knockout mice with MMTV-Cre mice, which were originally generated by Drs. Deng and Hennighausen, respectively [14] [15].
Prolonged effects of estrogen were assessed by subcutaneously injecting 9-month-old Brca1co/co MMTV-Cre mice under the skin between the neck and shoulder with E2 pellets (1.7 mg in 90-day release pellets; Innovative Research of America). Three months later, grafted mice were euthanized by CO2 inhalation and their mammary glands were collected for further analysis.
For long-term administration of fulvestrant, mice were assigned to vehicle control or fulvestrant treatment (250 mg/kg, subcutaneously [sc], biweekly) groups after reaching 9 months of age, and were examined weekly up to 14 month of age for the occurrence of tumors. Fulvestrant (Abmole Bioscience) was dissolved in ethanol and further mixed with sesame oil and benzyl alcohol (20% ethanol/70% sesame oil/10% benzyl alcohol).
For tumor allografts, 18 different primary tumors obtained from 18 tumor-bearing Brca1co/coMMTV-Cre mice were orthotopically implanted into 5-week-old female HsdCpb:NMRI- Foxn1nu mice (Orient-Harlan Laboratories). After each grafted tumor reached ~1,000 mm3, the tumor tissue was excised, trimmed with a tissue slicer, and re-implanted into recipient mice. After implantation, the recipient mice were treated biweekly with vehicle or fulvestrant (250 mg/kg, sc). Tumor growth was assessed as the ratio of the tumor volume at a given time to that recorded at the initiation of treatment. For histology, tissues were fixed in 10% (v/v) formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin (H&E), and examined by light microscopy. All procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee of the National Cancer Center of Korea.
Western blotting, cell staining, and histological analysis
Western blot analysis was carried out according to standard procedures using enhanced-chemiluminescence detection (Amersham). Tumor tissue lysates were prepared as previously described [16, 17]. The following antibodies were used: anti-β-actin (#8459), anti-phospho-AKT (#4060), anti-phospho-ATM (#4526), anti-phospho-cyclin D1 (#2921), anti-MAPK (#9102), anti-phospho-MAPK (#4376), anti-PDK1 (#3062), anti-phospho-PDK1 (#3438), anti-PI3K (#4257), anti-phospho-PI3K (#4228), anti-PTEN (#9188), anti-phospho-PTEN (#9549), and anti-phospho-Rb (#9308) (Cell Signaling Technology); anti-β-actin (#SC-47778), anti-AKT (#SC-8312), anti-BRCA1 (#SC-6954), anti-cyclin D1 (#SC-20044), anti-phospho-ER-α (#SC-101675), anti-ER-α (#SC-542), anti-Rb (#SC-50) and anti-p53 (#SC-126) (Santa Cruz); anti-Ki67 (Novus, #NB500-170); and anti-PCNA (Atlas Antibodies, #HPA030522). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and anti-mouse antibodies (Jackson Immuno Research), as appropriate, were used as secondary antibodies.
Cells grown on chamber slides (BD Biosciences) were fixed in 3% paraformaldehyde and incubated with an antibody against γ-H2AX (Millipore, #05-636). Immunoreactivity was detected with an Alexa Fluor 647-conjugated secondary antibody (Molecular Probes), and cells were counterstained with DAPI (4,6-diamidino-2-phenylindole) to label nuclei. Numbers and intensities of γ-H2AX foci were estimated in 10 randomly selected fields for each condition using measuring tools in the Leica application suite (Ver. 4.2.0; Leica Microsystems). Signal accumulation (SA) per cell was calculated according to the following formula: SA = Σ(foci number × average foci area × average intensity in field)/total number of cells. Quantification of γ-H2AX foci was performed using at least 75 cells for each condition.
Antigenic proteins were histologically detected using a Zymed Histostain kit (Invitrogen), according to the manufacturer's instructions. The primary antibodies used were anti-cleaved caspase-3 (#9644), anti-cyclin D1 (#2978) (Cell Signaling Technology), anti-cytokeratin 5 (905504, BioLegend), and anti-PCNA (#HPA030522, Atlas Antibodies,). Apoptotic cells were assessed using terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick-end labeling (TUNEL) assays (Millipore), and accumulation of collagen in tissues was monitored using Masson's trichrome staining (Sigma). All comparisons of histochemical analyses were performed on a manually generated tissue microarray (Quick-Ray, UNITMA) containing control and comparable samples on the same slide.
Comet assay
DNA breakage was assessed using a comet assay, a single-cell electrophoretic method that produces a fluorescent comet-like image with a tail whose length and fluorescence intensity is proportional to the degree of DNA breakage. All comet assays were carried out under denatured conditions using a Comet assay kit (Trevigen), according to manufacturer's protocol. MCF7 cells transfected with siRNA targeting BRCA1 or scrambled (control) siRNA were irradiated (10 Gy) in the absence or presence of E2 (100 nM), harvested by treating with trypsin immediately or after a 2-hour incubation, mixed with low‑melting agarose at 37°C, and plated onto comet slides. The slides were immersed in a pre‑chilled lysis solution and incubated at 4°C overnight. Thereafter, slides were electrophoresed and stained in a 2.5-μg/ml propidium iodide solution. The lengths of comet tails were measured and analyzed using a Komet 4.0.2 image analysis system (Andor Technology). Tail moment, defined as the product of the tail length and the fraction of total DNA in the tail, incorporates measures of both the smallest detectable size of migrating DNA (reflected in the comet tail length) and the number of relaxed/broken pieces (represented by the intensity of DNA in the tail). Olive Tail Moment was calculated from at least 30 cells for each condition according to the following formula: Olive Tail Moment = (Tail Lengthmean-Head Lengthmean) % of Tail DNA/100.
Expression analysis
Tumor tissues were dissected free of surrounding normal tissues, and immediately frozen using a pre-chilled aluminum block. Total RNA was purified from tumor tissues using an RNeasy mini kit (Qiagen) and subjected to genome-wide RNAseq analyses, performed by eBiogen (Seoul, Korea). The raw data obtained from duplicate analyses of 8 samples were normalized using Cufflinks RNAseq workflow [18]. Spearman's rank correlations were performed to select genes that were highly correlated with tumor volume. Highly correlated genes (HCG) were selected as markers, and a heat map was generated using the z-scores of their normalized expression, in fragments per kilobase per million mapped fragments (FPKM). The samples were sorted so as to highlight correlations of their ratio of tumor volume (RTV) with their gene expression pattern. Normalized FPKM values for HCGs in each adjuvant-based experiment were used as input to the heat map function of the Superheat R open source package to generate a heat map.
