Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(7):1153-1165. doi:10.7150/ijbs.41587 This issue Cite
Research Paper
Development and Validation of a Prognostic Nomogram for Gastric Cancer Based on DNA Methylation-Driven Differentially Expressed Genes
Yi Bai1,4*, Chunlian Wei2*, Yuxin Zhong3*, Yamin Zhang4, Junyu Long1, Shan Huang2, Fucun Xie1, Yantao Tian3, Xi Wang2 ![]() , Haitao Zhao1
, Haitao Zhao1 ![]()
1. Department of Liver Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College (CAMS & PUMC), Beijing, China.
2. Department of Immunology, Beijing Key Laboratory for Cancer Invasion and Metastasis, Advanced Innovation Center for Human Brain Protection, School of Basic Medical Sciences, Capital Medical University, Beijing, China.
3. Department of Pancreatic and Gastric Surgery, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
4. Department of Hepatobiliary Surgery, First Central Hospital, Tianjin, China.
*These authors contributed equally to this work.
Received 2019-10-29; Accepted 2020-1-16; Published 2020-2-10
Abstract

Background/Aims: The incidence of gastric cancer (GC) ranks fifth among common tumors and GC is the third leading cause of cancer-related death worldwide. The aim of this study was to develop and validate a nomogram for predicting the overall survival (OS) of patients with GC.
Methods: DNA methylation (DNAm)-driven genes were identified by integrating DNAm and gene expression profiling analyses from The Cancer Genome Atlas (TCGA) GC cohort. Then, a risk score model was built based on Kaplan-Meier (K-M), least absolute shrinkage and selector operation (LASSO), and multivariate Cox regression analyses. After analyzing the clinical parameters, a nomogram was constructed and assessed. Another cohort (GSE62254) was used for external validation.
Results: Thirteen differentially expressed DNAm-driven genes were narrowed down to a six-gene signature (PODN, NPY, MICU3, TUBB6 and RHOJ were hypermethylated, and MYO1A was hypomethylated), which was associated with OS (P < 0.05) after survival and LASSO regression analyses. These differentially expressed genes (DEGs) with altered DNAm statuses were included in the prognostic risk score model. The univariate Cox regression analysis indicated that risk score, age, and number of positive lymph nodes were significantly associated with survival time in GC patients. The multivariate Cox regression analysis also indicated that these variables were significant prognostic factors for GC. A nomogram including these variables was constructed, and its performance in predicting the 1-, 3- and 5-year survival outcomes of GC patients was estimated through time-dependent receiver operating characteristic (ROC) curves. In addition, the clinical benefit of this model was revealed by decision curve analysis (DCA). Pathway enrichment analysis suggested that these DNAm-driven genes might impact tumor progression by affecting signaling pathways such as the “ECM RECEPTOR INTERACTION” and “DNA REPLICATION” pathways.
Conclusions: The altered status of the DNAm-driven gene signature (PODN, MYO1A, NPY, MICU3, TUBB6 and RHOJ) was significantly associated with the OS of GC patients. A nomogram incorporating risk score, age and number of positive lymph nodes can be conveniently used to facilitate the individualized prediction of OS in patients with GC.
Keywords: nomogram, risk score, gastric cancer, DNA methylation, prognosis
Introduction
The incidence and cancer-related death of gastric cancer (GC) rank fifth and third, respectively, among those of common tumors [1, 2]. Curative surgery, chemotherapy that combines platinum with fluoropyrimidines or paclitaxel (PTX) plus ramucirumab [3-5] and target therapies remain the most common treatment options. Because GC is frequently diagnosed in advanced stages, the prognosis of GC is still not satisfactory [6]. Aside from progress in treatment approaches, exploring efficient biomarkers for early identification is another important precaution to improve the prognosis of GC patients. Compared to traditional diagnostic methods, more specific and sensitive biomarkers demonstrate promising value in early diagnosis, predicting prognosis and even therapeutic responses.
In pursuit of predictive factors for patients with GC, an increasing number of studies have identified some valuable biomarkers, such as fibroblast growth factor receptor (FGFR) [7] and disrupted in renal cancer 1 (DIRC1) [8]. However, prognostic biomarkers for GC are still limited, and due to a lack of specificity and sensitivity, few markers have been adopted for application.
DNA methylation (DNAm) is an important epigenetic event that can influence pretranscriptional gene silencing, genetic imprinting, X-chromosome inactivation (XCI), genome stability, and cell fate determination [9]. De novo methyltransferases, namely, DNMT3A and DNMT3B [10, 11], play a vital role in tumorigenesis mainly by methylating CpG dinucleotides [12]. Aberrant DNAm in the promoter regions is generally believed to be a hallmark of tumors, which often leads to the transcriptional silencing of tumor suppressor genes (TSGs) and the abnormal activation of oncogenes in tumor cells [13]. There is evidence that abnormal DNAm frequently occurs in early-stage tumors [14], and these alterations are relatively stable and potentially reversible therapeutically [15-17]. Hence, the deregulated DNAm status shows prospective utility as a biomarker for early diagnosis, prognosis and clinical decision-making for a variety of tumors.
Despite extensive studies on the relationship between abnormal DNAm and the prognosis of patients with GC, individualized prognostic models considering the DNAm-driven gene signature have rarely been reported. By integrating methylation and mRNA expression profile data, we identified prognosis-related differentially expressed genes (DEGs) with altered DNAm status and established a risk score model after Kaplan-Meier (K-M) and LASSO analyses. Finally, we established a nomogram via an integrated analysis of both the DNAm signature and clinicopathologic risk factors to predict overall survival (OS) in patients with GC, which was then validated in another Gene Expression Omnibus (GEO) cohort.
Materials and Methods
Patient Population and Clinical Data
All The Cancer Genome Atlas (TCGA) data are available through the NIH Genomic Data Commons (GDC). Here, TCGA level-3 molecular data and the corresponding clinical files were obtained from the GDC (2019/1/21 analysis archive). The methylation levels of genes were scored using β values ranging from 0 to 1 (unmethylated to totally methylated).
Identification of DEGs between GC and Nontumorous Tissues
We identified the DEGs between 343 GC tissues and thirty adjacent nontumorous gastric tissues from the training dataset (HTSeq-Counts of TCGA-STAD transcriptome profiling with complete prognostic information and diagnosed as adenomas and adenocarcinomas) using the DESeq package [18]. An absolute log2-fold change (|FC|) of > 1 and an adjusted P value of < 0.05 were set as cutoff criteria. Visualization of the six DNAm-driven gene expression patterns between GC and noncancer gastric tissues was performed with Prism 8.0 (GraphPad, San Diego, CA, USA).
Approach used to Identify DNAm-Driven Genes
Gene expression data and DNAm data were integrated with the same TCGA barcode structure. DNAm-driven genes are those genes whose DNAm levels are negatively correlated with the mRNA expression level after linear regression analysis. Simultaneously, the differential DNAm state between GC tissues and adjacent nontumorous gastric tissues was compared by employing the Wilcoxon rank-sum test method as described previously [19].
Functional Enrichment Analysis
Seventy-one DNAm-driven genes were subjected to Gene Ontology (GO) and pathway enrichment analyses, with the help of the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Tool (version 6.8) and ConsensusPathDB (http://cpdb.molgen.mpg.de/), respectively.
Feature Selection and Building the Predictive Signature
Initially, K-M analysis was utilized to evaluate the relationship between DNAm-driven genes and the survival time of GC patients. To further narrow the scope of the candidate DNAm-driven genes, we adopted the LASSO binary logistic regression model and multivariate Cox regression after primary filtration. The linear combination of the regression coefficient derived from the multivariate Cox regression model (β) multiplied by its mRNA level generated a prognostic risk score with six genes.
Development and Validation of the Risk Score Model
Employing X-tile [20] to determine appropriate cut-off values, we separated patients into low- and high-risk groups, after which the K-M survival curves were plotted using the survival data of the two groups of GC patients. The potential of the predictive signature was assessed in the primary cohort and validated in the GSE62254 cohort.
Screening of Prognostic Factors
The significance of the risk score model and other traditional clinical characteristics to predict OS in GC patients was evaluated by univariate Cox regression analysis. Then, confounding factors were excluded through multivariable logistic regression analysis. The statistical significance levels were all two-sided at 0.05, and the hazard ratio (HR) and its 95% confidence interval (CI) were also calculated.
Development and Assessment of the Nomogram in the TCGA Dataset
Multivariate Cox regression analysis distinguished significant predictive factors, from which we built a predictive model. To evaluate the performance of the nomogram in the primary cohort, we assessed the calibration of OS probability at different years for patients with GC by applying the Hosmer-Lemeshow test to plot calibration curves.
Harrell's concordance index (C-index) was measured to quantify the discrimination performance of the nomogram. The nomogram was validated with 1000 bootstrap resamples to calculate a robust C-index. The value of the C-index ranged from 0.5 (indicates random chance) to 1.0 (indicates perfect capacity to correctly distinguish the outcome via this model).
We also conducted a time-dependent ROC analysis [21] to measure the predictive performance of the nomogram. Then, decision curve analysis (DCA) [22] was employed to quantify the clinical utility with clinical consequences of a decision considered.
External Validation of the Nomogram
In the validation phase, we verified the nomogram in the GEO by using another GC cohort, GSE62254.
Copy Number Variation (CNV), Mutation Characteristics and Gene Set Enrichment Analysis (GSEA) of Six DNAm-Driven Genes
Graphic illustrations of CNV and the six-gene mutation profiles in all GC tissues from the TCGA dataset were obtained from cBioPortal (http://www.cbioportal.org/). GSEA was performed using gsea-3.0.jar software according to the methods described in the user guide (http://software.broadinstitute.org/gsea/index.jsp).
Statistical Analysis
All statistical analyses were conducted with R software (version 3.5.2). All statistical tests were two-sided, and P values less than 0.05 were considered statistically significant.
Results
Identification of DEGs in GC
The study flowchart describing the process is shown in Figure 1. After the comparison of mRNA expression between GC tissues (n = 343) and adjacent nontumorous gastric tissues (n = 30), 2737 DEGs (|logFC| > 1, adjusted P value < 0.05) remained for further analysis. Among these genes, 649 DEGs were upregulated, and 2088 DEGs were downregulated (Table S1).
Flowchart depicting how prognostic genes were identified.
Identification of DNAm-Driven Genes in GC
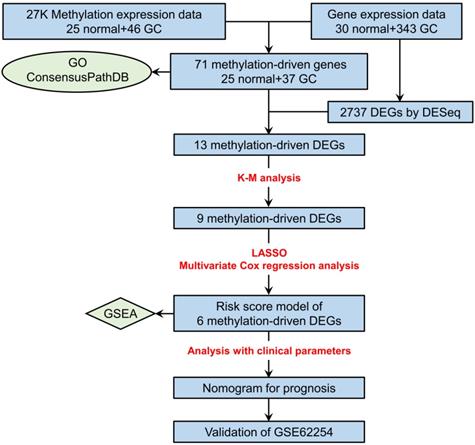
To identify DNAm-driven genes in GC, we performed MethylMix analysis[19] on data from seventy-one clinical samples (Illumina Human Methylation 27 platform) downloaded from the TCGA. A total of seventy-one DNAm-driven genes (forty-seven hypermethylated and twenty-four hypomethylated) with an adjusted P value < 0.05 between the hyper- and hypomethylation groups and a correlation between DNAm and gene expression less than -0.3 were screened, and their methylation levels were visualized via a heatmap (Figure 2A, Table S2). GO analyses were performed with the aim of elucidating the functional characteristics of the identified DNAm-driven genes, and we obtained nine GO terms (P < 0.05; Figure 2B; Table S3). We found that the GO functions of these DEGs were significantly enriched in the following categories: “regulation of transcription, DNA-templated”, “metal ion binding”, “transcription factor activity, sequence-specific DNA binding”, “nucleic acid binding”, and “embryonic skeletal system morphogenesis” (P < 0.001). However, the Reactome database pathway analysis from ConsensusPathDB showed that the genes were enriched in Pink/parkin mediated mitophagy and mitophagy (P < 0.001) and six other pathways, namely, generic transcription pathway, signaling by ERBB2, RNA polymerase II transcription, peroxisomal protein import, gene expression (transcription), and regulation of PLK1 activity at G2/M transition (P < 0.05; Figure 2C; Table S4).
Candidate DNAm-driven genes screened by the Wilcoxon test. (A) Heatmap of the candidate DNAm-driven genes (n=71) in GC and nontumorous gastric tissues. (B) GO analysis of seventy-one DNAm-driven genes. (C) Pathway analysis based on multiple databases.