Functional enrichment analysis
Functional GO (Gene Ontology) enrichment analyses were performed using a right-sided hypergeometric test (Bonferroni corrected P-value < 0.05) based on the Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg/pathway.html), Wikipathway (https://www.wikipathways.org), and GO biological process. Similar GO terms were grouped by calculating the number of related genes that shared any two GO terms (Cohen's kappa score > 0.4). Functional groupings of resultant terms were visualized using Cytoscape v.3.5.1 [19].
Statistical analyses
Student's t test (http://www.physics.csbsju.edu/stats/t-test.html) was used to compare differences in means between two groups, as specified in the text. Spearman's rank correlation (http://http://sites.utexas.edu/sos/guided/inferential/numeric/bivariate/rankcor/) was used to correlate gene expression with tumor volume (|Rho| >0.6). A P-value < 0.05 was considered statistically significant.
Results
Estrogen restores the proliferation of BRCA1-deficient mammary epithelial cells
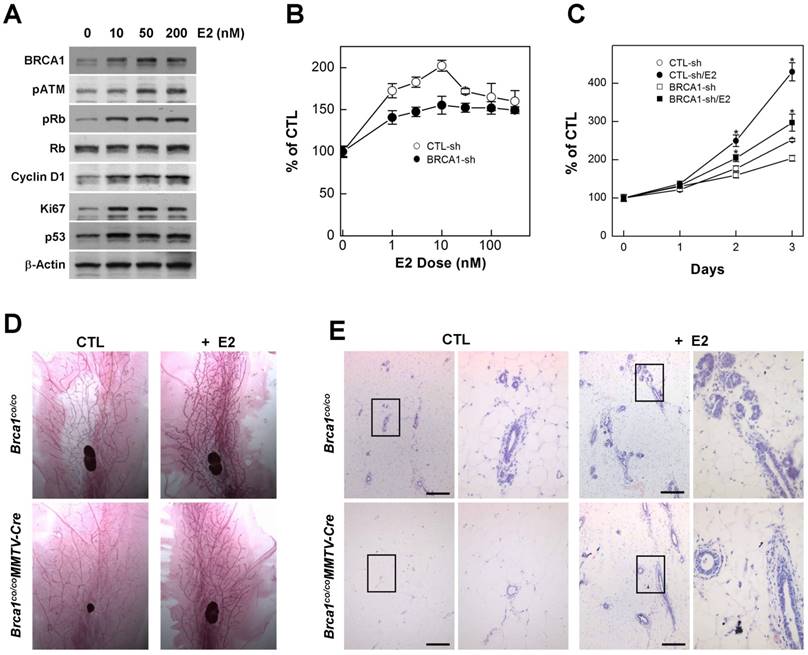
Estrogen is an endogenous hormone that is required for the development and maintenance of reproductive organs [20]. It is also a potent mitogen for breast epithelial cells that acts via the ER [21] [22]. However, high levels of estrogen signaling induce excess proliferation and alter the proteins responsible for DNA-damage repair, including BRCA1, ATM, and p53 [23]. In addition, cytochrome P450-mediated estrogen metabolites are capable of causing DNA damage [24], suggesting that excess estrogen interrupts DNA damage-repair processes. Consistent with this, we found that treatment with estradiol (E2), an endogenous estrogen, induced proteins that regulate proliferation, cell cycle progression and apoptosis in MCF7 cells (Fig. 1A), suggesting that estrogen signaling is involved in several important pathways related to carcinogenesis. Although mutations in BRCA1 eventually contribute to tumorigenesis, upon acquisition of a BRCA1 deficiency, cells immediately exhibit genetic instability, senescence, growth arrest and apoptosis, indicating that BRCA1-associated tumors require survival and growth stimuli [1]. Importantly, treatment with E2 induced the proliferation of BRCA1-depleted cells, generated by transfecting with siRNA against BRCA1, in a concentration- and time-dependent manner, as it did in normal cells (Fig. 1B and 1C).
In mice, the loss of BRCA1 attenuates the proliferation of mammary gland epithelial cells and decreases or delays the development of mammary glands during puberty [14]. Considering that E2 acts as a stimulator of mammary epithelial cell proliferation, we examined whether E2 restores proper development of mammary glands in adolescence Brca1-mutant mice. To this end, we administrated E2 (1 μg) by subcutaneous injection and collected mammary glands 2 days after the injection. Consistent with our in vitro results, adolescent female Brca1-mutant mice treated with E2 showed elongation of mammary gland ducts compared with sham controls (Fig. 1D). A further histological analysis also revealed that ductal linings became thickened through accumulation of epithelial cells (Fig. 1E), suggesting that estrogen is capable of supporting the survival and proliferation of BRCA1-deficient cells.