Generation of a Prognostic Risk Score Model for GC
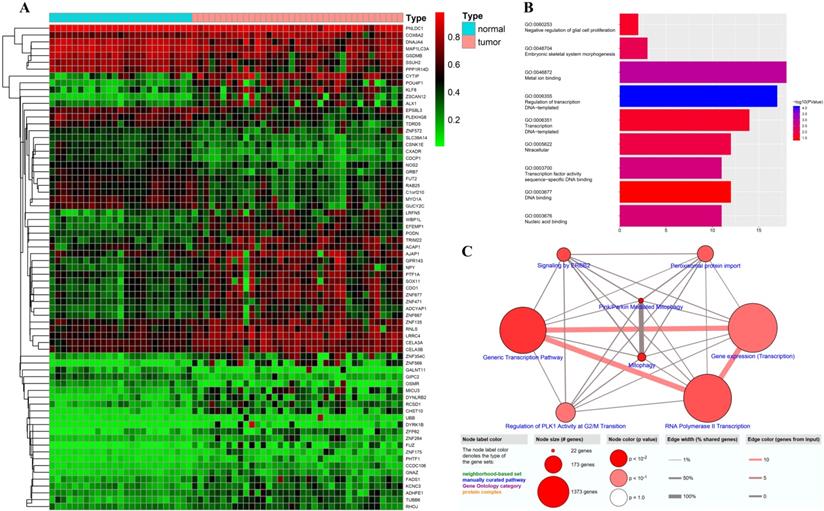
Next, we intersected the DEGs and DNAm-driven genes. Then, we explored the relationship between the gene expression of thirteen DNAm-driven DEGs and OS by utilizing K-M analysis (Table S5). Of note, the X-tile approach was used to determine the optimal cut-off value. Among the thirteen included DNAm-driven DEGs, nine met the criteria for statistical significance via the log-rank test (P < 0.05) (Figure S1). The nine selected candidate DNAm-driven DEGs featured coefficients (not zero) in a further LASSO logistic regression model in which the selected genes were required to appear 1000 times of 1000 repetitions (Figure 3A). Finally, six DNAm-driven DEGs (PODN, MYO1A, NPY, MICU3, TUBB6 and RHOJ) were selected as prognostic genes and presented in the risk score calculation formula. The predictive model was established by adding the product of the expression level and relative coefficient of each gene in the LASSO regression as follows: risk score = (0.2159037 * NPY mRNA level) + (0.2069438* MICU3 mRNA level) + (-0.2337186 * MYO1A mRNA level) + (0.1574830 * RHOJ mRNA level) + (0.1584843 * TUBB6 mRNA level) + (0.3310443 * PODN mRNA level). Positive coefficients of PODN, NPY, MICU3, TUBB6 and RHOJ in the LASSO regression implied that their high expression represented poor OS in GC patients. Nevertheless, a high value of MYO1A indicated prolonged OS in both the training and validation datasets (Figure S1; Figure S2). For the 300 patients with full clinical data, we calculated the risk score based on the six-gene expression signature and identified the most compatible cut-off value with X‐tile diagrams. Those with a risk score over the cut-off value, 163 patients in total, were classified as the high-risk group, while the remaining 137 patients were classified as the low-risk group. The K-M analysis of the two groups showed that the OS of the high-risk score group was significantly shorter (P < 0.0001; Figure 3B). The gene expression profiles of all patients and the correlated risk scores were visualized as a heatmap (Figure 3C).
Texture feature selection and six-gene risk score model construction in the TCGA cohort. (A) Tuning parameter (λ) selection in the LASSO model used ten-fold cross-validation via the maximum criteria. The dotted vertical lines were drawn at the optimal values using the maximum criteria and the one standard error of the maximum criteria (the 1-SE criteria). (B) Comparison of OS between the high-risk score and low-risk score groups. (C) Heatmap of the six-gene expression profiles and distribution of corresponding risk scores in the high-risk and low-risk subgroups in the TCGA database.
Development and Evaluation of a Nomogram for OS Prediction in GC
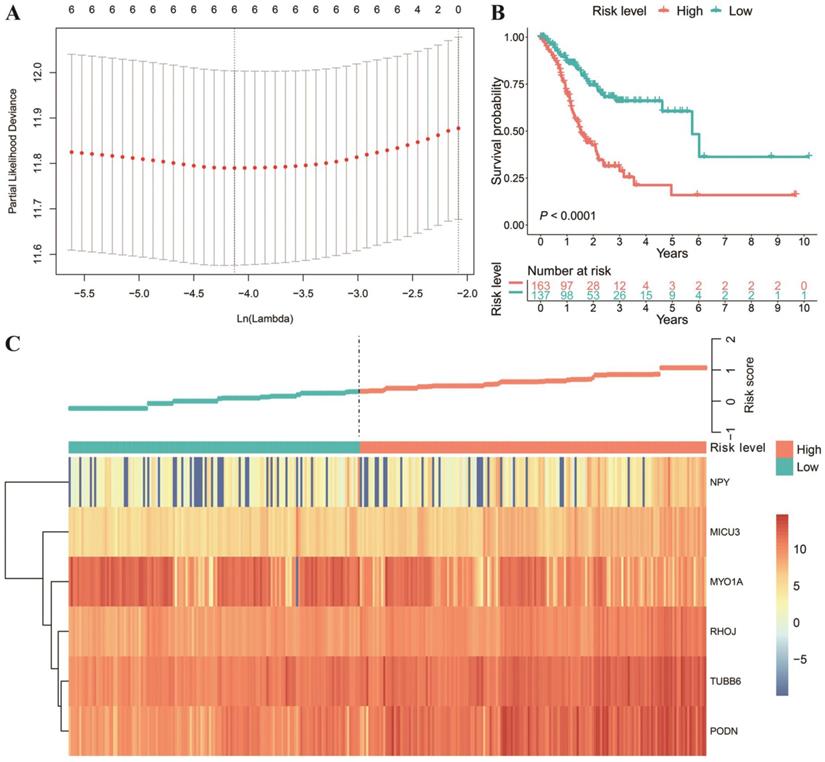
Age, quantity of positive lymph nodes and risk level were regarded as significant predictive factors in univariate and multivariate regression analyses (Figure 4A). Taking all the above significant predictive factors into consideration, we generated a comprehensive nomogram (Figure 4B). In addition, Schoenfeld model residuals vs age, number of positive lymph nodes and risk level were plotted to obtain a preliminary assessment of whether these predictive factors should be incorporated into the model. Schoenfeld residuals suggested that this model met the equally proportional risk hypothesis (Figure 4C). The C-index and the robust C-index were 0.701 and 0.695, respectively. The calibration curve of the model for the possibility of OS at 3 years and 5 years demonstrated accurate predictive ability (Figure 4D). In addition, the prognostic capacity of the six-gene signature was demonstrated by the area under the curve (AUC) of the time-dependent ROC curve. Compared with age and number of positive lymph nodes, the AUC of the nomogram was increased (Figure 4E), indicating that the acuity of the nomogram was predominantly better than that of age or number of positive lymph nodes. DCA was performed to assess the clinical meaning. As demonstrated by the favorable probability, the combined model showed better net benefit than the age or number of positive lymph nodes only model, which indicates that the nomogram can help clinicians make more accurate assessment of patient prognosis. (Figure 4F). Because the nomogram was built based on more than one prognostic factor, it works better than each single factor alone. The model performed well in both the short- and long-term OS prediction, therefore, we have confidence in its potential to assist doctors in making medical decisions and GC patients in planning their follow-up schedules.
Nomogram to predict 1-, 3- and 5-year OS. The OS nomogram was developed in the TCGA cohort, with age, number of positive lymph nodes and risk level (DNAm signature) incorporated. (A) Univariate and multivariate analyses of the risk level, clinical factors and pathological characteristics with OS. The statistical significance level is indicated by different colors; red indicates statistical significance, and blue indicates no significance. (B) Nomogram to predict the 1-, 3- and 5-year OS of GC patients. (C) The Schoenfeld residual suggested that this model met the equally proportional risk hypothesis. Schoenfeld model residuals vs age, number of positive lymph nodes and risk level were plotted to obtain a preliminary assessment of which of these predictive factors should be incorporated into the model. (D) Calibration curves of 1‐, 3‐ and 5‐year OS. Blue dotted lines represent the ideal predictive model, and the red solid line represents the observed model. (E) Time‐dependent ROC analysis was used to evaluate the accuracy of the OS nomograms. The red, blue and green solid lines represent the combined model, age, and number of positive lymph nodes, respectively. (F) DCA curves evaluate OS nomograms from the perspective of clinical benefit and scope of clinical benefits. The y-axis represents the net benefit. The x-axis represents the predicted OS probability. The black dotted line represents the condition that all patients survive in 5 years, while the gray solid line represents the condition that none of the patients survive for more than one year. In the current study, the decision curve showed more benefit with a threshold probability > 0.0% using the OS nomogram.
External Validation of the Nomogram
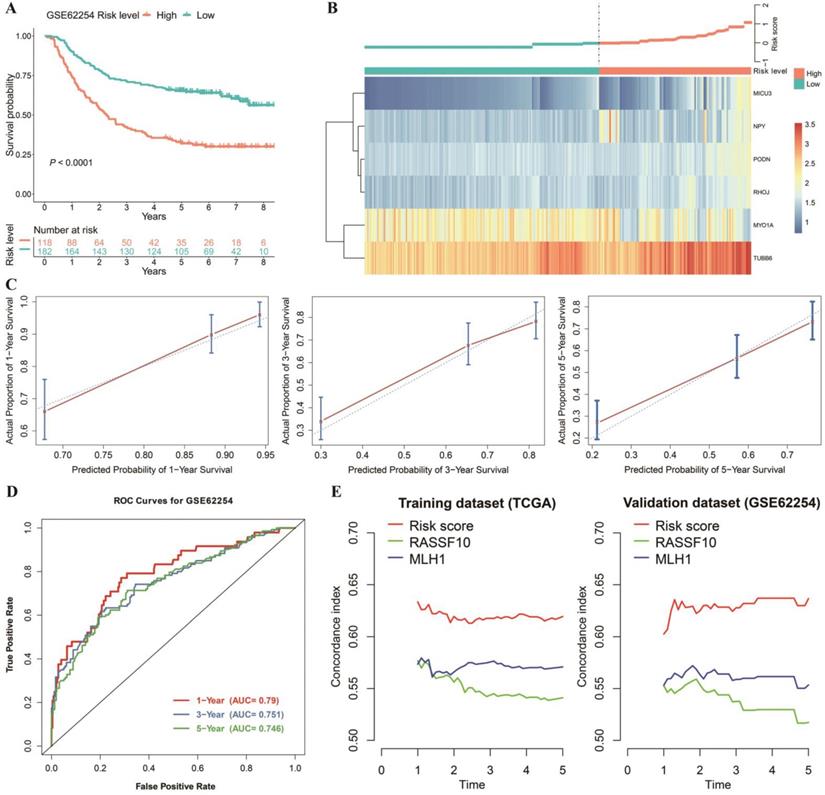
The nomogram mentioned previously was further validated in the GEO dataset GSE62254 (Figure 5A, B, C and D). The nomogram calibration curves for the possibility of 1-, 3- and 5-year OS displayed obvious concordance between the predicted results and observations in the GEO cohort (Figure 5C). Similar to the performance in the TCGA cohort (Figure 4E), the AUCs were 0.79, 0.751, and 0.746 for 1-, 3- and 5‐year survival times, respectively, in our validation dataset (Figure 5D). In addition, we compared the existing DNAm-driven gene prognostic models[23, 24] with our risk score model based on the C-index from the first year to the fifth year. The results show that the risk score model performs best in both the training and the validation sets (Figure 5E).
Validation of the prediction model. (A) OS was significantly lower in the high‑risk score group than in the low‑risk score group. (B) Heatmap and distribution of the six gene expression profiles in the high-risk and low-risk subgroups in the GEO database. (C) Calibration curve for the risk score model in the validation cohort. The blue dotted line represents the ideal predictive model, and the blue solid line represents the observed model. (D) ROC of the survival prediction model with the combined model, age, and number of positive lymph nodes in the validation dataset. (E) Concordance index of the indicated prognostic model in the training and validation datasets.