Estrogen alters DNA damage sensing, cell cycle regulation, and proliferation
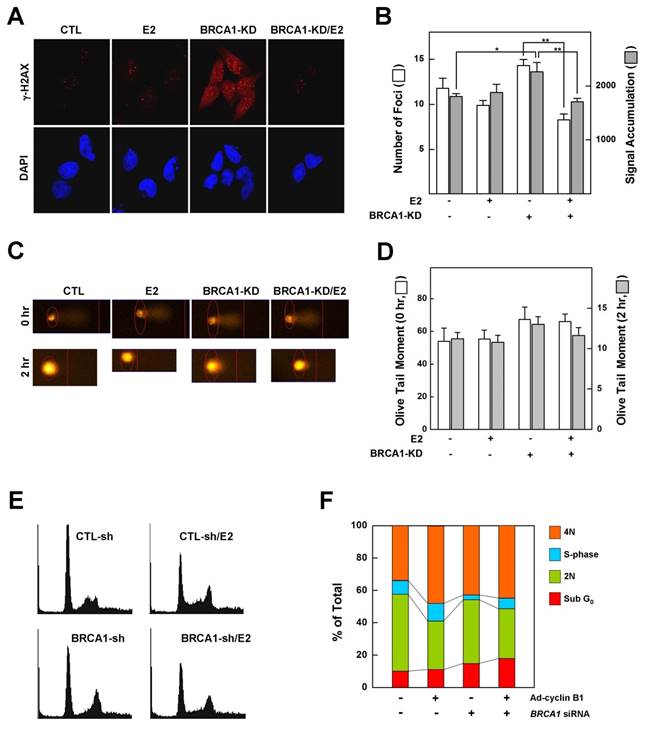
Phosphorylation of histone H2AX (γ-H2AX) is the earliest ATM-dependent response to DNA double-strand breaks [25]. Loss of BRCA1 triggers the formation of γ-H2AX foci, indicating an accumulation of unrepaired DNA double-strand breaks in the Brca1 mutant [6]. Since our data showed that treatment with E2 rescued the growth arrest associated with a BRCA1 deficiency, we examined whether it also alters the DNA-damage response and changes in cell cycle progression caused by a deficiency of BRCA1. MCF7 cells, which express wild-type BRCA1, were transfected with siRNA against BRCA1 or with scrambled siRNA (control), exposed to low-dose irradiation (0.3 Gy) with or without E2 (100 nM) pretreatment, and assessed for γ-H2AX foci formation as described in Materials and Methods. Our analysis showed that pretreatment with E2 did not alter the number or intensity of DNA-damage-induced γ-H2AX foci in BRCA1-replete MCF7 cells. However, DNA damage increased the number and intensity of γ-H2AX foci in MCF7 cells transfected with siRNA against BRCA1 compared with parental MCF7 cells, an effect that was significantly attenuated by pretreatment with E2 (Fig. 2A and 2B). Overall, however, the extent of DNA strand breakage was not strikingly different among conditions (Fig. 2C and 2D). These results suggest that treatment of BRCA1-deficient cells with estrogen prevents the proper sensing of DNA damage required for subsequent DNA repair and cell cycle arrest. Because E2 relieves hypersensitization of DNA damage and growth suppression in BRCA1-deficient cells, we examined the effect of E2 on cell cycle regulation in BRCA1-knockdown MCF7 cells. As shown in Figure 2E and 2F, E2 treatment reduced the G1-phase cell population and increased the S-phase population, suggesting that E2 leads to escape from G1/S arrest and survival of BRCA1-deficient cells.
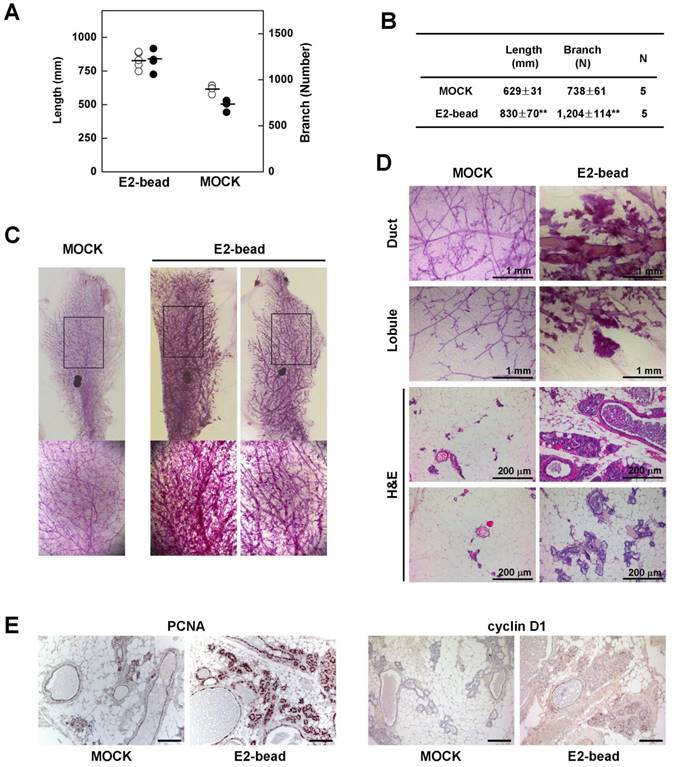
Next, we tested whether induction of estrogen signaling facilitates the development of epithelial cell abnormalities in mammary glands of Brca1-mutant mice. To examine the effect of estrogen in the mammary gland, we implanted estrogen pellets (1.7 mg E2 for 90-day release) into 9-month-old Brca1co/coMMTV-cre mice (N = 5). After 90 days, we quantified mammary gland density in E2 pellet-implanted and control mice using Branch software (Supplementary Fig. 1). Administration of estrogen induced a significant increase in the density of mammary glands in association with an increase in total ductal length (32% induction) and branch number (63% increase) (Fig. 3A and 3B). Additionally, a histological analysis of mammary glands showed that E2 treatment induced duct thickening, lobule enrichment, and foci formation in mammary glands (Fig. 3C and 3D). Further examination of control and E2-bead-treated mammary glands showed that treatment with E2 increased the levels and incidence of proliferating cell nuclear antigen (PCNA)- and cyclin D1-positive epithelial cells (Fig. 3E). Taken together, these results suggest that estrogen stimulation proceeds through cell-cycle checkpoints, despite their genetic instability and allows the proliferation BRCA1-deficient epithelial cells.
Effects of fulvestrant, an ER inhibitor, on the initiation of BRCA1-associated tumorigenesis
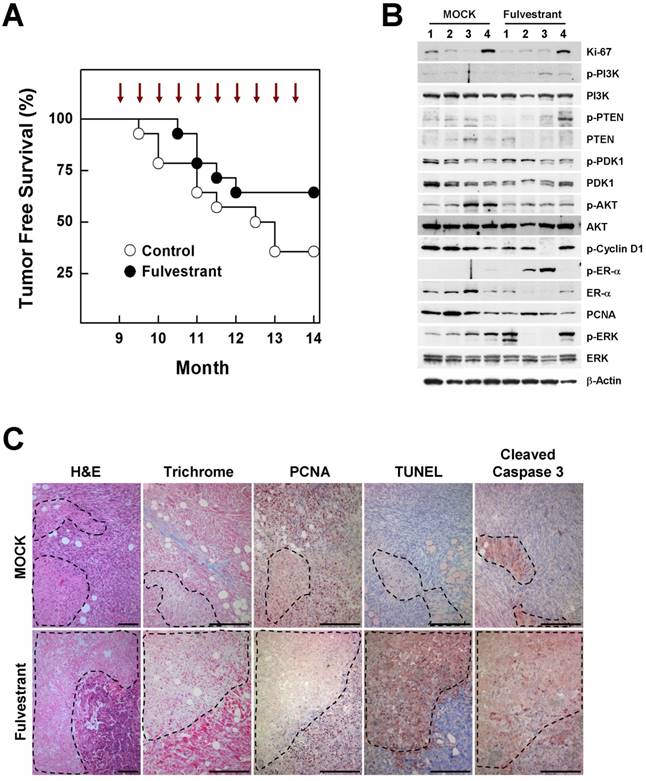
The contribution of estrogen signaling to the onset of BRCA1-associated breast cancer remains a matter of controversy. Conflicting results have been obtained using the same mouse model (Brca1co/coMMTV-creTp53+/-), with one study showing that oophorectomized Brca1-mutant mice exhibit a much lower tumor incidence than sham controls, and a second showing that treatment of Brca1-mutant mice with tamoxifen produces no chemopreventive effect against mammary cancer development [7] [8]. To test whether inhibition of estrogen signaling prevents BRCA1-deficient breast cancer, we injected 9-month-old Brca1co/coMMTV-cre mice biweekly for 5 months with fulvestrant, which binds to the ER and promotes its degradation. At the end of the study period (age, 14 months), palpable mammary tumors were detected in 36% (5 of 14) of fulvestrant-treated mice compared with 64% (9 of 14) of mice in the control group (Fig. 4A). A further analysis of tumor proteins revealed that half of fulvestrant-treated tumors (2 of 4) showed detectable levels of phosphorylated ER-α (phospho-ER-α), and exhibited an inverse correlation between ER-α and phospho-ERK (extracellular signal-regulated kinase) levels in the same tumors (Fig. 4B). In addition, fulvestrant-treated tumors displayed large necrotic areas with a lower level of the proliferation marker, and higher levels of cleaved caspase-3 and positive TUNEL staining compared with untreated tumor tissues (Fig. 4C), suggesting that estrogen signaling contributes to altered aggressiveness of Brca1-mutant tumors.
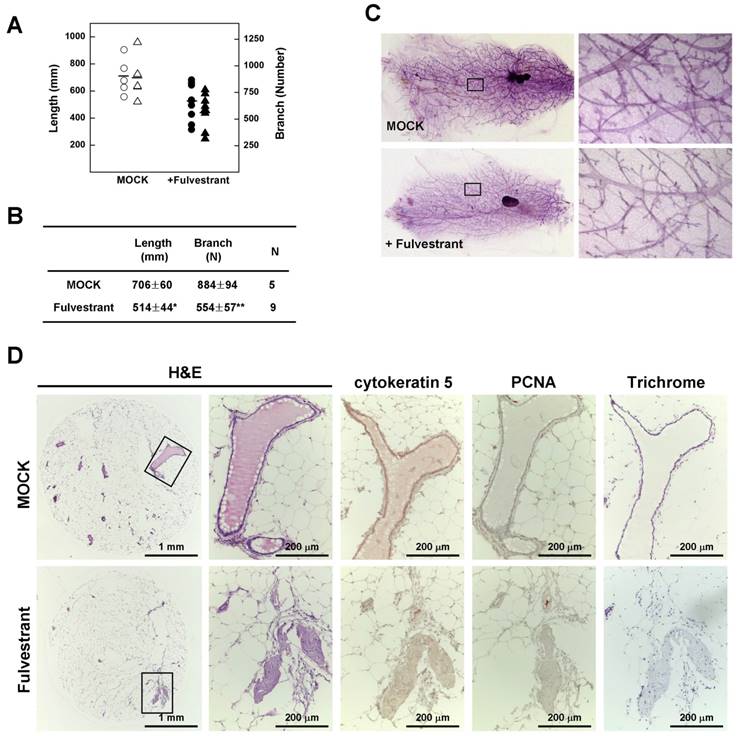
To confirm the relationship between estrogen and mammary epithelial cell proliferation, we measured mammary gland density in 14-month-old non-tumor-bearing mice using Branch software. Inhibition of estrogen signaling by treatment with fulvestrant significantly reduced the density of mammary glands in association with a decrease in total ductal length (27% reduction) and branch number (37% decrease) (Fig. 5A and 5B) with a reduction in ductal thickness (Fig. 5C). In addition, histological analyses revealed that the number and size of lobular and ductal tubes were reduced in fulvestrant-treated mammary glands and their structure showed evidence of degeneration and shrinkage (Fig. 5D). Therefore, our results suggest that inhibition of estrogen signaling prevents the proliferation of mammary epithelial cells and reduces tumor formation in Brca1-mutant mice.