Methylation Degree and Gene Expression of the Six DNAm-driven DEGs in Cancer and Normal Samples
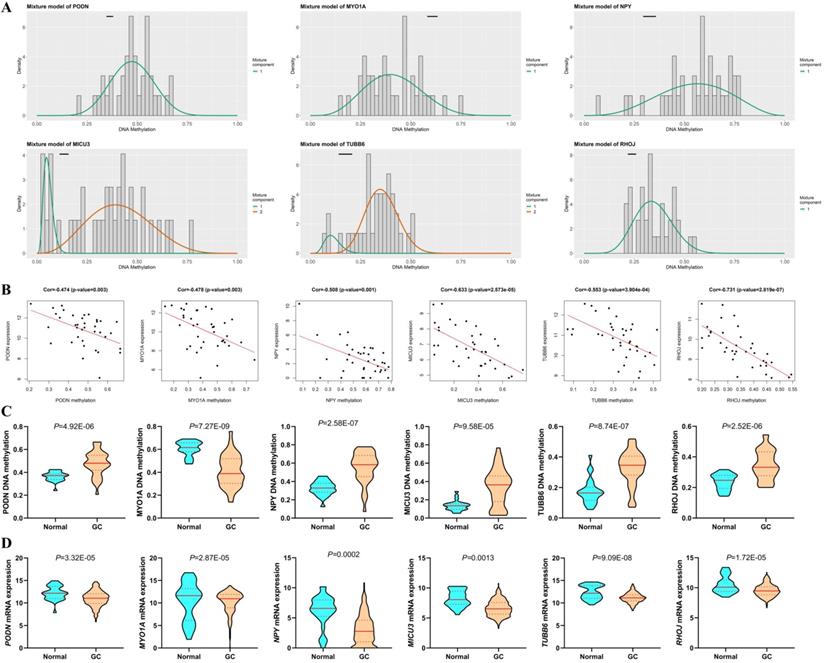
Among the six DNAm-driven DEGs, five (PODN, NPY, MICU3, TUBB6 and RHOJ) were hypermethylated, while MYO1A was hypomethylated (Figure 6A and C) based on the TCGA GC cohort. As shown in Figure 6B, there is a significant inverse correlation between methylation and mRNA levels (|R|>0.3, P < 0.05). Moreover, the mRNA expression of hypermethylation-driven DEGs was decreased significantly in GC tissues compared with that in adjacent nontumorous gastric tissues (P < 0.05, Figure 6D).
DNAm-driven genes. (A) Differential methylation statuses. The histogram demonstrates the distribution of PODN, MYO1A, NPY, MICU3, TUBB6 and RHOJ methylation in GC samples. Beta values represent the methylation level (range from 0 to 1), and the horizontal black bar indicates the distribution of methylation values in the nontumorous gastric samples. (B) Regression analysis between the mRNA level and DNAm level of the six DNAm-driven DEGs. The vertical axis and the horizontal axis denote the mRNA level and DNAm level, respectively. (C) DNA methylation of the six DNAm-driven DEGs. (D) mRNA expression of the six DNAm-driven DEGs.
CNV, Mutation Characteristics and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment
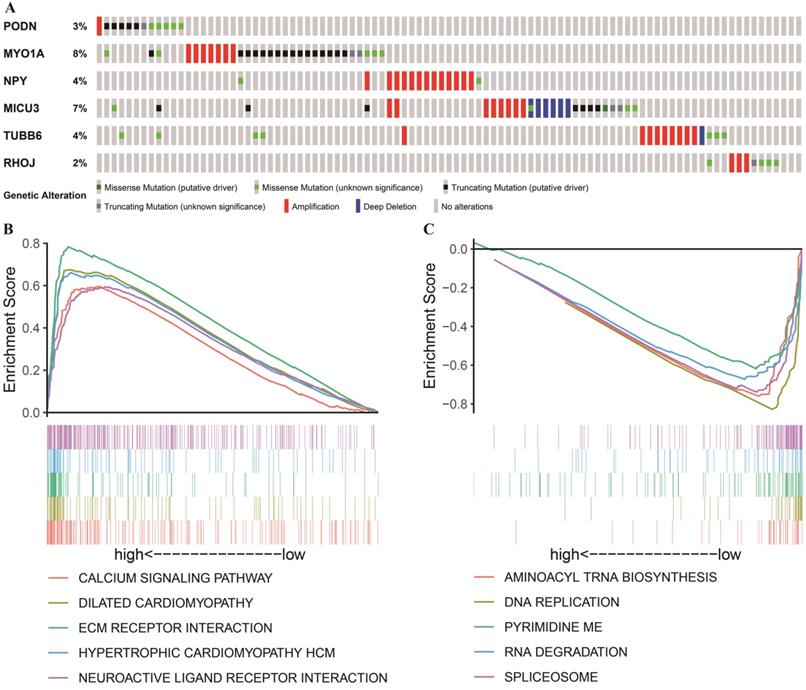
Apart from being affected by methylation, the selected DNAm-driven DEGs (PODN, MYO1A, NPY, MICU3, TUBB6 and RHOJ) are also affected concurrently by gene amplification, deletion and mutations (Figure 7A). By utilizing the GDC TCGA Stomach Cancer (STAD) database, we observed that the genetic alteration percentages of six genes ranged from 2%-8%, which had little contribution to mRNA expression. For example, there was no correlation between CNV and the mRNA expression of each gene after regression analysis (Figure S3). When the existing alterations that increase mRNA levels were ignored, the five hypermethylated genes still exhibited a relatively downward trend of mRNA levels compared to their expression in adjacent nontumorous gastric tissues. This finding indicates that DNAm plays a more critical role in GC in this study. However, the role of these alterations in determining the MYO1A mRNA level is difficult to estimate at present, and a high proportion of putative truncating mutations may attenuate its gene expression. To further characterize the potential signaling pathways involved in the influences on the risk score model, GSEA was performed to enrich the KEGG pathways in the two groups. A false discovery rate (FDR) less than 0.05 and an absolute value of the enrichment score (ES) greater than 0.5 were defined as the cutoff criteria. As shown in Figure 7B, the top five signaling pathways in the high-risk score group (risk score > 0.314) were “CALCIUM SIGNALING PATHWAY”, “DILATED CARDIOMYOPATHY”, “ECM RECEPTOR INTERACTION”, “HYPERTROPHIC CARDIOMYOPATHY HCM” and “NEUROACTIVE LIGAND RECEPTOR INTERACTION” while the top five signaling pathways in the low-risk score group (risk score < 0.314, Figure 7C) were “AMINOACYL TRNA BIOSYNTHESIS”, “DNA REPLICATION”, “PYRIMIDINE METABOLISM”, “RNA DEGRADATION” and “SPLICEOSOME”. The vast majority of the above signaling pathways are reported to be involved in tumor progression, laying the foundation for further exploring the molecular mechanisms of GC.
Genetic alterations and mutation characteristics of DNAm-driven DEGs and GSEA. (A) Genetic alterations of DNAm-driven DEGs in GC samples. The rows and columns indicate the genes and tumor samples, respectively. (B, C) Enrichment plots of the top five KEGG pathways in the high-risk score (Figure 7B) and low-risk score (Figure 7C) groups in GC.