Effects of fulvestrant on the progression of BRCA1-associated tumorigenesis
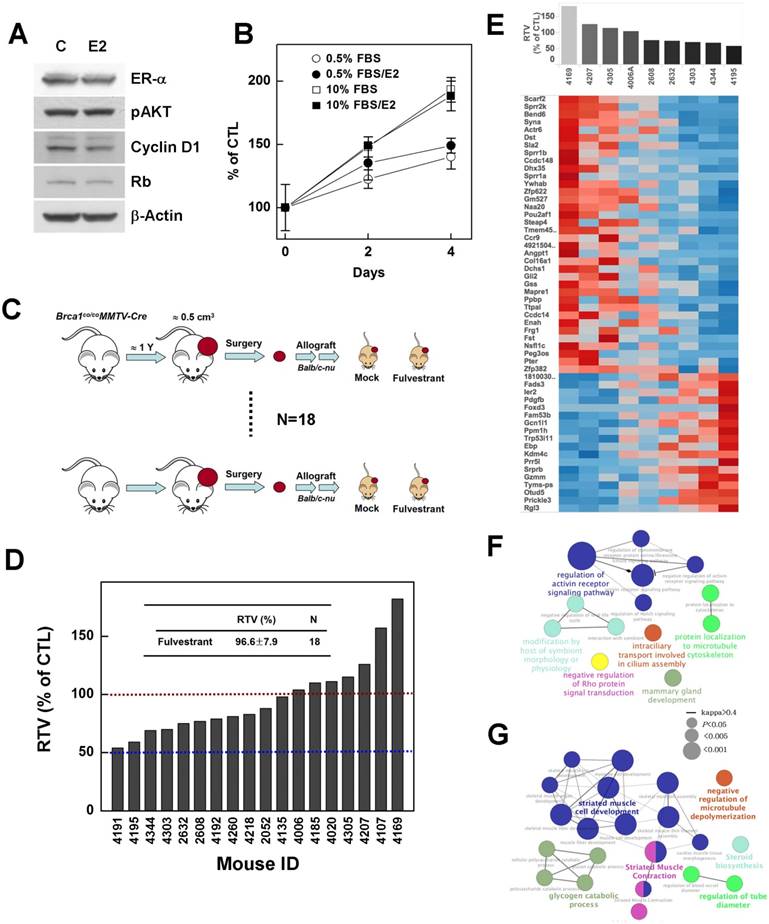
BRCA1-mutant breast cancer is traditionally considered a triple-negative cancer. However, a recent large-scale human pathological study reported identification of ER-positive BRCA1 mutant tumors in more than 20% of examined tumor tissues, and showed that these tumors developed to a greater extent in elderly (>50 years) women and showed a less aggressive tendency [26]. Thus, we tested whether suppression of estrogen signaling could effectively control the progression of ER-positive BRCA1-mutation-induced mammary tumors. To this end, we examined the effect of estrogen on the proliferation of the ER-positive, BRCA1-mutated epithelial cell line, HCC1937. However, although ER-α was readily detected in HCC1937, E2 treatment did not alter the expression levels of phospho-AKT, cyclin D1 or Rb, which are responsible for cell cycle regulation and proliferation (Fig. 6A), and failed to induce phosphorylation of ER-α and Rb protein (data not shown). In addition, E2 failed to induce proliferation of HCC-1937 cells (Fig. 6B), suggesting that estrogen signaling is not active or is disrupted in this cell line.
Next, to examine whether inhibition of estrogen signaling attenuates the progression of ER-positive BRCA1-mutant tumor, we tested fulvestrant in an in vivo allograft model, transplanted with ER-positive tumors identified during post-menopausal periods (Fig. 4B). The chemotherapeutic efficacy of fulvestrant was examined in 18 sets of allograft mice produced from tumors from 18 individuals (Fig. 6C). The results showed that none of these cases exhibited more than a 50% reduction in the ratio of tumor volumes (RTV), and further that the average size of fulvestrant-treated tumors was similar to that of vehicle-treated tumors (RTVmean = 96.6%; Fig. 6D). These findings suggest that preventing estrogen signaling is not an effective strategy for controlling BRCA1-deficient breast cancer, regardless of ER status.
Estrogen induces proliferation of BRCA1-deficient mammary gland epithelial cells. (A) Protein expression patterns in MCF7 cells treated with different concentration of E2; β-actin was detected as a loading control. MCF7 cells were transfected with control siRNA or siRNA targeting BRCA1, and treated with the indicated concentrations of E2 (B) for the indicated durations (C), after which survival was estimated using the MTT assay. The numbers represent mean values ± SD. (D) Whole-mount and (E) H&E staining of abdominal mammary glands from 2-month-old Brca1co/coMMTV-cre and age-matched wild-type (Brca1co/co) female mice treated with vehicle or E2. The panels at right are magnifications of the boxed areas in the adjacent panels (E). Scale bars: 200 μm.
Estrogen reduces the ability to sense DNA damage and alters G1/S phase transition in BRCA1-knockdown MCF7 cells. (A) MCF7 cells transfected with control siRNA or siRNA targeting BRCA1 were irradiated (0.3 Gy) in the absence or presence of E2 (100 nM), then immunostained for γ-H2AX foci (red) and counterstained with DAPI to detect nuclei (blue). (B). Foci numbers and signal accumulation per cell are shown in the histogram (*P < 0.05, **P < 0.01). (C) MCF7 cells transfected with control siRNA or siRNA targeting BRCA1 were irradiated in the absence or presence of E2 (100 nM), and DNA breakage was measured using Komet software (Andor Technology). (D) Olive tail moments of comet assays were calculated from at least 30 cells for each condition. (E) Representative histograms showing the DNA content of MCF7 cells. MCF7 cells transfected with control siRNA or siRNA targeting BRCA1 were treated with E2 (100 nM) for 1 day and analyzed by flow cytometry with propidium iodide staining. (F) Percentages of cells in each phase of the cell cycle are shown.
Fulvestrant response-associated genes
Although, overall, fulvestrant caused no significant reduction in the progression of ER-positive BRCA1-mutant tumors, there was an enormous variation in response among individuals. To increase the potential clinical efficacy of fulvestrant in breast cancer, it would be useful to be able to predict the response in advance of treatment initiation. In an effort to identify underlying causes of the different responses, we classified treated tumors based on their responsiveness to fulvestrant treatment, and examined gene expression patterns in the corresponding baseline tumor tissue samples. Nine non-treatment allograft mammary tumors were collected for this purpose. The point at which the size of the tumor in any given recipient reached 3 cm3 was used as the endpoint for examining the RTV (drug response) and gene expression pattern. The whole transcriptome was screened using mRNA sequencing data, and the Cufflinks computational pipeline was used to identify genes whose expression correlated with RTV following fulvestrant treatment of Brca1-deleted tumors. A total of 442 genes that appeared to be highly correlated at a significance level of 0.05 (Spearman's rank correlation > 0.6) were identified (Fig. 6E, Supplementary Tables 1 and 2).
To interrogate downstream effects of the 442 putative marker genes—238 positively correlated and 204 negatively correlated—in response to fulvestrant, we identified downstream pathways that might reveal a more specific effect on the regulation of resistance or sensitivity to fulvestrant. We performed a functionally grouped ontology (GO) analysis of marker genes using KEGG and Wikipathway databases. These analyses revealed six groups of pathways connected to various biological functions (corrected P < 0.05). Drug-resistance marker genes (positive correlation) were connected to various pathways, including regulation of ctiving receptor signaling pathway (GO:0032925, P < 0.0001), which is associated with the Notch signaling pathway; modification by host of symbiont morphology or physiology (GO:0051851, P = 0.02); intraciliary transport involved in cilium assembly (GO:0035735, P = 0.03); protein localization to microtubule cytoskeleton (GO:0072698, P = 0.03); and negative regulation of Rho protein signal transduction (GO:0035024, P = 0.03) (Figure 6F, Supplementary Table 3). Drug-sensitive marker genes (negative correlation) were involved in striated muscle cell development (GO:0055002, P = 0.0007), striated muscle contraction (wikipathway, P = 0.0007), glycogen catabolic processes (GO:0005980, P = 0.02), regulation of tube diameter (GO:0035296, P = 0.046), and negative regulation of microtubule depolymerization (GO:0007026, P = 0.046) (Fig. 6G, Supplementary Table 4).
Estrogen increases the rigidity of mammary glands in Brca1-mutant mice. (A) Branch software (ver. 1.1) was used to estimate the total length (open) and branch numbers (filled) of ducts between the lymph node and end tip in mammary glands of 12-month-old Brca1co/coMMTV-Cre mice in the absence or presence of E2 (1.7 mg) for 90 days. (B) The density of the mammary gland was significantly increased in E2-treated mice compared with control mice (**P < 0.01). (C) Representative whole-mount stainings of mammary glands from 12-month-old Brca1co/coMMTV-Cre mice in the absence or presence of E2 for 90 days. (D) Histological analysis of mammary glands in the absence or presence of E2-beads. (E) Levels of PCNA and cyclin D1 in dense mammary glands were examined in the absence or presence of E2-beads by immunohistochemical analysis. Scale bars: 100 μm.
Inhibition of estrogen signaling by fulvestrant treatment reduces mammary tumor formation in Brca1co/coMMTV-cre mice. (A) Kaplan-Meier curves showing tumor-free survival of Brca1co/coMMTV-cre mice. Nine-month-old Brca1co/coMMTV-cre mice were treated with vehicle (n = 14) or fulvestrant (n = 14) every other week for 5 months. Nine of 14 (64%) vehicle-treated mice and five of 14 (36%) fulvestrant-treated mice spontaneously developed palpable mammary tumors during this period. (B) Protein expression patterns and (C) histological analysis of tumors from vehicle- and fulvestrant-treated mice. Areas highlighted by dotted lines represent necrotic regions. Scale bars: 100 μm.
Discussion
We herein show that estrogen signaling plays an important role in the tumorigenesis attributable to Brca1 mutation, and provide evidence that targeting ER-α could be therapeutically relevant for preventing BRCA1-associated breast cancer. BRCA1-deficient breast cancer is a very well-known hereditary cancer. However, except for resection of normal mammary glands, there is no acceptable measure for proactively preventing tumor development. Mutations of BRCA1 are known to confer increased risk for breast, ovarian and prostatic cancers, but it is unclear why these hormone-responsive tissues are particularly sensitive to such genetic alterations.
A previous report showed that estrogen and its metabolites cause DNA double-strand breaks and that BRCA1 is required for regulation of estrogen metabolism and repair of this DNA damage to prevent estrogen-induced genomic instability [27]. Indeed, early events in BRCA1-associated tumorigenesis remain poorly understood, but loss of BRCA1 in mammary epithelial cells induces senescence in association with activation of several mediators, including ATM, p53, CHK2, RAS and Rb [28] [29] [30], thereby preventing the growth of error-prone epithelial cells. Interestingly, induction of estrogen signaling inhibits cellular senescence and improves proliferation of mammary epithelial cells and endothelial progenitor cells [31] [32], suggesting that estrogen signaling is able to serve as a pre-initiator of mammary epithelial cells that are growth-arrested by virtue of loss of BRCA1.
In this study, we found that estrogen suppressed the formation of irradiation-induced γ-H2AX foci in BRCA1-deficient cells, disturbing the ability of these cells to sense DNA damage. In addition, estrogen facilitated the progression of G1/S transition and induced the proliferation of BRCA1-knockdown cells, leading to the survival and proliferation of cells harboring genetic instabilities. These findings provide insight into why tumors caused by mutations of BRCA1 predominantly develop in estrogen-targeted tissues, despite the fact that BRCA1 is required to maintain genetic stability throughout the body.
Fulvestrant treatment decreases the rigidity of mammary glands in Brca1-mutant mice. (A) Branch software (ver. 1.1) was used to estimate the total length (circles) and branch numbers (triangles) of ducts between the lymph node and end tip in mammary glands of 14-month-old non-tumor-bearing mice. (B) The density of the mammary gland was significantly lower in fulvestrant-treated mice than in vehicle-treated mice (*P < 0.05, **P < 0.01). (C) Representative whole-mount stainings of non-tumor-bearing mammary glands from 14-month-old Brca1co/coMMTV-Cre mice treated with vehicle or fulvestrant for 5 months. (D) Histological analysis of mammary glands from control and fulvestrant-treated mice. Areas highlighted by boxes (left panels) were further analyzed by H&E staining, immunohistochemistry, and trichrome staining.
Inhibition of estrogen signaling fails to suppress the growth of Brca1-mutant breast tumors. (A) Protein expression patterns in BRCA1-deficient HCC1937 cells treated with E2 (100 nM); β-actin was detected as a loading control. (B) HCC1937 cells were treated with the indicated concentrations of FBS, with or without E2 (100 nM), and survival was estimated using the MTT assay. (C) Overview of the allograft model and fulvestrant treatments. Eighteen spontaneously developed mammary tumors were collected from Brca1co/coMMTV-Cre mice and transplanted into nude mice. Corresponding tumors grown under mock conditions were compared with those treated with fulvestrant. After tumors in any mouse implanted with the same original tumor reached 3 cm3, all mice implanted with that tumor were sacrificed and examined. (D) Graph shows calculated RTVs (RTV of treated tumor/RTV of control tumor X 100) for tumors treated with fulvestrant. (E) Analysis of fulvestrant response-associated biomarkers. Heat map shows selected downregulated and upregulated genes according to the responsiveness to fulvestrant (|R| > 0.6, P < 0.05). Tumor samples are sorted with respect to the fulvestrant response to highlight the correlation between response and gene expression. Highly correlated genes are ranked high in the heat map. Top-ranked genes only were selected for presentation (see Supplementary Tables 1 and 2 for the full gene list). (F and G) Functional GO enrichment analysis of genes that were positively correlated (F) and negatively correlated (G) with fulvestrant responsiveness. A node is an enriched GO term (Bonferroni corrected P < 0.05), and two associated terms connected by a line share many responsive genes (Cohen's kappa score > 0.4). The node label with the highest significance among associated terms is colored.