Discussion
The lack of specific and sensitive biomarkers for predicting prognosis remains an urgent problem to be solved in the management of GC patients. Some prognostic models for GC patients have been reported. Recently, an ISGC classifier based on the ImmunoScore (IS) signature [25] was proposed to effectively predict patients with GC who would benefit from adjuvant chemotherapy. In tumor cells, alterations in the genome and epigenome can always be detected and have proven to be associated with certain tumor characteristics, such as oncogenic transformation and cellular proliferation [26]. Considering that genome methylation is highly specific, herein, we first developed and validated a prognostic risk score model based on the DNAm signature and then combined this model with age and number of positive lymph nodes to construct an OS nomogram for predicting the prognosis of individual patients with GC. Of note, this model also has the potential to be widely applied after external validation and performed better than similar models reported previously [23, 24].
Aberrant methylation changes occur frequently in tumors. Among these deregulated DNAm-driven genes, some may promote malignant transformation via the overexpression of oncogenes or the knockdown of TSGs, which make up a new balance in the tumor microenvironment and have the potential to be predictive biomarkers for prognosis. With the advance of methylation sequencing, epigenetic changes are easy to identify with high sequencing depth and accuracy. Therefore, we utilized a model-based instrument (MethylMix)[19] to identify DNAm-driven genes with aberrant methylation and linked these data to RNA-seq data that reflected gene expression. This integrative analysis has been performed in another cancer type[27]. It is worth mentioning that in our study, seventy-one preliminarily screened DNAm-driven genes were mainly enriched in gene expression-related signaling pathways, such as “RNA polymerase II transcription”, “generic transcription pathway”, “gene expression (transcription)”, and “regulation of PLK1 activity at G2/M transition”, which suggests that methylation changes in GC regulate gene expression. In our risk model, the expression of five genes (PODN, NPY, MICU3, TUBB6 and RHOJ) was decreased in tumor tissues, and the greater the degree of downregulation was, the better the prognosis was, indicating that the hypermethylation of these genes may play a protective role in GC patients. In detail, the downregulation of DNAm-driven genes is a compensatory response to protect the organism. A high degree of downregulation is achieved through hypermethylation; if downregulation is insufficient under this condition, the stronger the protection will be, and the worse the prognosis will be. MYO1A may be a typical TSG because its hypomethylation always predicts a good prognosis. It is easy to understand that the downregulation of methylation levels in oncogenes and the upregulation of methylation levels in TSGs contribute to tumorigenesis. To clarify their potential mechanisms in affecting OS, GSEA was conducted to identify the relevant KEGG pathways in the high-risk and low-risk groups. Risk factors triggering the six dysregulated genes are enriched in several pathways, such as “ECM RECEPTOR INTERACTION”, “NEUROACTIVE LIGAND RECEPTOR INTERACTION” and “HYPERTROPHIC CARDIOMYOPATHY HCM”. Previous studies have found that some genes enriched in “dilated cardiomyopathy” and “hypertrophic cardiomyopathy HCM” signaling pathways closely related to multiple cancer types. For instance, TGFβ1 stimulates THBS1 expression in oral squamous cell carcinoma (OSCC) cells. THBS1 promotes the expression of matrix metalloproteinases (MMPs) partly through integrin signaling, thereby favoring OSCC invasion [28]. Another gene, TGFβ3, directly induces the upregulation of stromal POSTN expression. Hence, the growth, migration and invasion of head and neck cancer cells are accelerated [29]. A comparative genomic analysis of oral versus laryngeal and pharyngeal cancer also found that LAMA2 (TCGA: 5% vs 19%) mutations are enriched in laryngeal and pharyngeal squamous cell carcinoma (L/P-SCC)[30], while other factors are enriched in pathways such as “DNA REPLICATION” and “PYRIMIDINE METABOLISM”. Overexpression of the key metabolite cytidine related gene ENTPD8, which is enriched in the “PYRIMIDINE METABOLISM” pathway, was reported to promote cell apoptosis and inhibit proliferation by promoting CTP metabolization into cytidine in pancreatic cancer tissue (PCT)[31].
Among the six DNAm-driven DEGs, podocan, a protein of the small leucine-rich proteoglycan (SLRP) family encoded by the PODN gene, was found to be a potent regulator of the cellular phenotype in the extracellular matrix (ECM). ECM molecules are highly effective modulators of cell functions, such as migration and proliferation [32]. Given the inhibitory effect of high podocan levels on smooth muscle cell (SMC) proliferation [33], PODN may also be involved in cell proliferation regulation, which requires further experimental validation. Ras homolog family member J (RHOJ), a member of the Rho GTPase family, acts as a molecular switch by regulating cell functions, such as migration and proliferation, correlating well with increased cell motility and invasiveness [34]. This finding is consistent with our results, and the specific regulatory mechanism by which this gene impacts GC is still unknown. As a sympathetic neurotransmitter highly relevant to tumor biology, neuropeptide Y (NPY) is released from activated peripheral sympathetic neurons under chronic stress or hypoxia. The release of NPY can regulate many bioprocesses (e.g., stimulate cell proliferation, migration and survival, and regulate cell differentiation) [35]. Aberrant NPY methylation is involved in tumorigenesis [36]. Mitochondrial calcium uptake family member 3 (MICU3), a paralog of MICU1, which likely arose by gene duplication and exhibits high expression levels in the brain, encodes an EF-hand-containing protein that functions by interacting with MICU1, forming a dimer and enhancing MCU-dependent mitochondrial Ca2+ uptake [37]. Mitochondrial Ca2+ regulates various cellular events, including tumorigenesis. Abnormal fatty acids, such as cis-9, 10-methyl-octadecanoic acid (MOA), caused by Helicobacter pylori (HP) infection, serve as activators of protein kinase C (PKC) in a Ca2+-dependent manner. Interestingly, PKC has been implicated in regulating the proliferation activity of gastric epithelial cells and the malignant transformation process, associated with the increased proliferation of gastric epithelial cells and linked with GC [38]. In this study, the MICU3 gene showed a hypermethylated state and relatively low expression in GC, which may disturb the mitochondrial Ca2+ uptake function, thus playing a role in regulating the cellular and molecular functions of GC cells. Tubulin beta 6 class V (TUBB6) was recognized as a potential mutation hot spot in human colorectal cancers accompanied by microsatellite instability [39] and serves as a biomarker for predicting GC peritoneal metastasis [40]. Brush border protein myosin Ia (MYO1A), which plays an essential role in polarization and differentiation in colon cancer, is highly expressed in normal gastric epithelial cells, suppressing intestinal tumors. In colorectal tumors, epigenetic regulation often inactivates its expression. Despite relatively sparse CpG islands, promoter methylation has been observed in several colon cancer cell lines and primary colorectal tumors [41]. Existing studies have also shown that MYO1A can suppress tumorous changes in the normal gastric epithelium, indicating that MYO1A may serve as an important protective factor [42]. Methylation was negatively correlated with MYO1A mRNA expression in our study, and MYO1A plays an inhibitory role in the progression of GC. In addition, MYO1A mRNA levels were simultaneously affected by an 8% mutation rate, which may contribute to nonsense expression in GC. Indeed, MYO1A is among the most frequently mutated genes in some types of GC.