In fact, following the demonstration of transcriptional inhibitory interactions between BRCA1 and ER-α [33], several studies tested whether inhibition of estrogen signaling ameliorated BRCA1-mutation-induced malignancy. In these studies, the contribution of estrogen signaling to BRCA1-deficient mammary tumor formation was assessed by examining mammary tumor formation in oophorectomized Brca1 conditional-knockout mice (Brca1co/coMMTV-CreTp53+/-) [7]. The authors of this study found that the incidence of tumors in oophorectomized mice at 4 months post-surgery was much lower than that in sham controls. However, pre-administration of tamoxifen, which is a well-known anticancer drug used to treat ER-positive breast cancer, exerted no chemopreventive effect on mammary cancer development in Brca1-mutant mice; instead, the partial agonist activity of tamoxifen was associated with significant increases in mammary epithelial cell proliferation and the prevalence of mammary hyperplasia at a younger age [8]. It was recently shown that an efficacious response to tamoxifen also requires Her-2 and PAX2 [9], suggesting that tamoxifen treatment may not be sufficient to inhibit endogenous estrogen signaling and prevent BRCA1-associated mammary tumor formation.
Other important results that bear on the cumulative risk for BRCA1-associated contralateral breast cancer have been reported. The cumulative risk of developing a tumor in the contralateral breast up to the age of 75 years is 30.4% in BRCA1-mutant carriers, a frequency significantly higher than that in BRCA1-proficient patients [34]. Retrospective multicenter-cohort studies of BRCA1 mutation carriers with unilateral breast cancer studies have shown that suppression of estrogen signaling by treatment with tamoxifen significantly reduces the risk of developing a contralateral breast cancer and increases the survival of mutation carriers, [35-37], suggesting that estrogen signaling is still an attractive target for prevention of BRCA1-associated breast cancer. Here, we tested the ability of fulvestrant, a different type of ER antagonist that lacks agonist activity [11], to prevent Brca1-mutant mammary tumors. Of the Brca1co/coMMTV-cre mice treated with fulvestrant, 36% (5 of 14) formed palpable mammary tumors; in contrast, tumors formed in 64% (9 of 14) of untreated control mice during the same period. Our analysis of mammary tissues from non-tumor-bearing mice in the same group also showed that fulvestrant treatment significantly reduced ductal length (27% reduction) and branching (37% decrease), suggesting that inhibition of ER signaling reduces the incidence of BRCA1-associated mammary tumors in association with a decrease in epithelial cell proliferation and mammary gland rigidity.
Of particular interest is our observation that, although BRCA1-associated mammary tumors are known as triple-negative breast cancers (TNBCs), expression of ER-α was readily detected in Brca1-mutant mammary tumors developed to a greater extent in elderly women [26]. To determine whether inhibition of estrogen suppressed the progression of ER-α-positive BRCA1-associated mammary tumors, we tested the efficacy of fulvestrant using an allograft mouse model. We found that administration of fulvestrant was not able to significantly suppress the progression of ER-α-expressing Brca1-mutant mammary tumors. Furthermore, although detectable levels of ER-α protein were maintained in BRCA1-deficient HCC1937 cells, no subsequent response or proliferation was detected upon treatment with E2. In addition, spontaneously developed mammary tumors from Brca1-mutant mice expressed considerable amounts of ER-α, but did not display the phosphorylated form of ER-α, suggesting that estrogen signaling is not active or is disrupted in ER-α-positive cells and tumors.
Notably, our histological analysis revealed that suppression of estrogen signaling altered the histopathological characteristics of developed tumors, as shown in tumors from mice administered preventative fulvestrant treatment. Specifically, H&E staining of tumor tissues revealed extensive necrotic regions in fulvestrant-pretreated tumors. In addition, immunohistochemical analyses showed that these areas exhibited higher levels of TUNEL and cleaved caspase-3 staining, but lacked PCNA-positive cells. Interestingly, 2 of 4 mammary tumors from fulvestrant-treated Brca1-mutant mice exhibited a different ER-α expression pattern, with detectable phospho-ER-α and a low level of ER-α. It has been reported that the loss of BRCA1 increases estrogen-induced phosphorylation of ERK (extracellular signal-regulated kinase) in association with estrogen-induced cell proliferation in vitro and mammary tumorigenesis in vivo [38]. Our analysis of estrogen signaling and ERK activation in Brca1-mutant tumors showed that phospho-ERK levels were positively correlated with ER-α levels, but were inversely correlated with phospho-ER-α levels. Indeed, the status of estrogen signaling in breast cancer is considered an indicator of anti-estrogenic therapeutic strategies and clinical outcomes. It has been reported that ER-positive cases are significantly associated with low-grade, small-size, early-stage tumors (P < 0.05) [39]. In addition, high expression levels of BCAR4 (breast cancer anti-estrogen resistance 4), which is involved in the resistance of breast cancer to anti-estrogen therapy, is associated with poor metastasis-free survival and overall survival, reflecting enhanced tumor aggressiveness [40]. Moreover, human pathological analyses have shown that ER-positive BRCA1-associated breast cancer tends to develop in older ages (>50 years) and is less aggressive than ER-negative BRCA1-associated breast cancer [26]. Taken together, these results imply that estrogen signaling in BRCA1-associated tumorigenesis not only contributes to the initiation of tumor formation, but also alters the histopathological characteristics that influence the aggressiveness of tumor progression.