To the best of our knowledge, the six-gene predictive model has not been previously published, and it will help to identify new prognostic biomarkers in GC from a clinical perspective. Moreover, our signature based on specific genes is easy to test routinely, considering its cost-effectiveness. There are also some shortcomings to this study. To detect GC outcomes, it was recently proposed to increase the amount of research on clinical biomarkers, such as epiregulin [43], the albumin-to-globulin ratio (AGR) and the lymphocyte-to-monocyte ratio (LMR) [44] in patients with GC. DNAm has expanded the field of cancer research, attracting an increasing number of scientists. However, although a favorable performance in external validation indicates its potential, it is too early to conclude that our two-dimensional model (epigenetic and transcriptional signatures) is preferable to traditional examinations, such as medical imaging evaluation, in directly predicting patient outcomes. Therefore, further experimental verification is required. In addition, although the nomogram incorporates age, number of positive lymph nodes and risk level to successfully predict the OS of GC patients, the clinical characteristics were considered insufficient due to limited information in the study cohorts. In the future, it will be necessary to construct a better prognostic nomogram derived from more centers with complete clinical information and sequencing data.
In summary, a risk score prediction model comprising six DNAm-driven DEGs was identified and validated, and this model combined with other clinical factors could produce a good prognostic nomogram for GC patients. Our findings support the assumption that genes tightly controlled by DNAm are likely related to tumor outcomes. Importantly, only six genes were used to build the prognostic model. Measuring the expression levels of these six DNAm-driven genes can provide a cost-effective and accurate prediction for the prognosis of GC in clinical practice.
In conclusion, our study established a nomogram that combined the DNAm signature, age and number of positive lymph nodes and is cost effective in clinical practice, advancing the individualized prediction of OS in GC patients with high sensitivity and specificity.
Abbreviations
AGR: albumin-to-globulin ratio; C-index: Harrell's concordance index; CNV: copy number variation; DAVID: Database for Annotation, Visualization and Integrated Discovery; DCA: decision curve analysis; DEG: differentially expressed gene; DIRC1: disrupted in renal cancer 1; DNAm: DNA methylation; DNMT3A: De novo methyltransferase; ECM: extracellular matrix; ES: enrichment score; FDR: false discovery rate; FGFR: fibroblast growth factor receptor; GC: gastric cancer; GDC: Genomic Data Commons; GEO: Gene Expression Omnibus; GSEA: Gene Set Enrichment Analysis; HP: Helicobacter pylori; IS: ImmunoScore; K-M: Kaplan-Meier; LASSO: least absolute shrinkage and selector operation; LMR: lymphocyte-to-monocyte ratio; MICU3: mitochondrial calcium uptake family member 3; MMP: metalloproteinase; MOA: methyl-octadecanoic acid; MYO1A: myosin Ia; NPY: neuropeptide Y; OS: overall survival; OSCC: oral squamous cell carcinoma; PCT: pancreatic cancer tissue; PKC: protein kinase C; PTX: paclitaxel; RHOJ: Ras homolog family member J; ROC: receiver operating characteristic; SLRP: small leucine-rich proteoglycan; SMC: smooth muscle cell; TCGA: The Cancer Genome Atlas; TSG: tumor suppressor gene; TUBB6: Tubulin beta 6; XCI: X-chromosome inactivation.
Supplementary Material
Supplementary figures and table legends.
Supplementary table 1.
Supplementary table 2.
Supplementary table 3.
Supplementary table 4.
Supplementary table 5.
Acknowledgements
We thank Zhisong Liu and Fei Miao for assistance with the statistical methods.
Funding
This work was supported by the International Science and Technology Cooperation Projects (2016YFE0107100), the Capital Special Research Project for Health Development (2014-2-4012), the Beijing Natural Science Foundation (L172055 and 7192158), the National Ten-thousand Talent Program, the Fundamental Research Funds for the Central Universities (3332018032), the CAMS Innovation Fund for Medical Science (CIFMS) (2017-I2M-4-003 and 2018-I2M-3-001), the Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 13th Five-year Plan (IDHT20190510), the Ministry of Science and Technology of People's Republic of China (2014CB910100), and the National Natural Science Foundation of China (81171899 and 81372230).
Author Contributions
YB, CLW and YXZ designed the research scheme and performed the bioinformatics analyses. JYL and SH downloaded and organized the clinical, methylation and gene expression data in GC. FCX and YMZ performed the statistical analyses. YB and CLW wrote the manuscript. YTT, XW and HTZ critically revised the article for essential intellectual content and provided administrative support. All authors read and approved the final version of the manuscript. All authors reviewed and revised the manuscript. XW and HTZ were the guarantors for this study.
Data Availability
The data analyzed in this study can be downloaded from the TCGA and GEO.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
3. Ajani JA, D'Amico TA, Almhanna K, Bentrem DJ, Chao J, Das P. et al. Gastric Cancer, Version 3.2016, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network: JNCCN. 2016;14:1286-312
4. Japanese Gastric Cancer A. Japanese gastric cancer treatment guidelines 2014 (ver. 4). Gastric cancer: official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 2017;20:1-19
5. Smyth EC, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D. et al. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology. 2016;27:v38-v49
6. Panarese I, De Vita F, Ronchi A, Romano M, Alfano R, Di Martino N. et al. Predictive biomarkers along gastric cancer pathogenetic pathways. Expert review of anticancer therapy. 2017;17:417-25
7. Ishiwata T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Frontiers in bioscience (Landmark edition). 2018;23:626-39
8. Li Z, Yang AJ, Wei FM, Zhao XH, Shao ZY. Significant association of DIRC1 overexpression with tumor progression and poor prognosis in gastric cancer. European review for medical and pharmacological sciences. 2018;22:8682-9
9. Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6-21
10. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247-57
11. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nature genetics. 1998;19:219-20
12. Cheng Y, Yan Z, Liu Y, Liang C, Xia H, Feng J. et al. Analysis of DNA methylation patterns associated with the gastric cancer genome. Oncology letters. 2014;7:1021-6
13. Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J. et al. Senescent cells harbour features of the cancer epigenome. Nature cell biology. 2013;15:1495-506
14. Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y. et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2011;60:499-508
15. Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR. et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63:317-25
16. Nielsen SN, Grell K, Nersting J, Abrahamsson J, Lund B, Kanerva J. et al. DNA-thioguanine nucleotide concentration and relapse-free survival during maintenance therapy of childhood acute lymphoblastic leukaemia (NOPHO ALL2008): a prospective substudy of a phase 3 trial. The Lancet Oncology. 2017;18:515-24
17. Weller M, Tabatabai G, Kastner B, Felsberg J, Steinbach JP, Wick A. et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2057-64
18. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106
19. Gevaert O. MethylMix: an R package for identifying DNA methylation-driven genes. Bioinformatics. 2015;31:1839-41
20. Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:7252-9
21. Kamarudin AN, Cox T, Kolamunnage-Dona R. Time-dependent ROC curve analysis in medical research: current methods and applications. BMC Med Res Methodol. 2017;17:53
22. Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Med Decis Making. 2006;26:565-74
23. Shigeyasu K, Nagasaka T, Mori Y, Yokomichi N, Kawai T, Fuji T. et al. Clinical Significance of MLH1 Methylation and CpG Island Methylator Phenotype as Prognostic Markers in Patients with Gastric Cancer. PloS one. 2015;10:e0130409
24. Deng J, Liang H, Ying G, Li H, Xie X, Yu J. et al. Methylation of ras association domain protein 10 (RASSF10) promoter negative association with the survival of gastric cancer. American journal of cancer research. 2014;4:916-23
25. Jiang Y, Zhang Q, Hu Y, Li T, Yu J, Zhao L. et al. ImmunoScore Signature: A Prognostic and Predictive Tool in Gastric Cancer. Annals of surgery. 2018;267:504-13
26. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38-55
27. Gao C, Zhuang J, Li H, Liu C, Zhou C, Liu L. et al. Exploration of methylation-driven genes for monitoring and prognosis of patients with lung adenocarcinoma. Cancer cell international. 2018;18:194
28. Pal SK, Nguyen CT, Morita KI, Miki Y, Kayamori K, Yamaguchi A. et al. THBS1 is induced by TGFB1 in the cancer stroma and promotes invasion of oral squamous cell carcinoma. J Oral Pathol Med. 2016;45:730-9
29. Qin X, Yan M, Zhang J, Wang X, Shen Z, Lv Z. et al. TGFbeta3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Scientific reports. 2016;6:20587
30. Vossen DM, Verhagen CVM, Verheij M, Wessels LFA, Vens C, van den Brekel MWM. Comparative genomic analysis of oral versus laryngeal and pharyngeal cancer. Oral Oncol. 2018;81:35-44
31. An Y, Cai H, Yang Y, Zhang Y, Liu S, Wu X. et al. Identification of ENTPD8 and cytidine in pancreatic cancer by metabolomic and transcriptomic conjoint analysis. Cancer Sci. 2018;109:2811-21
32. Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. Journal of cell science. 2008;121:255-64
33. Hutter R, Huang L, Speidl WS, Giannarelli C, Trubin P, Bauriedel G. et al. Novel small leucine-rich repeat protein podocan is a negative regulator of migration and proliferation of smooth muscle cells, modulates neointima formation, and is expressed in human atheroma. Circulation. 2013;128:2351-63
34. Kim C, Yang H, Park I, Chon HJ, Kim JH, Kwon WS. et al. Rho GTPase RhoJ is Associated with Gastric Cancer Progression and Metastasis. Journal of Cancer. 2016;7:1550-6
35. Son MY, Kim MJ, Yu K, Koo DB, Cho YS. Involvement of neuropeptide Y and its Y1 and Y5 receptors in maintaining self-renewal and proliferation of human embryonic stem cells. Journal of cellular and molecular medicine. 2011;15:152-65
36. Mendoza-Perez J, Gu J, Herrera LA, Tannir NM, Zhang S, Matin S. et al. Prognostic significance of promoter CpG island methylation of obesity-related genes in patients with nonmetastatic renal cell carcinoma. Cancer. 2017;123:3617-27
37. Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell death and differentiation. 2019;26:179-95
38. Beil W, Obst B, Wagner S, Sewing KF. The Helicobacter pylori fatty acid cis-9,10-methyleneoctadecanoic acid stimulates protein kinase C and increases DNA synthesis of gastric HM02 cells. British journal of cancer. 1998;77:1852-6
39. Gylfe AE, Kondelin J, Turunen M, Ristolainen H, Katainen R, Pitkanen E. et al. Identification of candidate oncogenes in human colorectal cancers with microsatellite instability. Gastroenterology. 2013;145:540-3 e22
40. Zhang J, Huang JY, Chen YN, Yuan F, Zhang H, Yan FH. et al. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Scientific reports. 2015;5:13750
41. Mazzolini R, Dopeso H, Mateo-Lozano S, Chang W, Rodrigues P, Bazzocco S. et al. Brush border myosin Ia has tumor suppressor activity in the intestine. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1530-5
42. Mazzolini R, Rodrigues P, Bazzocco S, Dopeso H, Ferreira AM, Mateo-Lozano S. et al. Brush border myosin Ia inactivation in gastric but not endometrial tumors. International journal of cancer. 2013;132:1790-9
43. Xia Q, Zhou Y, Yong H, Wang X, Zhao W, Ding G. et al. Elevated epiregulin expression predicts poor prognosis in gastric cancer. Pathology, research and practice. 2019;215:873-9
44. Ma M, Wang J, Hu Y, Weng M, Liu X, Wang Y. Prognostic Value of Inflammatory Biomarkers in Gastric Cancer Patients and the Construction of a Predictive Model. Digestive surgery. 2018:1-10
Author contact
![]() Corresponding authors: Xi Wang: xiwangedu.cn or Haitao Zhao: zhaohtcn
Corresponding authors: Xi Wang: xiwangedu.cn or Haitao Zhao: zhaohtcn