Clinical investigations of prevention and treatment options for BRCA1-associated breast cancers are constrained by many difficulties. Thus, preclinical simulation with a mouse model is a good experimental stand-in for testing treatment efficacy in BRCA1-associated breast cancer. Here, using an endogenous-tumor-bearing Brca1-mutant mouse model, we found that inhibition of estrogen signaling could be a useful strategy for reducing BRCA1-associated breast cancer. Further studies are needed to evaluate the potential of this strategy to progress to human clinical trials.
Supplementary Material
Supplementary figures and tables.
Abbreviations
BRCA1, breast cancer type 1 susceptibility protein; ER, estrogen receptor; MMTV, mouse mammary tumor virus; PCNA, proliferating cell nuclear antigen; RTV, ratio of tumor volumes; TNBC, triple-negative breast cancer.
Acknowledgements
This work was supported by the National Cancer Center of Korea (NCC-1610030/1710900), and the National Research Foundation of Korea (2014R1A2A1A11049935/2018R1A2B6001216). We would like to thank the Bioinformatics Cores at National Cancer Center Korea for support with bioinformatics analysis.
Ethics Approval
All procedures involving animals and their care were approved by the Institutional Animal Care and Use Committee of the National Cancer Center of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416-26
2. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
3. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329-33
4. Venkitaraman R. Triple-negative/basal-like breast cancer: clinical, pathologic and molecular features. Expert Rev Anticancer Ther. 2010;10:199-207
5. Valentin MD, da Silva SD, Privat M, Alaoui-Jamali M, Bignon YJ. Molecular insights on basal-like breast cancer. Breast Cancer Res Treat. 2012;134:21-30
6. Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201-13
7. Bachelier R, Xu X, Li C, Qiao W, Furth PA, Lubet RA. et al. Effect of bilateral oophorectomy on mammary tumor formation in BRCA1 mutant mice. Oncol Rep. 2005;14:1117-20
8. Jones LP, Li M, Halama ED, Ma Y, Lubet R, Grubbs CJ. et al. Promotion of mammary cancer development by tamoxifen in a mouse model of Brca1-mutation-related breast cancer. Oncogene. 2005;24:3554-62
9. Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M. et al. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663-6
10. Addo S, Yates RA, Laight A. A phase I trial to assess the pharmacology of the new oestrogen receptor antagonist fulvestrant on the endometrium in healthy postmenopausal volunteers. Br J Cancer. 2002;87:1354-9
11. Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90(Suppl 1):S2-6
12. Carlson RW. The history and mechanism of action of fulvestrant. Clin Breast Cancer. 2005;6(Suppl 1):S5-8
13. Valachis A, Mauri D, Polyzos NP, Mavroudis D, Georgoulias V, Casazza G. Fulvestrant in the treatment of advanced breast cancer: a systematic review and meta-analysis of randomized controlled trials. Crit Rev Oncol Hematol. 2010;73:220-7
14. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37-43
15. Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L. et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323-30
16. Baek HJ, Lee YM, Kim TH, Kim JY, Park EJ, Iwabuchi K. et al. Caspase-3/7-mediated Cleavage of beta2-spectrin is Required for Acetaminophen-induced Liver Damage. Int J Biol Sci. 2016;12:172-83
17. Baek HJ, Lim SC, Kitisin K, Jogunoori W, Tang Y, Marshall MB. et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology. 2008;48:1128-37
18. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562-78
19. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S. et al. A travel guide to Cytoscape plugins. Nat Methods. 2012;9:1069-76
20. Gibson DA, Saunders PT. Estrogen dependent signaling in reproductive tissues - a role for estrogen receptors and estrogen related receptors. Mol Cell Endocrinol. 2012;348:361-72
21. Mester J, Redeuilh G. Proliferation of breast cancer cells: regulation, mediators, targets for therapy. Anticancer Agents Med Chem. 2008;8:872-85
22. Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347-53
23. Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014;4:106
24. Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99:61-6
25. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462-7
26. Spurdle AB, Couch FJ, Parsons MT, McGuffog L, Barrowdale D, Bolla MK. et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: a large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014;16:3419
27. Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ. et al. BRCA1 deficiency exacerbates estrogen-induced DNA damage and genomic instability. Cancer Res. 2014;74:2773-84
28. Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X. et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006;25:2167-77
29. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638-42
30. Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P. et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat Commun. 2015;6:7505
31. Liu Z, Wang L, Yang J, Bandyopadhyay A, Kaklamani V, Wang S. et al. Estrogen receptor alpha inhibits senescence-like phenotype and facilitates transformation induced by oncogenic ras in human mammary epithelial cells. Oncotarget. 2016;7:39097-107
32. Imanishi T, Hano T, Nishio I. Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J Hypertens. 2005;23:1699-706
33. Fan S, Wang J, Yuan R, Ma Y, Meng Q, Erdos MR. et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284:1354-6
34. Graeser MK, Engel C, Rhiem K, Gadzicki D, Bick U, Kast K. et al. Contralateral breast cancer risk in BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2009;27:5887-92
35. Metcalfe K, Lynch HT, Ghadirian P, Tung N, Olivotto I, Warner E. et al. Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2004;22:2328-35
36. Gronwald J, Tung N, Foulkes WD, Offit K, Gershoni R, Daly M. et al. Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: an update. Int J Cancer. 2006;118:2281-4
37. Phillips KA, Milne RL, Rookus MA, Daly MB, Antoniou AC, Peock S. et al. Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2013;31:3091-9
38. Li W, Xiao C, Vonderhaar BK, Deng CX. A role of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. Oncogene. 2007;26:7204-12
39. Bulut N, Altundag K. Does estrogen receptor determination affect prognosis in early stage breast cancers? Int J Clin Exp Med. 2015;8:21454-9
40. Godinho MF, Sieuwerts AM, Look MP, Meijer D, Foekens JA, Dorssers LC. et al. Relevance of BCAR4 in tamoxifen resistance and tumour aggressiveness of human breast cancer. Br J Cancer. 2010;103:1284-91
Author contact
![]() Corresponding author: Sang Soo Kim, Phone: (8231) 920-2491; Fax: (8231) 920-2494; Email: sangsookimre.kr
Corresponding author: Sang Soo Kim, Phone: (8231) 920-2491; Fax: (8231) 920-2494; Email: sangsookimre.kr